DNA methylation analysis on a droplet-in-oil PCR array
Yi
Zhang
a,
Vasudev
Bailey
ab,
Christopher M.
Puleo
a,
Hariharan
Easwaran
b,
Elizabeth
Griffiths
b,
James G.
Herman
b,
Stephen B.
Baylin
b and
Tza-Huei
Wang
*ac
aDepartment of Biomedical Engineering, Johns Hopkins University, Baltimore, MD 21218, USA. E-mail: thwang@jhu.edu; Tel: +1-410-516-7086
bCancer Biology Program, The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, MD 21210, USA
cDepartment of Mechanical Engineering, Johns Hopkins University, Baltimore, MD 21218, USA
First published on 6th March 2009
Abstract
We performed on-chip DNA methylation analysis using methylation-specific PCR (MSP) within an arrayed micro droplet-in-oil platform that is designed for more practical application of microfluidic droplet technologies in clinical applications. Unique features of this ready-to-use device include arrayed primers that are pre-deposited into open micro-reaction chambers and use of the oil phase as a companion fluid for both sample actuation and compartmentalization. These technical advantages allow for infusion of minute amounts of sample for arrayed MSP analysis, without the added complexities inherent in microfluidic droplet-based studies. Ease of use of this micro device is exemplified by analysis of two tumor suppressor promoters, p15 and TMS1 using an on-chip methylation assay. These results were consistent with standard MSP protocols, yet the simplicity of the droplet-in-oil microfluidic PCR platform provides an easy and efficient tool for DNA methylation analysis in a large-scale arrayed manner.
Introduction
Although various methods are available for analyzing nucleic acids, polymerase chain reaction (PCR) remains one of the most widely used techniques. Many molecular biology protocols today still heavily rely on PCR or modified PCR. For example, allele-specific PCR is used for identifying single nucleotide polymorphisms,1 asymmetric PCR is used for generating single stranded DNA hybridization probes or targets,2 real-time quantitative PCR is used for DNA/RNA quantification,3 BEAMing PCR is incorporated in next-generation sequencing,4,5 and methylation-specific PCR (MSP) is used for DNA methylation detection.6 Currently, one of the major trends in biological and chemical analysis has been the development of microfabricated miniaturized platforms.7–12 In particular, great effort has been put into the miniaturization of genetic tests13–15 as they provide fast analysis, reduced reagent and sample consumption, and high portability. The most straightforward approach used involves fabricating an array of reaction chambers of micro, nano or even picoliter volumes for parallel PCR reactions.16–21 Alternatively, miniaturization is realized by performing PCR in free micro droplets on a modified surface. The droplets function as the virtual reaction chambers by providing fluidic confinement.22–24 However, many current array based devices are designed for performing multiple PCR reactions in identical conditions with the same primers and DNA templates, thus limiting their use to singleplex analysis. Multiple gene screening with different primer sets still relies on separate sample loading via complex fluidic networks. Furthermore, most designs concentrate on engineering perspectives, focusing on the miniaturization and system integration, while often overlooking practicality issues. As a result, very complicated microfluidic control modules are usually incorporated in order to cope with pressurization and micro bubble formation and expansion during thermal cycling.25,26 Unfortunately, such delicate devices often require operation by experienced microfluidic personnel and thus are difficult to fit into routine biological laboratories and clinical settings. Hence they are not commonly adopted for use in PCR based diagnostic assays such as MSP.MSP (Fig. 1) is most extensively used for analyzing DNA methylation that is closely associated with tumorigenesis, a multi-step process resulting from gain-of-function (oncogenes) or loss-of-function (tumor suppressor) gene alterations. These changes could be caused either by genetic or epigenetic changes. One of the most well studied epigenetic changes is the heritable transcriptional silencing of tumor suppressor genes (TSG) by aberrant CpG DNA hypermethylation of their promoters.27–31 In higher order eukaryotes, DNA methylation only occurs at 5′ cytosines in CpG dinucleotides, which when observed in high frequency at many transcription promoter regions are termed CpG islands.32 There is clear evidence that the down regulation of tumor suppressor genes in cancer is tightly associated with DNA hypermethylation in the promoter regions, where the transcription of DNA to RNA is initiated33 Therefore, the assessment of DNA methylation status has great clinical implications, offering another important parameter for early cancer diagnosis and prognosis, as well as responsiveness to cancer therapy.34–36MSP takes advantage of the change in DNA sequence after bisulfite modification, wherein only unmethylated cytosines are converted to uracils, while methylated cytosines remain unaltered. Primers specific to the modified DNA sequences are then introduced to amplify and distinguish methylated from unmethylated DNA. Generally, each bisulfite treated DNA sample is run in two separate MSP reactions. Each reaction contains either a methylated specific primer set or an unmethylated specific primer set, which means the total number of reactions is doubled compared to common PCR based assays. In a single DNA methylation study of clinical samples, hundreds or even thousands of analyses are carried out at different promoter regions.34MSP assays with such a great number of samples are cumbersome and can often take months. Therefore, a higher throughput approach would be desirable.
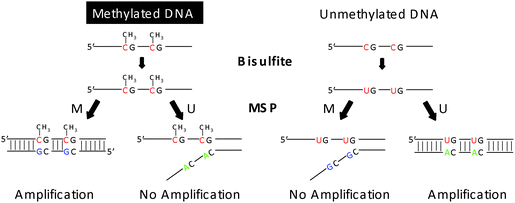 | ||
| Fig. 1 Principle of methylation-specific PCR (MSP). Genomic DNA is treated with bisulfite, converting unmethylated cytosines to uracils, while methylated cytosines remains unaltered. Methylated specific (M) primers can only amplify methylated sequence. Unmethylated specific (U) primers can only amplify unmethylated sequence. If M primer is introduced to unmethylated sequence, no amplification is observed due to sequence difference and vice versa. | ||
To address the increasing interest in high throughput DNA methylation analysis, we have developed an easy to handle droplet-in-oil microfluidic MSP platform for DNA methylation detection. The proposed device has nine snowflake-like functional units arranged in a circular array. Each functional unit consists of 12 open reaction chambers which are also arranged in a circular array and connected to sample access port through a microfluidic network. Methylation specific primers are pre-deposited into reaction chambers. The device can perform 108 MSP reactions in parallel. Each functional unit is capable of DNA methylation analysis of multiple genes with single sample dispensing, thereby significantly reducing the sample preparation time, improving throughput and allowing for automation. The simple and disposable device can easily be handled by individuals who do not have microfluidic experience because it takes the advantage of a novel sample loading scheme.37,38 The unique scheme uses the mineral oil as a companion fluid for sample actuation, in addition to reaction compartmentalization and evaporation prevention during the thermal cycling. It generates micro droplets by simple injections with a syringe and avoids the use of infusion pumps or pressure regulators, making the array an ideal platform for high throughput point-of-care detection. The MSP device was validated by testing with multiple gene analyses and comparing the results with conventional in-tube MSP assay.
Experimental
Device design, fabrication and preparation
The device contains nine functional units arranged in a circular array (Fig. 2a). The layout of each functional unit resembles a snowflake where six channels of 500 µm width extend from a center sample input port to peripheral reaction chambers (Fig. 2b). Each channel was symmetrically bifurcated and terminated at two different chambers. The 12 reaction chambers were equally separated from each other and evenly distributed around the center circle. The size of each functional unit is 32 mm in diameter and the overall size of the system is 12 cm. The prototype device was fabricated by casting polydimethylsiloxane (PDMS) (Corning Inc.) over a lithographically patterned 50 µm thick SU-8 master on silicon substrate. The sample access port was created by punching a through-hole at the origin of the center circle with a dispensing needle of 0.64 mm (0.25 inch) diameter. The reaction chambers were made by punching through the peripheral circles using a hollow puncher of 4 mm diameter. The punched PDMS was then bonded to a thin glass slide (Fisher Scientific Inc.) after oxygen plasma treatment. Bonded chips were baked at 75 °C overnight. To prevent possible enzyme adsorption and improve PCR efficiency, the surface of the reaction chambers was dip coated with 0.1% (wt%) bovine serum albumin (BSA). Methylation specific primers (Table 1) that target the promoter regions of two different tumor suppressor genes (p15 and TMS1) were pre-deposited into designated reaction chambers. Primer deposition was accomplished by pre-metering primers and allowing full-solvent evaporation on the bottom surface of the chambers. The primer deposition process could be automated using inkjet technology.39 Prepared chips were thus primed for simultaneous methylation detection and stored at 4 °C for future use. | ||
| Fig. 2 Device pre-deposited with methylation specific primers. (a) Device overall layout. Nine functional units are arranged in a circular array. A total of 108 reactions can be prepared with nine sample injections, which significantly improves the throughput. (b) Functional unit layout. The blue areas indicate open structures, while the black areas represent closed channels. The sample inlet is punched with a dispensing needle (0.64 mm diameter) at the center of the interior circle. Reaction chambers at the peripheral are created by punching through-holes with a hollow puncher (4 mm diameter). Primers specific for tumor suppressor promoters (p15 and TMS1) are deposited onto the bottom surface of designated reaction chambers by pipetting. | ||
| Primer set | Sense primer, 5′-3′ | Antisense primer, 5′-3′ | Amplicon size, bp |
|---|---|---|---|
| a U represents unmethylated specific primers. b M represents methylated primers. | |||
| P15-Ua | GGTTGGTTTTTTATTTTGTTAGAGTGAGGT | AACCACTCTAACCACAAAATACAAACACA | 80 |
| P15-Mb | GGTTTTTTATTTTGTTAGAGCGAGGC | TAACCGCAAAATACGAACGCG | 68 |
| TMS1-U | GAAGGTGGGGAGTTTAGGTTTTGTTTT | AAATTCTCCAACACATCCAAAATAACAT | 140 |
| TMS1-M | GCGGGGAGTTTAGGTTTCGTTTC | CCAACGCATCCAAAATAACGTCG | 130 |
DNA isolation and bisulfite modification
Human genomic DNA (gDNA) was extracted from human peripheral blood, which was drawn from a volunteer after the receipt of informed consent. All chemicals used in the sample preparation were purchased from Sigma Aldrich Inc. unless otherwise stated. Normal leukocytes (NL) were extracted by centrifugation. The extract was digested by 0.5 mg mL−1proteinase K in 50 µL PK buffer, 19.5 mM ammonium sulfate, 78.8 mM Tris, 7.9 mM magnesium chloride and 11.7 nM 2-mercaptoethanol at 60 °C. Proteinase K was then inactivated by heating to 100 °C for 10 min. DNA was purified by phenol–chloroform extraction. In vitro methylated DNA (IVD) was obtained by treating NL DNA with SSSI methyltransferase and purified using Wizard DNA CleanUp System (Promega). The purified DNA was subjected to bisulfite modification. 1 µg DNA was first denatured by adding NaOH (final concentration of 0.2 M) and incubated at 37 °C for 10 min. 520 µL of sodium bisulfite (concentration of 3M) and 30 µL of hydroquinone (concentration 10 mM), freshly prepared before each bisulfite treatment, were added to the denatured DNA, gently vortexed and incubated at 55 °C for 16 hours. Subsequently, the bisulfite-modified DNA was purified using Wizard DNA CleanUp System (Promega) following the manufacturer's instructions and eluted into 50 µL water. Lastly, the purified DNA was incubated with 0.3 M NaOH at room temperature, followed by ethanol precipitation. The final modified DNA product was suspended in 20 µL of Tris EDTA buffer (pH 8.5) and used immediately or kept at −20 °C for long term storage.MSP protocol
The principle of MSP has been previously described.6 In brief, unmethylated cytosines are chemically converted to uracils by bisulfite treatment, whereas methylated cytosines remain as cytosines. The sequence difference between the methylated and unmethylated DNA after bisulfite modification are recognized and amplified by methylation specific primers. Primer sequences are selected from regions containing CpG islands, thus providing maximal discrimination between methylated and unmethylated DNA. Primer pairs listed in Table 1 have been previously validated.6,40 A total of two sets of four primer pairs were purchased from the Integrated DNA Technology (IDT DNA). Each set includes unmethylated (U) and methylated (M) primers which are specific for bisulfite modified unmethylated sequence and methylated sequence respectively. M primers can only amplify the bisulfite modified methylated sequence and vice versa. The MSP mixture (50 µL) contains dNTPs (each at 1.5 mM), 1X PCR buffer (67 mM Tris, 16.6 mM ammonium sulfate, 6.7 mM MgCl2, 10 nM 2-mercaptoethanol), 0.05 U µL−1 HotStar Taq polymerase (Qiagen Inc.), 1X DNA intercalating dye EvaGreen (Biotium Inc.), 0.1% (wt%) BSA, bisulfite treated DNA as template, and PCR grade water to adjust the final volume. The thermal cycling conditions are as follows: enzyme activation and pre-denaturation at 95 °C for 15 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s and elongation at 72 °C for 30 s. A final elongation step is performed at 72 °C for 5 min.Conventional MSP assays were performed using NL (unmethylated DNA sample) and IVD (methylated DNA sample) as templates. Four pairs of primers, namely p15-U, p15-M, TMS1-U and TMS1-M (Table 1) were used to assess the methylation status of the DNA samples. Reactions were carried out in a conventional thermal cycler (Bio-Rad Inc.) with 50 µL reaction volume. Final MSP products were loaded onto 2% agarose gel, stained with GelStar®nucleic acid gel stain (Lonza Group Ltd.) and imaged with an UV illumination system. On-chip MSP reactions were performed under the identical condition as conventional MSP reactions. MSP mixtures also contained 0.1% (wt%) BSA as a dynamic coating against enzyme adsorption.9 EvaGreen served as a fluorescence reporter for imaging purposes. MSP mixture was loaded onto the chips using an injection needle. The total amount of pre-deposited primers in each reaction chamber was metered according to the final volume, so that the final primer concentration was 300 nM. The thermal cycling was performed with a flat-bed Peltier heating plate. The MSP chip was thermally coupled to the heating plate by mineral oil and slightly pressed against the heating plate to ensure full contact. The direct fluorescence image of the micro MSP chip was taken using a Typhoon™ scanner (GE Healthcare) with 488 nm excitation. The fluorescence emission was filtered by a band pass filter, which centers at 520 nm with a bandwidth of 40 nm, before it was collected. The gain of the internal photomultiplier tube was set to 230 with normal sensitivity. Images were processed with ImageJ (NIH) to remove background noise and enhance contrast.
Results and discussion
The sample loading scheme is demonstrated in Fig. 3. Primers were deposited onto the bottom surface of the designated reaction chambers, near the outlets of the microchannels connected to the reaction chambers, during the device fabrication and preparation stage. Oligonucleotides were adsorbed to the surface after full-solvent evaporation. The chips were stored at 4 °C until use. For each functional unit, first the sample was injected from the center sample access port through a dispensing needle. The sample liquid was guided to the reaction chambers through the microfluidic network (Fig. 3a). The aqueous solution dissolved the primers, thus completing the ingredient list of the MSP mixture. Because each reaction chamber contains a unique set of primers, multiple MSP reactions could be prepared with single sample injection. For the current circular snowflake array, only nine injections were required for preparing 108 distinct MSP reactions, which significantly reduced the sample preparation time and improved the throughput. Next, metering and compartmentalization were accomplished using immiscible mineral oil as companion fluid. The mineral oil forced the sample inside the microchannels into reaction chambers, allowing resuspension and mixing of the pre-deposited primers with the DNA sample and MSP reagent (Fig. 3b). Excess oil was then injected to fill the volume in the microchannels and prevent possible backflow and cross contamination (Fig. 3c). Finally, the MSP mixture formed droplets in the mineral oil and sat on the bottom of the reaction chambers due to differences in solvent polarities and densities. Consequently, the oil provided insulation for the reaction chambers against direct evaporation (Fig. 3d). To aid visualization, this sample loading scheme was monitored using aqueous food dye. Pictures of the device with food dye before and after the infusion of the mineral oil are shown in Fig. 4.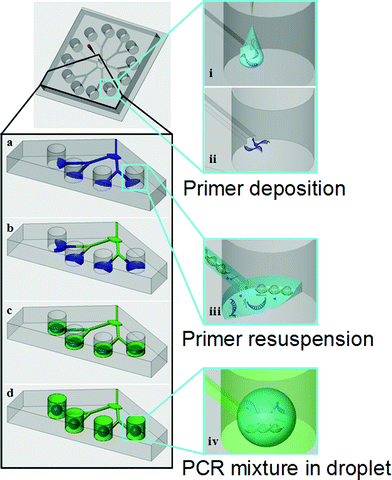 | ||
| Fig. 3 Illustration of sample loading and primer deposition on an individual functional unit. (a) The sample was injected and guided into each reaction chamber. (b) The companion fluid was subsequently injected from the same inlet, driving the sample into reaction chambers. (c) Excess mineral oil occupied the residual volume inside the microchannels, preventing possible backflow and cross contamination. (d) The companion fluid filled the entire chamber and covered the sample droplets. (i) Primers were deposited onto the bottom of reaction chambers. (ii) The solvent evaporated, leaving primers on the glass substrate. (iii) Resuspension of primers due to infusion of sample containing human gDNA and PCR mix. (iv) All the components required for PCR were trapped inside the droplet that was encapsulated by the mineral oil. The sample (in blue) is compartmentalized into droplets by the companion fluid (in green). | ||
 | ||
| Fig. 4 Demonstration of the sample loading scheme with food dye. (a) The entire circular snowflake array was loaded with a total of nine injections. (b) 108 reactions were prepared after infusion of mineral oil. (c) For each functional unit, 50 µL aqueous food dye was injected from the center sample access port. The dye was guided into 12 reaction chambers through the microfluidic network. (d) The mineral oil forced all the dye into the reaction chambers, leaving no dead volume. | ||
This sample loading scheme has several advantages. First, multiple reactions are prepared with a single sample injection, which improves the throughput and allows for automation. Second, since eventually all the sample will be pushed into each reaction chamber, there is no dead volume, enabling the loading of minute amounts of sample. Last, unlike other micro PCR arrays, the reaction volume of the droplet-in-oil MSP chip is not defined by the size of the reaction chamber, and, thus the assay is more flexible in terms of reaction volume. MSPs with different reaction volumes can be performed on the same device.
The gel image of the conventional reactions (Fig. 5a) revealed the methylation status of the input DNA templates. NL are extracted from healthy individuals, hence only U primers give rise to positive amplification. The in vitromethylation process converts the unmethylated DNA to methylated DNA, resulting in positive amplification with M primers. The U bands in IVD are due to incomplete conversion, which is frequently observed.41
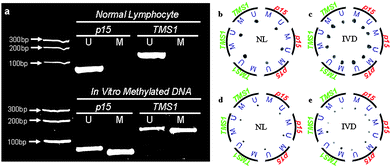 | ||
| Fig. 5 (a) Gel electrophoresis images of in-tube control MSP reactions. Only U primers gave rise to positive amplification in NL sample. M primers resulted in positive amplification with IVD. U bands with IVD sample were due to partial in vitromethylation conversion. Product sizes were 80 bp (p15-U), 69 bp (p15-M), 140 bp (TMS1-U) and 130 bp (TMS1-M). (b–e) Direct fluorescence images of MSP on-chip with dark spots indicating positive amplification. NL sample was only positive with U primers (b,d). IVD sample was positive with both U and M primers due to incomplete conversion (c,e). A total of 50 µL MSP mixture was loaded from the center access port and guided into 12 reaction chambers through the microfluidic network, corresponding to ∼4 µL per reaction (b,c). The open chamber design allowed for flexible reaction volume. A reduced sample, 10 µL, was injected into reaction chambers, which corresponded to ∼0.8 µL per reaction (d,e). The results agreed with control reactions. | ||
The on-chip MSP assay was analyzed by fluorescence imaging (Fig. 5b–5e). Dark spots in the images indicated DNA amplification. Positive amplification of NL was only observed with U primers (Fig. 5b). In contrast, IVD was successfully amplified by M primers. Since the in vitromethylation conversion was partial, both U and M reactions gave positive results (Fig. 5c). With the reaction volume being substantially reduced, the results obtained using our micro MSP chip agreed with the results from control reactions (Fig. 5a).
The majority of the micro PCR chips were closed chamber systems, which required filling up the entire reaction chamber with sample mixture. Therefore, the reaction volume was defined by the chamber size and fixed once the device was fabricated. In contrast, our open chamber device was more flexible and allowed for a wide range of reaction volumes. The reaction chamber did not define the size of the droplet but merely served as containment. To demonstrate that the reaction volume could be further reduced using the same device, a total of 10 µL MSP mixture was used in this case which resulted in submicroliter reaction volume. The MSP results from NL and IVD again agreed with control reactions (Fig. 5a) although smaller spots were observed due to the reduction in reaction volumes (Fig. 5d and 5e). Further downscaling of sample volume can be performed by using finer needles and syringes for sample injection, yet care needs to be taken to avoid null detection when analyzing the samples containing DNA of very low concentrations.
The open chamber design has other advantages. The open access facilitates primer deposition. With pre-deposited primers, the sample preparation procedure is greatly simplified and the throughput is significantly improved. Currently, the design allows for preparing 108 reactions with nine sample injections. It is possible to further reduce the number of injections by integrating more reactions chambers into a single snowflake-like functional unit. The open chamber design also provides easy access to surface modification and post PCR sample retrieval for downstream analysis. Bubble formation and expansion were the prominent problems that complicate many closed chamber PCR devices.40,42–46 Bubbles can easily expand and push the sample out of the chamber during heating. Therefore, bubble containment modules are often incorporated for successful PCR reactions.19,40,47,48 Open chamber design, on the other hand, circumvents this problem, not by eliminating bubbles but by tolerating bubble formation. The tolerance to bubbles eases the sample loading process. Since no additional equipment is required for operating bubble containment module and microfluidics, our micro MSP chip is compatible with conventional bench-top thermal cyclers with minor modification and can be easily handled by individuals who are not experienced in microfluidics. The simple design also provides easy portability. Along with chemicals and reagents, the micro MSP chips can be provided in the form of diagnostic kits for clinical laboratory applications. Although the reaction chambers are open, the MSP reactions are performed in a sealed format. The companion fluid compartmentalizes the samples by encapsulating them into droplets. These sample droplets are completely covered by the companion fluid and isolated from the environment, therefore the chance of contamination is minimized.
Droplet-in-oil (emulsion) PCR has recently attracted increasing attention.4,22,49,50 Our platform provides a simple and relatively high throughput method to prepare droplets for biochemical reactions. It also has been demonstrated that the droplet-in-oil format is compatible with sample preparation methods such as cell isolation and nucleic acid extraction.23,24,51 Applications of the device are not solely limited to DNA methylation detection. With different pre-deposited target primer sets, it could be used for many other applications such as pathogen detection, mutation analysis, and personal genotyping.
Conclusion
The results presented above all indicate that our constructed platform confers unprecedented ease in sample handling due to the novel sample loading scheme, which offers a simple way to prepare multiple droplets for biochemical reactions. By pre-depositing primers and using immiscible oil as companion fluid, it allows multiple reactions with just single sample injection. MSP assays are performed in sealed micro droplets that isolate the reactions from the environment and prevent contamination. The open chamber design facilitates easy surface modification, primer deposition and sample retrieval. It also bypasses the need for a bubble containment mechanism. In addition, the proposed platform allows reliable MSP reactions in a submicroliter volume as opposed to the typical volume of 50 µL for MSP. The downscaling of MSP assays by nearly two orders of magnitude greatly reduces the consumption of reagent and valuable samples. Furthermore, this platform allows multiple DNA methylation analyses performed in an array-based manner. We expect the device to be implemented as a versatile miniaturized PCR platform applicable to many other PCR-based genomic analysis assays. The aforementioned advantages may make the proposed platform widely adopted by both clinical and research laboratories.Acknowledgements
Authors appreciate and credit funding from NIH (1R21CA120742-01 and SPORE P50-CA058184-10), NSF (0546012, 0730503 and 0725528) and Micro/Nano Fluidics Fundamentals Focus (MF3) Center.References
- C. R. Newton, A. Graham, L. E. Heptinstall, S. J. Powell, C. Summers, N. Kalsheker, J. C. Smith and A. F. Markham, Nucleic Acids Res., 1989, 17, 2503–2516 CAS.
- J. E. Rice, J. A. Sanchez, K. E. Pierce, A. H. Reis, A. Osborne and L. J. Wangh, Nature Protocols, 2007, 2, 2429–2438 Search PubMed.
- C. A. Heid, J. Stevens, K. J. Livak and P. M. Williams, Genome Research, 1996, 6, 986–994 CrossRef CAS.
- F. Diehl, M. Li, Y. P. He, K. W. Kinzler, B. Vogelstein and D. Dressman, Nat. Methods., 2006, 3, 551–559 CrossRef CAS.
- M. Margulies, M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y. J. Chen, Z. T. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. I. Alenquer, T. P. Jarvie, K. B. Jirage, J. B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. McDade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. G. Yu, R. F. Begley and J. M. Rothberg, Nature, 2005, 437, 376–380.
- J. G. Herman, J. R. Graff, S. Myohanen, B. D. Nelkin and S. B. Baylin, Proc. Natl. Acad. Sci. U.S. A, 1996, 93, 9821–9826 CrossRef CAS.
- H. Craighead, Nature, 2006, 442, 387–393 CrossRef CAS.
- A. J. deMello, Nature, 2006, 442, 394–402 CrossRef CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- D. Janasek, J. Franzke and A. Manz, Nature, 2006, 442, 374–380 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 442, 412–418 CrossRef CAS.
- P. A. Auroux, Y. Koc, A. deMello, A. Manz and P. J. R. Day, Lab Chip, 2004, 4, 534–546 RSC.
- L. J. Kricka and P. Wilding, Analytical and Bioanalytical Chemistry, 2003, 377, 820–825 CrossRef CAS.
- C. S. Zhang and D. Xing, Nucleic Acids Res., 2007, 35, 4223–4237 CrossRef CAS.
- M. A. Burns, B. N. Johnson, S. N. Brahmasandra, K. Handique, J. R. Webster, M. Krishnan, T. S. Sammarco, P. M. Man, D. Jones, D. Heldsinger, C. H. Mastrangelo and D. T. Burke, Science, 1998, 282, 484–487 CrossRef CAS.
- M. Krishnan, D. T. Burke and M. A. Burns, Analytical Chemistry, 2004, 76, 6588–6593 CrossRef CAS.
- D. S. Lee, S. H. Park, H. S. Yang, K. H. Chung, T. H. Yoon, S. J. Kim, K. Kim and Y. T. Kim, Lab on a Chip, 2004, 4, 401–407 RSC.
- J. Liu, C. Hansen and S. R. Quake, Analytical Chemistry, 2003, 75, 4718–4723 CrossRef CAS.
- M. A. Northrup, B. Benett, D. Hadley, P. Landre, S. Lehew, J. Richards and P. Stratton, Analytical Chemistry, 1998, 70, 918–922 CrossRef CAS.
- A. T. Woolley, D. Hadley, P. Landre, A. J. deMello, R. A. Mathies and M. A. Northrup, Analytical Chemistry, 1996, 68, 4081–4086 CrossRef CAS.
- P. Neuzil, C. Y. Zhang, J. Pipper, S. Oh and L. Zhuo, Nucleic Acids Research, 2006, 34 Search PubMed.
- J. Pipper, M. Inoue, L. F. P. Ng, P. Neuzil, Y. Zhang and L. Novak, Nat. Med., 2007, 13, 1259–1263 CrossRef CAS.
- J. Pipper, Y. Zhang, P. Neuzil and T. M. Hsieh, Angewandte Chemie-International Edition, 2008, 47, 3900–3904 CrossRef CAS.
- H. B. Liu, H. Q. Gong, N. Ramalingam, Y. Jiang, C. C. Dai and K. M. Hui, J. Micromech. Microeng., 2007, 17, 2055–2064 CrossRef CAS.
- T. Nakayama, Y. Kurosawa, S. Furui, K. Kerman, M. Kobayashi, S. R. Rao, Y. Yonezawa, K. Nakano, A. Hino, S. Yamamura, Y. Takamura and E. Tamiya, Anal. Chem., 2006, 386, 1327–1333 CAS.
- A. Bird, Genes Dev., 2002, 16, 6–21 CrossRef CAS.
- J. G. Herman and S. B. Baylin, New England Journal of Medicine, 2003, 349, 2042–2054 CrossRef CAS.
- P. A. Jones and S. B. Baylin, Nat. Rev. Genet., 2002, 3, 415–428 CAS.
- P. A. Jones and P. W. Laird, Nature Genetics, 1999, 21, 163–167 CrossRef CAS.
- V. E. A. Russo, R. A. Martienssen and A. D. Riggs, Epigenetic mechanisms of gene regulation, Cold Spring Harbor Laboratory Press, Plainview, N.Y., 1996 Search PubMed.
- R. Holliday and G. W. Grigg, Mutation Research, 1993, 285, 61–67 CAS.
- S. B. Baylin and J. G. Herman, Trends in Genetics, 2000, 16, 168–174 CrossRef CAS.
- M. V. Brock, C. M. Hooker, E. Ota-Machida, Y. Han, M. Z. Guo, S. Ames, S. Glockner, S. Piantadosi, E. Gabrielson, G. Pridham, K. Pelosky, S. A. Belinsky, S. C. Yang, S. B. Baylin and J. G. Herman, New England Journal of Medicine, 2008, 358, 1118–1128 CrossRef CAS.
- M. V. Brock, J. G. Herman and S. B. Baylin, New England Journal of Medicine, 2008, 358, 2514–2514 CrossRef CAS.
- M. Esteller, J. Garcia-Foncillas, E. Andion, S. N. Goodman, O. F. Hidalgo, V. Vanaclocha, S. B. Baylin and J. G. Herman, New England Journal of Medicine, 2000, 343, 1350–1354 CrossRef CAS.
- Y. Zhang, V. Bailey, C. M. Puleo, C. Chen and T. H. Wang, microTAS 2008 conference proceeding, 2008 Search PubMed.
- Y. Zhang, Vasudev J. Bailey, Hariharan Easwaran, Elizabeth Griffiths, James G. Herman, Stephen B. Baylin, Hetty E. Carraway, Tza-Huei Wang, in AACR conference “Molecular Diagnostics in Cancer Therapeutic Development”, Philadelphia, 2008 Search PubMed.
- P. Cooley, D. Wallace and B. Antohe, Journal of the Association for Laboratory Automation, 2002, 7, 33–39 Search PubMed.
- E. O. Machida, M. V. Brock, C. M. Hooker, J. Nakayama, A. Ishida, J. Amano, M. A. Picchi, S. A. Belinsky, J. G. Herman, S. Taniguchi and S. B. Baylin, Cancer Res., 2006, 66, 6210–6218 CrossRef CAS.
- O. G. a. J. G. Herman, Multiple Myeloma, Humana Press, 2005 Search PubMed.
- Z. Q. Niu, W. Y. Chen, S. Y. Shao, X. Y. Jia and W. P. Zhang, Journal of Micromechanics and Microengineering, 2006, 16, 425–433 CrossRef CAS.
- N. M. Toriello, C. N. Liu and R. A. Mathies, Analytical Chemistry, 2006, 78, 7997–8003 CrossRef CAS.
- J. F. Chen, M. Wabuyele, H. W. Chen, D. Patterson, M. Hupert, H. Shadpour, D. Nikitopoulos and S. A. Soper, Analytical Chemistry, 2005, 77, 658–666 CrossRef CAS.
- Z. Y. Chen, J. Wang, S. Z. Qian and H. H. Bau, Lab on a Chip, 2005, 5, 1277–1285 RSC.
- Y. S. Shin, K. Cho, S. H. Lim, S. Chung, S. J. Park, C. Chung, D. C. Han and J. K. Chang, Journal of Micromechanics and Microengineering, 2003, 13, 768–774 CrossRef CAS.
- N. C. Cady, S. Stelick, M. V. Kunnavakkam and C. A. Batt, Sensors and Actuators B-Chemical, 2005, 107, 332–341 CrossRef.
- E. T. Lagally, I. Medintz and R. A. Mathies, Analytical Chemistry, 2001, 73, 565–570 CrossRef CAS.
- T.-M. Hsieh, Y. Zhang, J. Pipper and P. Neuzil, microTAS 2006 conference proceeding, 2006 Search PubMed.
- K. D. Dorfman, M. Chabert, J. H. Codarbox, G. Rousseau, P. de Cremoux and J. L. Viovy, Analytical Chemistry, 2005, 77, 3700–3704 CrossRef CAS.
- U. Lehmann, C. Vandevyver, V. K. Parashar and M. A. M. Gijs, Angewandte Chemie-International Edition, 2006, 45, 3062–3067 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |