Cell lysis and DNA extraction of gram-positive and gram-negative bacteria from whole blood in a disposable microfluidic chip
Madhumita
Mahalanabis
a,
Hussam
Al-Muayad
a,
M. Dominika
Kulinski
b,
Dave
Altman
b and
Catherine M.
Klapperich
ac
aDepartment of Biomedical Engineering, Boston University, Boston, MA 02115, USA
bManufacturing Engineering, Boston University, Boston, MA 02115, USA
cMechanical Engineering, Boston University, Boston, MA 02115, USA
First published on 29th June 2009
Abstract
Sepsis caused by gram positive and gram negative bacteria is the leading cause of death in noncoronary ICUs and the tenth leading cause of death in the United States. We have developed a microfluidic sample preparation platform for rapid on-chip detection of infectious organisms for point-of-care diagnostics. The microfluidic chips are made of a robust thermoplastic and can be easily multiplexed for high throughput applications. Bacteria are lysed on-chip via hybrid chemical/mechanical method. Once lysed, the bacterial DNA is isolated using a microscale silica bead/polymer composite solid-phase-extraction (SPE) column. Lysis was confirmed using off-chip real time PCR. We isolated and detected both gram-negative (Escherichia coli) and gram-positive (Bacillussubtilis and Enterococcus faecalis) bacterial genomic DNA from microliter scale spiked whole human blood samples. The system performs better for gram-negative bacteria than it does for gram-positive bacteria, with limits of detection at 102CFU/ml and 103–104CFU/ml, respectively. Total extraction times are less than one hour and can be further decreased by altering the channel geometry and pumping configuration.
Introduction
Sepsis is the result of the progression of a systemic inflammatory response syndrome (SIRS) caused by an infection or presumed infection.1 It is a cascade of events starting with an infection and ending in dysfunction of the microcirculation that can result in organ failure and/or death. In almost all cases, infection by the causative agent spreads to the blood.2Sepsis was the 10th leading cause of death in 2004 in the United States3 which increased from the 13th leading cause in 1993–1994.3–5 It is estimated that annually, 751,000 cases of severe sepsis occur in the United States and the mortality rate ranges from 28%–50% depending on country and institution.6,7 Each sepsis case has an average cost of $22,100, amounting to nearly $17 billion nationally.8 The expectation is that cases of sepsis will continue to rise due to an aging American population, increase in antimicrobial resistance, increased immunosuppressed populations, increased use of invasive catheters and prosthetic materials.9
The leading causes of sepsis include the common gram-negative bacteria Escherichia coli, Klebsiella species, Pseudomonas species, Enterobacter species, and Haemophilus influenzae; while the most common gram-positive bacteria include Staphylococcus aureus (and other Staphylococcus species), Streptococcus pneumoniae (and other Staphylococcus species), and the Enterococcus species.2,5 Current gold standard diagnostics include a broad spectrum of blood testing, other bodily fluid testing and imaging, which are often not specific for the infectious organism. For specific diagnoses, the blood is cultured for 24–72 hours or more, during which time, patients are treated with broad range therapies out of necessity, resulting in higher health care costs and increasing antibiotic resistance of pathogenic bacteria.10
The availability of a rapid microfluidic test for bacterial infection of whole blood would greatly accelerate this process and improve the sensitivity of detection of the bacterial pathogen. The main challenge in miniaturizing the preparation of blood samples for molecular detection assays in a microfluidic platform is the considerable difficulty encountered in lysing gram-positive bacteria to liberate the DNA targets for diagnostic molecular assays. Of the two main types of gram-positive and gram-negative bacterial cell walls, the gram-positive cell wall provides greater strength and rigidity while maintaining elasticity and flexibility to counter intracellular turgor pressure. Both cell types contain a peptidoglycan layer, a covalent multilayered structure of rigid glycan chains crosslinked by flexible peptide bridges, resulting in a tough mesh that maintains cell shape and prevents osmotic lysis. The glycan chains are composed of two amino sugars N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) connected by β-1,4 glycosidic bonds with crosslinking peptides between the glycan layers.11 Though both gram-positive and gram-negative cell walls have peptidoglycan, the quantity and thickness, the length distribution, and the degree of crosslinking in gram-positive cells is more extensive than in gram-negative cells.11,12 The lysis methods for gram-positive bacteria must overcome these factors that result in a strong peptidoglycan barrier in order to cause disruption of the cell wall.
Several techniques for disrupting cell walls can be carried out at the bench either by EDTA-lysozyme lysis, sonication, thermal incubation, glass-bead grinding or with a French pressure cell.13,14 The mechanical lysis techniques require large-scale specialized laboratory equipment and are time-consuming.14,15 The gold standard non-mechanical lysis method is enzymatic lysis by muramidases which are enzymes such as lysozyme and mutanolysin that hydrolyze the β-1,4 glycoside linkages between GlcNAc and MurNAc of the glycan backbone.16 Several groups have demonstrated methods for on-chip lysis. A previous mechanical microchip technique includes a nanoscale barb design which uses pressure driven flow with sharp nanostructures to mechanically lyse human cells.17 Other methods include a microfluidic incubation chamber where bacteria were incubated with chemical lysing agents (chaotropic buffer, detergents, lysozyme)18 and a chamber coupled with heating elements to thermally lyse bacteria.19 These and other chip-based techniques are reviewed by Bhattacharyya and Klapperich.20
We have previously demonstrated the extraction of nucleic acids from mammalian cells and gram-negative bacteria in crude cell lysates,20,21 hematuric urine,22 and viral RNA from mammalian tissue culture supernatant23 using a thermoplastic microfluidic platform. Here we extend this previous work to the detection of simulated sepsis samples with common, difficult to lyse gram-positive bacteria in a complex biological sample of human whole blood. Our approach is to mechanically shear bacterial cell walls by passage through a porous polymer monolith assisted with detergent lytic conditions. We report the fabrication and characterization of a microfluidic sample preparation module capable of isolating PCR quality bacterial DNA from both gram-negative Escherichia coli and gram-positive Bacillus subtilis and Enterococcus faecalis inoculated in a sample of human whole blood. A small (100 µL) volume of human whole blood is processed on-chip by lysing the bacterial cells, followed by isolating, cleaning and concentrating the DNA from the lysate. The concentrated DNA eluted from the chip is pure enough for PCR diagnostic assays and other downstream applications.
Materials and methods
Materials
Cyclic polyolefin (Zeonor 690R) was obtained as a gift from Zeon Chemicals Inc. (Louisville, KY). Butyl methacrylate (99%, BuMA), ethylene glycol dimethacrylate (98%, EGDMA), methyl methacrylate (99%, MMA), 1-dodecanol (98%), cyclohexanol (99%), benzophenone (99%), and 2,2-dimethoxy-2-phenylacetophenone (99%, DMPAP), 6M guanidine thiocyanate (GuSCN), sodium dodecyl sulfate (SDS), hen egg white lysozyme, and mutanolysin were purchased from Sigma-Aldrich (St. Louis, MO). Nitric and sulfuric acids were purchased from Fisher Scientific (Fairlawn, NJ). A Qiagen DNeasy Blood and Tissue kit, proteinase K and guanidine thiocyanate (GuSCN) containing lysis buffer (buffer RLT) were purchased from Qiagen Inc. (Valencia, CA). Luria-Bertani broth and agar, brain-heart-infusion broth, and tryptic soy agar were purchased from DIFCO (Franklin Lakes, NJ). PicoGreen was purchased from Invitrogen (Carlsbad, CA). SYBR®Green PCR master mix (2X) was purchased from Applied Biosystems (Foster City, CA). PCR primers were ordered from Integrated DNA Technologies (Coralville, IA). Nalgene 4 mm syringe 0.22 µm filters, Excel 1 cc disposable syringes, and PCR grade nuclease free water were purchased from Fisher Scientific (Fairlawn, NJ). Silica microspheres (0.7 µm) were purchased from Polysciences, Inc. (Warrington, PA). Polyetheretherketone (PEEK) capillaries of 360 µm-i.d. and NanoPort assemblies for device-based fluidic connections were purchased from Upchurch Scientific (Oak Harbor, WA).Microchip fabrication
The microchannels were formed by hot embossing with a nickel-cobalt electroplated mold (NiCoForm, Inc., Rochester, NY) made from a silicon master as detailed elsewhere.22,24 The channels were 2 cm in length, 400 µm wide and 100 µm deep. Hot-embossing was performed with a hot press (Heated Press 4386, Carver, Wabash, IN) at 176 °C (30 degrees above the glass transition temperature (Tg) of Zeonex 690R) at a pressure of 500 psi for 4 minutes. Wells were drilled at the ends of the hot-embossed channels of 1.5 mm diameter and the embossed channels were sealed with another Zeonex wafer of the same dimensions by thermally bonding at the Tg. Nanoports (Upchurch Scientific, Oak Harbor, WA) were epoxied to the chip at the location of the wells to provide secure attachment of PEEK tubing attached to a syringe on a syringe pump (KDS 100, KD Scientific, Holliston, MA).Monolith formation
The hot-embossed and sealed channels were surface modified with MMA and 3% benzophenone to allow for covalent attachment of the monolith. After surface-modification, the monolith was formed using in situphotopolymerization of monomers in the presence of porogenic solvents. To enable the solid phase extraction of nucleic acids, the monoliths were impregnated with silica particles. Here, a BuMA and EGDMA monomer system with porogenic solvents of 1-dodecanol and cyclohexanol were used.21 The monolith solution consisted of BuMA (15 wt %), EGDMA (10 wt %), 1-dodecanol (52.5 wt %), and cyclohexanol (22.5 wt %) with 1.13% w/v of DMPAP added as an initiator. The silica particles were purchased in aqueous solution and a 100µL of silica-water suspension was used for 100 µL of monolith solution. The silica particles were spun down and resuspended in the monolith solution prior to photopolymerizarion. Detailed protocols are available in previous work.21–23Bacterial strains and culture
We used neomycin resistant E. coli DH5α as our gram-negative test organism (transfected with neomycin resistance on plasmid pEGFP-N2; GenBank Accession U57608, a gift from Dr. Satish Singh, Boston University Medical Center), non-chain forming B. subtilis strain 168 (a gift from Shigeki Moriya, Institute for the Biotechnology of Infectious Diseases, Sydney, Australia), and E. faecalis (ATCC catalog #29212, Manassas, VA) as model gram-positive organisms. Device capability was demonstrated with E. coliK-12 MG1655 and B. subtilis in 0.85% NaCl (to prevent chemical lysis) in 3M GuSCN buffer. PCR amplification of the eluted DNA was assessed with E. coli DH5α, B. subtilis, and E. faecalis inoculation of human whole blood. E. coli and B. subtilis were cultured in 3 ml of Luria Bertani (LB) broth at 37 °C for 16–18 hrs at 250 rpm. E. faecalis was grown in 3 ml of brain-heart-infusion (BHI) media at 37 °C for 16–18 hrs at 250 rpm. Bacterial cultures were grown until mid-late exponential phase in liquid media. To quantify the concentration of the bacterial liquid cultures, viable spread plate counts were determined by serial dilution plating on solid agar media (LB agar for E. coli and B. subtilis or tryptic soy agar for E. faecalis) and incubated overnight at 37 °C. E. coli, B. subtilis, and E. faecalis counts were on average 1.7 × 109, 2 × 108, and 2 × 109 colony forming units (CFUs) per milliliter of the overnight culture.Lysis controls
The Qiagen DNeasy Blood and Tissue Kit was used as the positive control for bacterial DNA extraction. As listed in the manufacturer's protocol, 1 ml of the bacterial overnight culture at the desired CFU/ml concentration was pelleted at 8000 RPM for 10 min. For gram-negative E. coli, the supernatant was aspirated and the cell pellet was resuspended in 180 µl Qiagen buffer ATL and processed without enzymatic lysis. Gram-positive B. subtilis and E. faecaliscell pellets were resuspended in 180 µl of an EDTA-enzyme-detergent solution and incubated at an elevated temperature of 37 °C for 90 min. The enzymes used in conjunction with the kit for positive controls were 40 mg/ml lysozyme for B. subtilis and 250 units/ml mutanolysin for E. faecalis. For both gram-negative and gram-positive bacteria, this step is followed by adding a chaotropic buffer with additional detergent and 0.8 mg/ml protease K incubation at another elevated temperature (56 °C) for 30–90 min. Bacterial suspensions for testing with the Qiagen kit were 1 ml volumes and bacterial DNA was eluted in two fractions of – 150 µl each or 300 µl total. For the E. coli cultures, the Qiagen protocol was followed with the minimum 30 min incubation at 56 °C for protease K digestion. For the more difficult to lyse B. subtilis and E. faecalis cultures, the Qiagen protocol incubation times were tripled to 90 min at both enzyme incubations (3 hours total) to increase DNA yield (as recommended by the manufacturer).Bacterial sample preparation, chip lysis, DNA isolation and RT-PCR amplification
Bacteria were cultured, diluted to 101–105CFU/ml, and pelleted as described earlier. Liquid culture media was aspirated and the cell pellets were resuspended in 100µl human whole blood (Innovative Research, Novi, MI). The bacteria-blood suspension was mixed with 150 µl of a 20 mM Tris-HCl, 2 mM EDTA, pH 8.0 buffer containing sodium dodecyl sulfate (SDS) and TritonX-100. In order to streamline the lysis and DNA isolation techniques, the bacterial suspension was finally mixed with 200 µl chaotropic buffer (3 M GuSCN) with 0.8 mg/ml proteinase K. For E. coli the final concentration of detergent was only 0.01% SDS and 1.2% TritonX-100. This was increased to 1% SDS with 2.4% TritonX-100 for the gram-positive bacteria. GuSCN is necessary for the preferential binding of the DNA to the silica particles in the SPE column.25 Using the GuSCN buffer as the initial suspension eliminates intermediate steps between lysis and nucleic acid isolation. The final volume of 450µl was loaded into a 1cc syringe and connected to the SPE column via PEEK tubing and NanoPort assemblies. A photograph of the experimental set up is included in Fig. 1. | ||
| Fig. 1 Photograph showing the pump, syringes and microchip system for nucleic acid extraction. The chip (shown in front) is enclosed in a fixture for convenient changes of the input fluid. | ||
After the sample completely passed through the SPE column, the column was washed with 300 µl 70% ethanol and the DNA was precipitated with 300 µl of 100% ethanol. Residual ethanol was removed by air drying the µSPE column for 10 min. The DNA was eluted with nuclease free PCR grade water in two fractions of 75 µl each. The syringe pump flow rate for all steps was 800–1000 µl/hr. Eluted samples were amplified using real-time PCR and compared to the Qiagen kit isolated samples. Additional negative control channels were run for each extraction experiment with sterile LB broth in blood mixed as above with GuSCN and detergent buffers.
For E. coli detection, we used published primers for the single copy chromosomal gene, d-1-deoxyxylulose-5-phosphate synthase (dxs).26 The PCR primers were designed to detect the 16 S rRNA in B. subtilis and the groES/EL chaperone gene in E. faecalis27 (Table 1). For each PCR reaction, 1X SYBR®Green PCR master mix was combined with 50–100 ng eluted DNA and 100 nM of each forward and reverse primer in a total volume of 25 µL. If the DNA yield was less than 50 ng, the maximum allowable volume of 12 µL of DNA was added per reaction. The PCR running conditions were 95 °C for 10 min followed by 45 cycles of 95 °C for 15 sec and 60 °C for 1 min. The reactions were run in 96 well plates on an Applied Biosystems 7300 real-time PCR machine. Positive control PCR reactions included on each PCR plate were DNA isolated with the Qiagen kit from 1 ml of undiluted overnight bacterial culture following the manufacturer's protocols for gram-negative and gram-positive bacteria, including pre-treatment with enzymes for up to 3 hours total as described above for gram-positive bacteria.
| Bacteria | Primer Name | Primer Sequence (5′ → 3′) | Target DNA |
|---|---|---|---|
| E. coli DH5α | Dxs F (forward) | CGAGAAACTGGCGATCCTTA | Dxs |
| Dxs R (reverse) | CTTCATCAAGCGGTTTCACA | ||
| B. subtilis 168 | L-bsub (forward) | CCTACGGGAGGCAGCAG | 16S rRNA |
| R-bsub (reverse) | CCAGTTTCCAATGACCCTCCCC | ||
| E. faecalis (ATCC 29212) | EfesF (forward) | GTGTTAAAACCATTAGGCGAT | GroES/EL chaperone |
| EfgsR (reverse) | AAGCCTTCACGAACAACAATGG |
Results
We examined the performance of the µSPE lysis and extraction column by assaying the eluent for PCR quality DNA from both gram-negative and gram-positive bacteria in the presence of human whole blood. For all bacterial strains, we compared the PCR amplification of DNA from 102–105CFU/ml (and up to 106CFU/ml for E. faecalis) from three µSPE channels per bacterial concentration. In addition, we included mock negative control channels loaded with sterile LB broth instead of bacterial cells. Bacterial samples processed with a commercial Qiagen DNeasy Blood and Tissue Kit, according to recommended bench-top protocols, served as positive controls. We collected two serial eluted fractions of 75 µl each and tested the fractions separately by RT-PCR.Only the fractions that resulted in amplification of the target DNA and thus gave a detectable CT value are plotted in Fig. 2. The negative control channels and the PCR water (no template) reactions did not amplify. For all three bacterial samples, gram-negative and gram-positive, and for the majority of the concentrations tested, the second fraction (F2) performed better than the first fraction (F1) with either lower CT values (Fig. 2A–C) or a detectable CT value in the case of the first fraction being undetectable for amplification (Fig. 2B). This is consistent with our previously reported results of E. coliDNA extraction and detection in hematuric urine.22
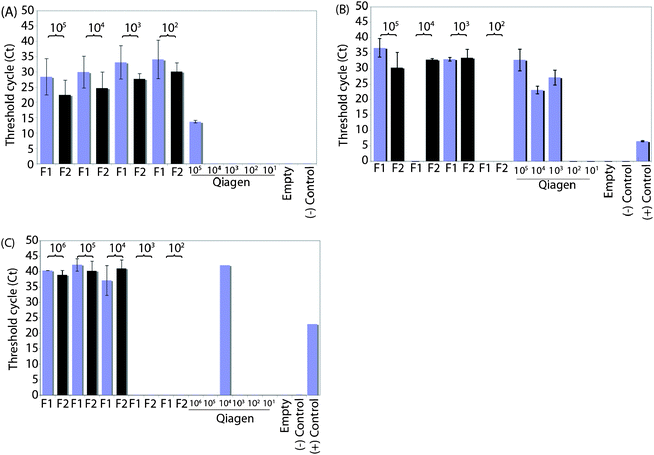 | ||
| Fig. 2 Amplification threshold (CT) cycles for E. coli DH5α (A), B. subtilis (B), and E. faecalis (C) at 102 to 105 or 106CFU/mL in human whole blood. Two 75 µl fractions of eluted DNA were collected from three channels per concentration; the first fraction collected is labeled as F1 and the second as F2. The positive controls were lysed with enzyme plus detergent (gram-positive B. subtilis and E. faecalis) or detergent alone (gram-negative E. coli) and extracted using the Qiagen DNeasy Blood and Tissue kit. The negative controls include empty channels loaded with sterile broth media (no bacteria) and PCR negative water (no DNA) controls. | ||
The performance of both the µSPE channels and the Qiagen kit varied in terms of the lowest bacterial concentration that was detected by PCR, depending on the bacteria tested. However, the µSPE channels performed similarly to or better than the Qiagen kit for all three bacteria, E. coli, B. subtilis, and E. faecalis. The channels produced a lower limit of detection of 102CFU/ml in the case of E. coliDNA extraction compared to 105CFU/ml for the kit (Fig. 2A). For B. subtilisextraction, the results from both methods were comparable for the lowest bacterial concentrations detected of 103CFU/ml, but the Qiagen kit had lower CT values at the lower concentrations of 104 and 103CFU/ml (Fig. 2B). Both the µSPE channels and the kit were able to extract E. faecalisDNA, but with detection at high CT values at around 40 amplification cycles, this bacteria was the most difficult to detect in our study (Fig. 2C).
However, the channels extracted DNA from E. faecalis more consistently over a range of bacterial concentrations from 104–106CFU/ml. We tested up to 106CFU/ml on for E. faecalis, since this gram-positive bacteria was negative at the lower concentrations that we tested. The kit was positive only at 104CFU/ml (CT = 42).
The best results obtained with the µSPE channels were with the gram-negative bacteria E.coli. This was the only bacterial strain tested that was detectable at all the concentrations tested, and as anticipated, amplified with decreasing CT values as cell numbers increased. This trend was also seen in gram-positive bacteria and is consistent with the expectation that the DNA concentration in the eluted samples is greater at higher bacterial loads, resulting in lower CT values. B. subtilis and E. coli associated CT values were consistently lower (25–33 cycles) than for E. faecalis, which, though detectable, had CT values near 40 cycles.
The highest concentration tested in common for all three bacteria was 105CFU/ml. At this concentration of E. coli, 6/6 (100%) of the channels extracted DNA that was detectable by RT-PCR. At this same high concentration both gram-positive bacteria were also consistently PCR positive with 5/6 (83.3%) channels being positive. The number of PCR positive channels decreased to 4/6 (66.7%) for the lowest concentration of 102CFU/ml for E. coli, and sharply declined to 1/6 (16.7%) for B. subtilis. Again E. faecalis was the most difficult to detect as 0/6 (0%) channels were PCR positive at 102 and 103CFU/ml.
Discussion
The data presented here demonstrate that the polymer monolith µSPE columns are able to rapidly lyse both gram-negative and gram-positive bacteria and extract genomic DNA that may be assayed by downstream molecular diagnostic methods. We demonstrated the ability of these monoliths to extract PCR quality DNA from the complex background of whole blood. From the PCR CT value readout, the order of most concentrated DNA eluted from the channels from greatest to lowest is E. coli, followed by B. subtilis, and then E. faecalis samples or gram-negative and then gram-positive bacteria. However, the detection of the two gram-positive bacteria we tested differed in the concentration of bacteria we could detect and the PCR threshold cycle (CT) at which the target DNA was amplified. A lower threshold value for the detection of amplicons in the case of E. faecalis as compared to B. subtilis suggests that the channel eluted DNA was less concentrated from E. faecalis samples. We believe that these results are directly due to the lysis capability of the polymer monolith in the channels. This extends from the reasoning that all experimental variables, except the type of bacterial cell wall, were constant between the experiments with the different bacteria; the channels were prepared identically with the same monolith and a constant mass of silica particles and used identically such that we expect minimal variation in the binding and elution of isolated DNA.22 Thus we postulate that the most important variable affecting the performance of the channels in extracting bacterial genomic DNA was the initial step of lysing the cell wall.In the case of mammalian cells and an enveloped virus (blood cells, fibroblasts, influenza), flow through the silica porous polymer monolith in the presence of a lysis agent resulted in membrane lysis and liberation of nucleic acid with sufficient pressure.21,23 We have also seen significant lysis in channels for gram-negative bacteria.22 We previously described the cell lysis scheme of our channels as primarily a mechanical shearing effect of the bacterial cell flowing through a path of tiny pores of different size (predominantly 1 µm diameter or less) within the porous polymer monolith.22 To further demonstrate this, we prepared triplicate 105CFU/ml dilutions of E. faecalis in blood in the same manner as those extracted with the µSPE channels (not treated with lytic enzymes) but instead extracted DNA using the Qiagen kit. Of the three samples tested, two were detected at CT values of 44.1 and 44.4, and the third sample was undetectable. These higher CT values compared to identical samples extracted using the µSPE columns (CT ≤ 40), suggest that while some of the lysis is likely due to the buffer action alone, it is augmented by the mechanical shearing effect. The backpressure inside of channels containing µSPE columns have been measured at 100–150 psi at steady state for the flow rates used in this work (data not shown). This mechanical lysis is likely enhanced by the addition of a chemical lysis buffer in which the bacteria is resuspended.
Although we were able to lyse and detect gram-positive bacteria without lengthy enzyme incubations by using a combination of detergents, it is clear that the sensitivity of our assay can be improved for microbes or particles with tough cell walls such as gram-positive Enterococci, Streptococci, and Staphylococci bacteria, yeast, fungal hyphae, and bacterial spores. In this study, we attempted to assist on-chip lysis of E. faecalis by using various concentrations of detergents (0.02% SDS, 0.05%, and 1% SDS in combination with 1.2% and 2.4% TritonX-100), three consecutive freeze-thaw cycles of bacteria suspended in blood (freezing at −80C and rapid thawing at 37C), and boiling bacterial cultures for 10–30 min. None of these methods yielded detectable E. faecalisDNA except 1% SDS with 2.4% TritonX-100, which we implemented as part of the bacterial resuspension buffer prior to loading the bacteria through the channels (see methods section). Strategies for the lysis of such tough membranes will involve the addition of sharp carbon nanotube structures or other sharp inclusions into the channel design. Challenges to implementing these inclusions involve fixing the spatial orientation of these objects within the monolith with maximum exposure on the surface of the pores for steric access to the particles to be lysed within the channel. The mechanical lysis structures we employ must be capable of breaking the macromolecular layered structures of these membranes. In the case of gram-positive bacteria, studies with B. subtiliscell wall peptidoglycan threads revealed that the cell wall is a viscoelastic polymer which behaves like a glass polymer at low relative humidity, but behaves like a rubbery polymer at relative humidities greater than 60%.28 This range of elasticity allows the cell wall in gram-positive bacteria to be flexible and withstand mechanical stress forces. Techniques in engineering stress analysis used to study the tensile strength of the peptidoglycan structure produced by B. subtilis revealed that the tensile strength is 300MPa with a modulus of 13GPa when dry and 13MPa with a modulus of 30MPa when wet, capable of withstanding 2.6MPa (26 atm) of turgor pressure.28,29 The lysis of gram-positive cells is difficult due to the thick multilayered and extensively crosslinked peptidoglycan layer in contrast to the single layer of peptidoglycan in gram-negative cells.11 The results of our study point to potential differences in the strength of the B. subtilis and E. faecaliscell walls as well. Though both are gram-positive, differences in the composition exist between these two bacteria as E. faecalis, a member of Group D Streptococci, is less susceptible to the muramidase lysozyme but is lysed well with a different muramidase, mutanolysin. The observed resistance to lysozyme is due to inhibition of lysozyme by O-acetylation modification to MurNAc, a glycan composing the peptidoglycan layer.30
Other groups have reported rapid detection of bacteria (between four to six hours) in human whole blood using real-time PCR. In contrast to our methods, lysis is achieved using traditional enzymes or mechanical forces (bead beating) and sensitivity is improved by large sample volumes and/or using primers against targets that have multiple copies within a bacterial cell to improve sensitivity.31,32 Jordan and Durso showed detection of 40 E.colicells from 0.3 to 1.0 ml of human blood using real-time PCR with primers against multi-copy targets of 16S rDNA which still rely on enzymatic lysis of gram-positive bacteria with the typical lengthy incubations similar to our positive control samples extracted with the Qiagen kit.33 Similarly, the SeptiFast assay by Roche Diagnostics uses multiplex PCR against multi-copy internal transcribed sequences (ITS) located between the 16S and 23S rRNA genes of bacteria from 3ml of blood lysed with ceramic beads.34,35 The most rapid (1 hour) and automated system, GeneXpert (Cepheid, Inc.), uses microfluidic cartridges for bacterial sepsis detection in a commercially available methicillin-resistant Staphylococcus aureus (MRSA) assay with a limit of detection of 610 CFU/ml.36,37
In our study, the standard commercial Qiagen kit protocol involves two enzyme incubations at elevated temperatures (37C and 56C) for 30–90 minutes each. Even at incubations for 90 minutes each, the kit was unable to detect E. faecalisDNA except at one bacterial concentration late in the PCR cycle. The kit protocol specifically indicates the use of the kit for gram-negative and gram-positive bacteria even in whole blood. The possibility of a negative result from the kit extracted DNA due to PCR inhibitors from whole blood is unlikely, since preliminary experiments confirmed that E. faecalisDNA extracted with the kit at the concentrations tested in the study (102–106CFU/ml) in the absence of blood were also undetectable by PCR. However, the positive control sample extracted with the kit contained 109CFU/ml and was easily detected (CT = 23). Clearly the concentrations tested from 102 to 106CFU/ml were below the detection limit of the kit for E. faecalis. The results were similar for B. subtilis and E. coli in that the concentrations detectable by the kit were either the same as or higher than those detected by the SPE channels. The Qiagen kit protocol recommends starting with 109 bacterial cells for optimal DNA yield. Qiagen states that (personal communication, Qiagen technical support) optimal yields from 109cells is approximately 20 µg of total DNA for yeast cells. If we were to extrapolate this to the final elution volume of 150 µl used in our experiments, it corresponds to 133 ng/µl concentration of eluted DNA with the kit. We achieved slightly better yields with the kit in our experiments, which averaged approximately 50 ng/µl from 108E. faecalis, 108E. coli and from 107B. subtilis. The maximum volume input of DNA in our PCR reaction was 12 µl, which is roughly 1600 ng from 109cells in the optimal case by Qiagen. In our extractions, 12 µl was equivalent to 500ng from the highest concentrations. As explained in the kit protocol, the DNA yield from the columns may be linearly extrapolated between different sample input concentrations; thus at 105cells, the DNA yield is expected to be between 0.5 and 5 ng per PCR reaction and presumably approaching the limit of detection of our PCR assays, dependent on the primers chosen, that typically require at least 50 ng of DNA. Thus the microfluidic lysis columns performed as well as or better than the kit and did not require additional incubation time or elevated temperature for the gram-positive species.
Conclusion
We have fabricated a microscale system that functions comparably to the standard benchtop bacterial and viral lysis and nucleic acid extraction kits. A porous polymer monolith can be used with pressure driven flow to perform mechanical/chemical bacterial lysis on a microfluidic device. The lysis column was packed with silica microspheres to streamline lysis and nucleic acid isolation for microliter scale sample preparation on a single platform. We established that this platform can be used with dilute, small volume starting samples of bacteria in human whole blood. Eluted samples were of sufficient quality for amplification and detection using real-time PCR.Acknowledgements
We thank the Wallace H. Coulter Foundation for funding this research.References
- J. M. O'Brien, Jr., N. A. Ali, S. K. Aberegg and E. Abraham, Am J Med, 2007, 120, 1012–1022 CrossRef.
- G. R. Bernard, J. L. Vincent, P. F. Laterre, S. P. LaRosa, J. F. Dhainaut, A. Lopez-Rodriguez, J. S. Steingrub, G. E. Garber, J. D. Helterbrand, E. W. Ely and C. J. Fisher, Jr., N Engl J Med, 2001, 344, 699–709 CrossRef CAS.
- A. D. Minino, M. P. Heron, S. L. Murphy and K. D. Kochanek, in National Vital Statistics Report, National Center for Health Statistics., Hyattsville, MD, 2007 Search PubMed.
- K. E. Sands, D. W. Bates, P. N. Lanken, P. S. Graman, P. L. Hibberd, K. L. Kahn, J. Parsonnet, R. Panzer, E. J. Orav, D. R. Snydman, E. Black, J. S. Schwartz, R. Moore, B. L. Johnson, Jr. and R. Platt, JAMA, 1997, 278, 234–240 Search PubMed.
- T. van der Poll and S. M. Opal, Lancet Infect Dis, 2008, 8, 32–43 CrossRef.
- D. C. Angus, W. T. Linde-Zwirble, J. Lidicker, G. Clermont, J. Carcillo and M. R. Pinsky, Crit Care Med, 2001, 29, 1303–1310 CrossRef CAS.
- D. C. Angus and R. S. Wax, Crit Care Med, 2001, 29, S109–116 CrossRef CAS.
- D. G. Remick, Am J Pathol, 2007, 170, 1435–1444 Search PubMed.
- S. P. Larosa, Cleve Clin J Med, 2002, 69, 65–73 Search PubMed.
- R. P. Peters, M. A. van Agtmael, S. A. Danner, P. H. Savelkoul and C. M. Vandenbroucke-Grauls, Lancet Infect Dis, 2004, 4, 751–760 CrossRef CAS.
- M. T. Cabeen and C. Jacobs-Wagner, Nature reviews, 2005, 3, 601–610 Search PubMed.
- W. Vollmer and J. V. Holtje, J Bacteriol, 2004, 186, 5978–5987 CrossRef CAS.
- A. P. Middelberg, Biotechnology advances, 1995, 13, 491–551 CrossRef CAS.
- J. Geciova, D. Bury and P. Jelen, International Dairy Journal, 2002, 12, 541–553 Search PubMed.
- C. Lamanna and M. F. Mallette, J Bacteriol, 1954, 67, 503–504 CAS.
- O. Salazar, Methods in molecular biology (Clifton, N.J), 2008, 424, 23–34 Search PubMed.
- D. Di Carlo, K. H. Jeong and L. P. Lee, Lab Chip, 2003, 3, 287–291 RSC.
- J. Wang, Z. Chen, P. L. Corstjens, M. G. Mauk and H. H. Bau, Lab Chip, 2006, 6, 46–53 RSC.
- R. H. Liu, J. Yang, R. Lenigk, J. Bonanno and P. Grodzinski, Anal Chem, 2004, 76, 1824–1831 CrossRef CAS.
- A. Bhattacharyya and C. M. Klapperich, in Encyclopedia of Micro- and Nano-Fluidics, ed. D. Li, Springer, 2008, p. 2226 Search PubMed.
- A. Bhattacharyya and C. M. Klapperich, Anal Chem, 2006, 78, 788–792 CrossRef CAS.
- M. D. Kulinski, M. Mahalanabis, S. Gillers, J. Y. Zhang, S. Singh and C. M. Klapperich, Biomed Microdevices, 2009 DOI:10.1007/s10544-008-9277-1.
- A. Bhattacharyya and C. M. Klapperich, Sensors and Actuators B: Chemical, 2008, 129, 693–698 CrossRef.
- A. Bhattacharyya and C. M. Klapperich, Lab Chip, 2007, 7, 876–882 RSC.
- R. Boom, C. J. Sol, M. M. Salimans, C. L. Jansen, P. M. Wertheim-van Dillen and J. van der Noordaa, J Clin Microbiol, 1990, 28, 495–503 CAS.
- C. Lee, J. Kim, S. G. Shin and S. Hwang, Journal of biotechnology, 2006, 123, 273–280 Search PubMed.
- A. Mahmoudpour, S. Rahimi, M. Sina, M. H. Soroush, S. Shahi and N. Asl-Aminabadi, Journal of oral science, 2007, 49, 221–227 Search PubMed.
- N. H. Mendelson and J. J. Thwaites, Journal of Bacteriology, 1989, 171, 1055–1062 Search PubMed.
- J. J. Thwaites and U. C. Surana, Journal of Bacteriology, 1991, 173, 197–203 Search PubMed.
- J. M. Pfeffer, H. Strating, J. T. Weadge and A. J. Clarke, J Bacteriol, 2006, 188, 902–908 CrossRef CAS.
- L. H. Chen, Q. J. Duan, M. T. Cai, Y. D. Wu and S. Q. Shang, Clin Pediatr (Phila), 2009 Search PubMed.
- S. Shang, G. Chen, Y. Wu, L. Du and Z. Zhao, Pediatr Res, 2005, 58, 143–148 CrossRef CAS.
- J. A. Jordan and M. B. Durso, J Mol Diagn, 2005, 7, 575–581 CAS.
- L. E. Lehmann, K. P. Hunfeld, T. Emrich, G. Haberhausen, H. Wissing, A. Hoeft and F. Stuber, Med Microbiol Immunol, 2008, 197, 313–324 CrossRef CAS.
- A. Vince, S. Z. Lepej, B. Barsic, D. Dusek, Z. Mitrovic, R. Serventi-Seiwerth and B. Labar, J Med Microbiol, 2008, 57, 1306–1307 CrossRef.
- A. S. Rossney, C. M. Herra, G. I. Brennan, P. M. Morgan and B. O'Connell, J Clin Microbiol, 2008, 46, 3285–3290 CrossRef CAS.
- E. M. Marlowe, Clinical Microbiology Newsletter, 2008, 30, 183–188 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |