Label-free quantification of asymmetric cancer-cell filopodium activities in a multi-gradient chip
Tsi-Hsuan
Hsu
a,
Meng-Hua
Yen
b,
Wei-Yu
Liao
c,
Ji-Yen
Cheng
*bd and
Chau-Hwang
Lee
*abe
aInstitute of Biophotonics, National Yang-Ming University, Taipei, 11221, Taiwan. E-mail: clee@gate.sinica.edu.tw; Fax: +886-2-27826680; Tel: +886-2-27898000 ext 18
bResearch Center for Applied Sciences, Academia Sinica, Taipei, 11529, Taiwan. E-mail: jycheng@gate.sinica.edu.tw; Fax: +886-2-27826680; Tel: +886-2-27898000 ext 34
cDepartment of Internal Medicine, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei, 10002, Taiwan
dDepartment of Mechanical and Mechantronic Engineering, National Taiwan Ocean University, Keelung, 20224, Taiwan
eGraduate Institute of Clinical Medicine, National Taiwan University, Taipei, 10002, Taiwan
First published on 15th January 2009
Abstract
We use novel super-resolution bright-field optical microscopy to observe the filopodium activities of human lung cancer cells in a multi-gradient cell culture chip. Temporal variations of the filopodium numbers are measured without fluorescent labelling. By carefully designing the fluidic field inside the culture chip, we establish stable concentration gradients of the injected reagents. The reagents are injected via a separated central inlet, and the concentration gradients are different at different positions in the chip. The same chip can be used for both control and treated experiments. Using epidermal growth factor as the treatment, we verify that the protrusions of filopodia indicate the direction of concentration gradients experienced by a living cancer cell; while the treatment of bovine serum albumin shows no specific effect on the growth of filopodia. The combination of label-free, high-resolution optical microscopy and a micro cell culture chip establishes a convenient and versatile platform for dynamical cancer-cell analyses.
Introduction
Micro cell culture chips have become popular for living cell analyses because of very small amounts of cell and reagent consumptions compared with experiments using traditional culture dishes.1,2 By providing a stable and controllable micro-environment, micro cell culture chips are especially useful for the assay of cell responses to external chemical or cytogenetic factors. For example, chemotaxis and polarization of epithelial cells induced by gradients of vascular endothelial growth factor in a cell culture chip were characterized by using fluorescence microscopy.3 An important prerequisite for studying cell activities is the capability of maintaining temperature, pH, nutrition, etc. during the observation period. Several microfluidic chips have been developed for this purpose,4,5 so that long-term and real-time observation can be carried out using an optical microscope without a cell incubator installed on it. Recently we also developed a cell culture chip capable of providing stable chemical gradients in addition to maintaining basic cell culture conditions.6 Such a platform allows us to observe cell dynamics as a response to asymmetric chemical stimulations.The observations of cell dynamics in micro culture chips are limited to only far-field optical microscopy because of the necessary package of the culture chips. The lateral resolution of far-field optical microscopy is known to be restricted by diffraction, estimated by R = 0.61λ/NA, where λ is the wavelength of light and NA is the numerical aperture of the objective. For common oil-immersion objectives and the visible spectral regime, R is generally above 200 nm. This size is just larger than the scales of many interesting cellular dynamics. For example, an important feature related to cancer-cell metastasis is the filopodia,7,8 of which the diameters are around 100–300 nm. Conventional far-field optical microscopy is thus difficult to conduct precise quantification or dynamical analyses of filopodia. In order to enhance the visibility of filopodia smaller than the optical resolution, fluorescence labelling is usually required. Fluorescent protein transfection is useful to see the filopodia of living cells with conventional fluorescence microscopy9,10 or total internal reflection fluorescence microscopy.11,12 However, because the efficiency of transfection is not constant for every individual cell, the quantification of filopodium numbers of living cells based on fluorescent protein transfection could be inaccurate. Moreover, if the filopodium activities are so fast that sufficiently long exposure time is not allowed, the dynamical analyses based on fluorescence microscopy would be inadequate.
Many techniques have been developed for improving the lateral resolution of far-field optical microscopy. For example, Carrington et al. demonstrated super-resolution fluorescence microscopy by using iterative algorithms to remove the blurring effect from diffraction.13 Structured-light illumination can also improve the lateral resolution to below 100 nm.14,15 Stimulated emission depletion microscopy has achieved lateral resolution smaller than 30 nm16 and been employed to reveal the dynamics of intracellular proteins.17 Although these fluorescence techniques overcome the resolution limit in the far field and can be applied on micro cell culture chips, the strong illumination required by fluorescence microscopy affects the cell viability18–20 and should be used carefully when cell activity or motility is the major content for the assay.
In this work we employ a novel super-resolution bright-field optical microscopy technique to observe cancer-cell filopodium dynamics in a cell culture chip with stable chemical concentration gradients. The resolution improvement relies on the topographic contrast21,22 rather than fluorescence dyes or proteins. Hence the observations reveal the natural activities of cancer cells. As an external stimulation, we establish concentration gradients of epidermal growth factor (EGF) around single cancer cells in this culture chip. EGF is known to enhance the metastasis for various types of cancer cells, and can also induce the growth of filopodia.8 We verify that the asymmetric protrusions of filopodia around a single cell indicate the direction of concentration gradients of the EGF. We also compare the effect of EGF to the growth of filopodia with that of bovine serum albumin (BSA). These works show that the combination of label-free, high-resolution observation with micro cell culture chips can be used for real-time analyses on the dynamical responses of cancer cells to small variations in the micro-environment.
Materials and methods
Design and fabrication of the multi-gradient cell culture chip
Generation of chemical gradients in microfluidic devices has been achieved by various approaches. For example, complex mixing channels provide a static linear gradient transversal to flow direction.3,23,24 The gradients generated at channel intersections have been shown by controlled flow distributions.25 An effusion-type generator that provides symmetric radial gradients has also been reported.26 Another approach is to utilize multiple injectors to generate gradients in a microfluidic chip.27 Microfluidic jets from parallel channels have also been used to generate gradients in an open reservoir that is about 200 µm wide.28Here we use an alternative approach that combines lateral flow and effusion to generate asymmetric concentration gradients in a relatively large area.6 The cell culture chip consists of a PMMA layer with microtrenches and flow equalization structures, a double-side adhesive tape layer for bonding, and a glass cover slip for seal [Fig. 1(a)]. Microtrenches, medium inlet/outlet, reagent inlet, and flow equalization structures are fabricated by a laser direct-writing technique.29 The culture medium is supplied by a syringe pump to flow into the chamber, through the medium inlet hole, and out of the chamber to a waste via the medium outlet. The cell culture region is 14 mm × 26 mm in area and 80 µm in depth. The thickness of the cover slip is 170 µm, and therefore the distance from the upper surface of the cover slip to the bottom of the flow chamber, 250 µm, is within the working distances of most objectives of high numerical apertures. A reagent inlet hole is fabricated at the centre of the chip bottom. This inlet is used for injecting reagents for the establishment of stable chemical concentration gradients.
 | ||
| Fig. 1 (a) Configuration of the cell culture chip used in this work. (b) Optical setup. The microscope employs a water-immersion objective (NA = 1.2) to observe the cells through the cover slip of the culture chip. | ||
Flow field and chemical gradients in the culture chip
The design of equalization gates could create a large fluidic resistance in the gate channels, which leads to a uniform flow profile inside the culture chip, producing a uniform micro-environment for cell culture. To verify this claim, we used the computational fluidic dynamics package, CFD-ACE+ (CFD Research Corp., Huntsville, AL), to model the two-dimensional velocity field inside the flow chamber. For medium replacement simulations, a fully developed velocity condition was first imposed at the inlet. A parabolic velocity profile with an average perfusion rate (50 µl/h) was used at the inlet. No-slip boundary conditions were set at the walls and zero back pressure conditions were set at the outlet. The fluidic density and dynamic viscosity of solution were set as 1000 kg/m3 and 8.55 × 10−4 kg/m-sec, respectively.The simulated velocity field is illustrated in Fig. 2(a). The equalization structure does yield a very uniform velocity profile inside the culture region. The actual flow velocity distribution was measured as follows. Dye solution was introduced into the flow chamber pre-filled with clear water and the flow front was photographed at two time points [Fig. 2(b)]. Assuming a negligible transversal flow, longitudinal flow velocity was measured by the horizontal displacement of the flow front. At the designated flow rate of 50 µl/h, we observe that over 90% width of the medium flow shows uniform flow velocity with CV of 6% [Fig. 2(c)]. Please notice that the curvatures in Fig. 2(b) reveal flow fronts rather than flow velocities. Although the flow fronts are nearly parabolic, in Fig. 2(c) we can see that the flow velocity profile is quite flat.
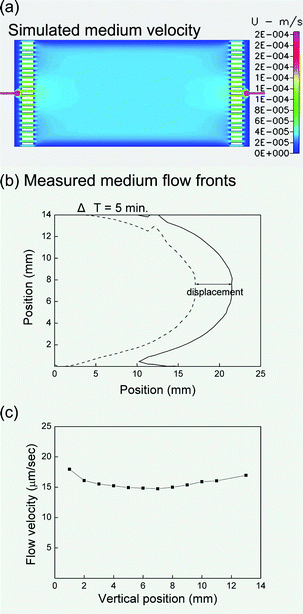 | ||
| Fig. 2 (a) Simulated medium velocity distribution in the flow chamber. (b) Measured flow front of the medium. The dashed and solid lines represent two measured flow fronts with a time interval of 5 minutes. (c) The actual flow velocities at various vertical positions calculated by the displacement and the time interval. The flow rate at the medium inlet is 50 µl/h for all the results in this figure. | ||
In order to confirm an adequate nutrient supply so that cell morphology does not change, a medium replacement experiment was also carried out. It was found that the medium is completely replaced after 2 hours. Since the average cell doubling time is about 48 hours, the simulations and experiments show that cells can be kept in a fresh medium continuously. Our previous cell culture experiment has shown that no observable morphology change under this medium replacement condition.6
When an additional reagent was introduced via the central reagent inlet, asymmetric gradients in the flow chamber were obtained. The CFD simulation and the experiment result of the gradient build-up are shown in Fig. 3. The effusion from the reagent inlet hole was flushed toward the right so that an asymmetric distribution was obtained [Fig. 3(a)]. The simulation clearly shows that steep gradients present at the upper stream of the reagent inlet while a gradual gradient builds up at the downstream side of the inlet. Experimentally the gradient build-up was observed by time-lapse photography of a dye solution injected from the reagent inlet. Although the concentration distributions in micro-fluidic channels are often measured by fluorescence microscopy,30 our approach provides the ability to measure the distribution in a relatively large area with a single exposure. The time lapse digital photographs (2592 × 3888 pixels for a 14 mm × 26 mm area) were used for the concentration plots. The lateral resolution of our digital photographs is about 6.7 µm, smaller than the average cell diameter in this work. The result in Fig. 3(b) shows that stable bell-shaped gradients appear at about 20 minutes after the injection of the dye solution. The distribution of the dye solution is almost identical to the CFD simulation result.
 | ||
| Fig. 3 Gradients of the injected reagent (dye solution) obtained by (a) simulation and (b) experiment, 20 minutes after the injection. The reagent effuses from the central reagent inlet at a rate of 50 µl/h. The flow rate of the culture medium is also 50 µl/h from the left to the right. (c) Measured concentration distributions at two positions A and B, as indicated in panel (b). The gradient at position A is steeper than that at position B. | ||
An important feature of this device is that there is no reagent in the region at the upper stream of the reagent inlet hole. Cells cultured in this “blank” region can readily be used as the control group. From the concentration distribution measured in Fig. 3(c), corresponding concentration gradients were calculated at individual positions where the cells were to be observed. Experiments of various concentration gradients can thus be conducted in a single device with the same flow condition.
Super-resolution bright-field optical microscopy
To achieve lateral resolution smaller than the diffraction limit, a prerequisite is to obtain high-contrast signal from tiny objects. Here we use a technique called non-interferometric wide-field optical profilometry (NIWOP)31 to provide nanometer topographic sensitivity from the filopodia. The principle of NIWOP is to use the sharp slope of the axial response curve of optical sectioning microscopy for nanometer sensing along the direction of optical axis. The depth sensitivity can be as high as 2 nm on solid-state samples. Using a maximum-likelihood estimation algorithm to restore the topographic images of NIWOP, we achieved lateral resolution around 70 nm on static specimens at an illumination wavelength of 365 nm.22Because the diameters of filopodia are usually larger than 100 nm, NIWOP can easily detect the topographic signal of filopodia relative to the flat bottom of the culture chip. Thus we are able to get high contrast images and then perform the super-resolution image restoration to improve the lateral resolution. We demonstrated this idea on cells cultured in a conventional culture dish.32 The resolution enhancement was performed by using the Huygens image-deconvolution software package (Scientific Volume Imaging BV, Hilversum, The Netherlands), and the final resolution on living cells was nearly 120 nm.
Fig. 1(b) shows the optical setup in this work. The NIWOP system was constructed on an upright optical microscope (Nikon Eclipse LV150, Kanagawa, Japan). The objective was a water-immersion one with a 1.2 numerical aperture (NA) and a 270-µm working distance (Nikon CFI Plan Apo VC60 × WI, Kanagawa, Japan). The reflection light from the filopodium surface and that from the chip bottom contain the topographic contrast required for the super-resolution image restoration. We placed the microscope into a temperature-controlled chamber such that mechanical instability caused by thermal expansion was minimized and the cells stayed in the physiological temperature. To preserve long-term viability of the cells, we used filters to set the illumination wavelength range as 550–700 nm. The optical sectioning ability was from a one-dimensional grid pattern (Edmund Optics W56608, Singapore) installed on the illumination light path. By moving the grid pattern across the optical axis and taking three images (I1, I2, I3) at the spatial phases 0, 2π/3, 4π/3, and then using the equation  to process the three images, one can obtain an optically sectioned image.33 At present the highest image-acquisition rate of this system is 20 frames/min (60 modulated images/min) and the exposure time of each modulated image is 0.5 seconds, such that most cellular dynamics can be captured in real-time and then analyzed. The grid pattern was installed on a motorized flipper mount such that we could also acquire conventional bright-field reflection images by removing the grid from the light path. For more details about this setup and the operation of the NIWOP system, please refer to our previous publications.22,31,32
to process the three images, one can obtain an optically sectioned image.33 At present the highest image-acquisition rate of this system is 20 frames/min (60 modulated images/min) and the exposure time of each modulated image is 0.5 seconds, such that most cellular dynamics can be captured in real-time and then analyzed. The grid pattern was installed on a motorized flipper mount such that we could also acquire conventional bright-field reflection images by removing the grid from the light path. For more details about this setup and the operation of the NIWOP system, please refer to our previous publications.22,31,32
We compare the difference of conventional bright-field reflection image and the super-resolution NIWOP image of the same area on a cell in Fig. 4. The super-resolution NIWOP image [Fig. 4(c)] possesses much better contrast and resolution than the bright-field reflection image, and therefore enables the quantification of filopodia without fluorescence labelling. In the following experiments, we used contrast larger than 20% and a protrusion length longer than 1 µm as the criterions for one filopodium count in the super-resolution NIWOP images.
 | ||
| Fig. 4 (a) Bright-field reflection image of a CL1-0 cell acquired when the grid pattern was removed from the light path. (b) Enlarged view of the region in the dashed square in (a). Some filopodia are identifiable but unclear. (c) Super-resolution NIWOP image of the same area in (b). A number of identifiable filopodia are indicated by arrows. | ||
Cell line and reagents
The cells used in this work were of human lung adenocarcinoma cell line CL1-0.34 We cultured the cells in Leibovitz L-15 Medium with 10% fetal bovine serum (FBS) and 1% antibiotic pen-strep-ampho in the cell culture chip. Before the observation we reduced the concentration of FBS to 0.02%. No other treatment to the cells was performed before and during the observation.The reagents for concentration gradient tests were EGF (molecular weight = 6045 Da) and BSA (molecular weight = 66 kDa). Both were from Sigma-Aldrich Co.
Results and discussion
Effect of the medium flow field on filopodium growth
Before the chemical concentration gradient assay, we first tested if there was any potential effect of the medium flow field on the dynamics of filopodia. We used the super-resolution NIWOP image as in Fig. 5(a) to count the filopodia of a single CL1-0 cell. The direction of flow in this image is from the top to the bottom. Therefore we divide the cell with a horizontal dashed line and compare the numbers of filopodia on the upper and lower sides. We set the image acquisition rate as one frame (3 modulated images) per 10 seconds. In 1000 seconds we obtained 100 super-resolution NIWOP images of one cell. Then we counted the filopodia on the upper and lower sides of the cell in each super-resolution image and plotted the filopodium numbers on the two sides along with time in Fig. 5(a). The temporal variations shown in Fig. 5(a) verify that the medium flow field does not induce different filopodium growths on the two sides of this cell. We repeated the same measurement on 30 cells, and calculated the difference between the filopodium numbers (average in 1000 seconds) on the upper and lower sides of each cell. The results in Fig. 5(b) show that the number differences of filopodia do not have observable responses to the medium flow field. This is an essential prerequisite for the temporal analyses of filopodium growth of the cells inside this culture chip. In the following treated and control experiments, we employed the same statistics for the analyses of dynamical filopodium growth.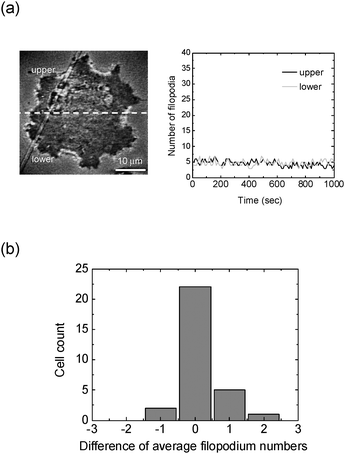 | ||
| Fig. 5 (a) Temporal variation of the numbers of filopodia when a CL1-0 cell is placed inside the culture chip with a stable medium flow field from the top to the bottom. The numbers of filopodia are counted on the super-resolution NIWOP image as shown in this figure. (b) The differences between the filopodium numbers (average in 1000 seconds) on the upper and lower sides of 30 cells. 22 cells show differences within ±0.5; and the largest difference is smaller than 2.5. | ||
Effect of EGF concentration difference on filopodium growth
Next we studied the effects of chemical concentration gradients on filopodium growth. We chose the cells close to the reagent inlet such that the gradients were more evident. On the basis of the experimental measurement of the concentration gradients in Fig. 3(c), we could calculate the concentration difference that the cell experienced at its position of observation.Fig. 6 shows the change in filopodium numbers of a CL1-0 cell before and after the treatment of EGF. The flow rate of EGF was 50 µl/h, while the concentration at the reagent inlet was 100 ng/ml. As shown in Fig. 3(b), we measured the two-dimensional concentration distribution C(x,y) in this chip. From the measured data we can calculate the concentration gradients Γ by using this equation, Γ(x,y) = ∇C(x,y), and therefore obtain the directions of gradients at every cell's position. The dashed lines in the images of Fig. 6 are normal to the direction of Γ at this observation position. In the image in Fig. 6(b), the filopodia at the edge toward the high-concentration area (the front side) are more than those at the edge toward the low-concentration area (the rear side). From the temporal variations of the filopodium numbers in Fig. 6(b), we see that the filopodia at both the front and rear sides increase after the treatment of EGF. However, the increase on the front side is much more significant than that on the rear side. The data in Fig. 6 also demonstrate that the numbers of filopodia are quite dynamic and hence temporal analyses on living cells are important for correct descriptions of cell responses to chemical concentration gradients.
 | ||
| Fig. 6 Super-resolution NIWOP images and temporal variations of the filopodia numbers of a CL1-0 cell. (a) Before the injection of EGF. The image shown here corresponds to the data points at 860 second in the temporal measurement. (b) After the injection of EGF. The starting point of the temporal measurement is the 20th minute after the injection. The image shown here corresponds to the data points at 670 second in the temporal measurement. The dashed lines in the images are normal to the direction of the calculated concentration gradient at this position. For convenience of description, we name the side facing the higher concentration as “front,” and the other as “rear.” Arrows mark the identifiable filopodia. | ||
In order to improve the confidence that filopodium growth indicates the direction of concentration gradients in a controlled micro-environment, we repeated the EGF treatment experiment on 30 cells at various positions in the micro culture chip. These cells were of similar diameters; and the different concentrations were mainly caused by various positions. Fig. 7(a) shows the increase of filopodia at the front sides (ΔNf) and rear sides (ΔNr) of each cell after the treatment of EGF. For each cell, the numbers shown in Fig. 7(a) are averages in 1000 seconds, starting from the 20th minute after the injection of EGF. Obviously the increase of filopodia on the front side is more than that on the rear side for every cell. However, because the original and increased filopodium numbers of individual cells were quite different, we cannot obtain a direct comparison among the measurements at these concentration differences. For a clearer exhibition of the data, we plot ΔNf/ΔNr versus the concentration differences in Fig. 7(b). This quantification shows that ΔNf/ΔNr of filopodia on a single cell is well proportional to the concentration differences established around the vicinity of this cell. This result suggests that the EGF concentration gradient efficiently triggers asymmetric filopodium activities.
 | ||
| Fig. 7 (a) Changes of filopodium numbers (average in 1000 seconds) at the front (ΔNf) and rear (ΔNr) sides of 30 cells at various positions inside the culture chip under the treatment of EGF. The EGF concentration differences are obtained as the products of the cell diameters and the concentration gradients at each cell's positions. Error bars represent the errors of the temporal average values. (b) The ratio ΔNf/ΔNr at various concentration differences of EGF. The straight line is a linear fit to the data points. | ||
Effect of BSA concentration difference on filopodium growth
As a control experiment, we injected BSA at a concentration of 100 ng/ml into the central reagent inlet and performed the same quantifications of filopodia on CL1-0 cells as the experiments of EGF. Fig. 8 shows the changes of average filopodium numbers at the front (ΔNf) and rear (ΔNr) sides of 30 CL1-0 cells at various positions in the micro culture chip. The values are averages in 1000 seconds, starting from the 20th minutes after the injection of BSA. We notice that all the values of changes are much smaller than those induced by EGF. In addition, the filopodium numbers do not show a correlated response to BSA concentration differences. Because the changes of filopodium numbers at both sides of all the cells are less than 3, we cannot reach a meaningful conclusion about the effects of BSA concentration gradients on the filopodium growth. Nevertheless, compared with the experimental results of EGF, it is very sure that the asymmetric filopodium growth is induced by the effect of EGF, rather than solely by the chemical concentration gradients. | ||
| Fig. 8 Changes of filopodium numbers (average in 1000 seconds) at the front (ΔNf) and rear (ΔNr) sides of 30 cells at various positions inside the culture chip under the treatment of BSA. The BSA concentration differences are obtained as the products of the cell diameters and the concentration gradients at each cell's positions. Error bars represent the errors of the temporal average values. Please note that all the changes in filopodium numbers are less than 3. | ||
Conclusions
In this work, we combined advanced far-field optical microscopy with resolution better than the diffraction limit and a well-designed multi-gradient cell culture chip for the study on filopodium dynamics of cancer cells. Stable chemical concentration gradients were established in the middle of the flow chamber by introducing a reagent via a central inlet hole of the culture chip. The upper stream of the central inlet was free of the injected reagents such that one could conduct control and treated experiments using a single culture chip. The micro cell culture chip allows long-term and real-time studies of cell dynamics under stable concentration gradients. The optical imaging with enhanced lateral resolution enables label-free observations of the filopodia of cancer cells at various concentration gradients, and hence temporal variations of filopodia could be quantified and analyzed according to their micro-environments. We used EGF as the treatment to induce the asymmetric growth of filopodia. We also compared the dynamics of filopodia induced by concentration gradients of BSA. The changes of filopodium numbers are much smaller than those induced by EGF, verifying that the asymmetric growth of filopodia responds to the growth factor, rather than merely to the concentration gradients in this culture chip.Although the local filopodium growth stimulated by growth factors has been known on the basis of observations using fluorescence microscopy, the studies in this work reveal filopodium activities on single cancer cells in controlled chemical concentration gradients with unprecedented resolution. We obtained the proportionality between the increase ratios of filopodium numbers on two sides of a cell and the concentration differences, which would be useful to characterize the movements of cancer cells induced by the gradients of trace bio-factors. In addition, because the period required for quantifying asymmetric growth of filopodia is much shorter than that for observing cell chemotaxis, the effects of specific chemicals or cytogenetic factors to cell motility can be assessed more quickly. The label-free, high-resolution observation technique of filopodium activities inside a controllable micro-environment would be extremely useful for the assays of cancer cell metastasis.
Acknowledgements
This work was financially supported by the National Science Council of Taiwan (contracts NSC 95-2113-M-001-037-MY3, NSC 96-2112-M-001-002 and NSC 96-2314-B-002-159).References
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- C. E. Sims and N. L. Allbritton, Lab Chip, 2007, 7, 423–440 RSC.
- A. Shamloo, N. Ma, M. Poo, L. L. Sohn and S. C. Heilshorn, Lab Chip, 2008, 8, 1292–1299 RSC.
- A. W. Blau and C. M. Ziegler, J. Biochem. Biophys. Methods, 2001, 50, 15–27 CrossRef CAS.
- M. Stangegaard, S. Petronis, A. M. Jørgensen, C. B. V. Christensen and M. Dufva, Lab Chip, 2006, 6, 1045–1051 RSC.
- J.-Y. Cheng, M.-H. Yen, C.-T. Kuo and T.-H. Young, Biomicrofluidics, 2008, 2 Search PubMed 024105.
- J. V. Small, T. Stradal, E. Vignal and K. Rottner, Trends Cell Biol., 2002, 12, 112–120 CrossRef CAS.
- L. M. Machesky, FEBS Lett., 2008, 582, 2102–2111 CrossRef CAS.
- T. M. Svitkina, E. A. Bulanova, O. Y. Chaga, D. M. Vignjevic, S. Kojima, J. M. Vasiliev and G. G. Borisy, J. Cell Biol., 2003, 160, 409–421 CrossRef CAS.
- D. S. Lidke, K. A. Lidke, B. Rieger, T. M. Jovin and D. J. Arndt-Jovin, J. Cell Biol., 2005, 170, 619–626 CrossRef CAS.
- M. A. Partridge and E. E. Marcantonio, Mol. Biol. Cell, 2006, 17, 4237–4248 CrossRef CAS.
- J. M. Schober, Y. A. Komarova, O. Y. Chaga, A. Akhmanova and G. G. Borisy, J. Cell Sci., 2007, 120, 1235–1244 CrossRef CAS.
- W. A. Carrington, R. M. Lynch, E. D. W. Moore, G. Isenberg, K. E. Fogarty and F. S. Fay, Science, 1995, 268, 1483–1486 CAS.
- M. G. L. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS.
- J. T. Frohn, H. F. Knapp and A. Stemmer, Proc. Natl. Acad. Sci. USA, 2000, 97, 7232–7236 CrossRef CAS.
- V. Westphal and S. W. Hell, Phys. Rev. Lett., 2005, 94, 143903 CrossRef.
- K. I. Willig, S. O. Rizzoli, V. Westphal, R. Jahn and S. W. Hell, Nature, 2006, 440, 935–939 CrossRef CAS.
- M. M. Knight, S. R. Roberts, D. A. Lee and D. L. Bader, Am. J. Physiol. Cell Physiol., 2003, 284, C1083–C1089 CAS.
- S. Landry, P. L. McGhee, R. J. Girardin and W. J. Keeler, Opt. Express, 2004, 12, 5754–5759 CrossRef.
- J. W. Dobrucki, D. Feret and A. Noatynska, Biophys. J., 2007, 93, 1778–1786 CrossRef CAS.
- C.-H. Lee, H.-Y. Chiang and H.-Y. Mong, Opt. Lett., 2003, 28, 1772–1774 Search PubMed.
- S.-W. Huang, H.-Y. Mong and C.-H. Lee, Microsc. Res. Tech., 2004, 65, 180–185 CrossRef.
- N. L. Jeon, H. Baskaran, S. K. W. Dertinger, G. M. Whitesides, L. V. D. Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–830 CAS.
- G. M. Walker, J. Sai, A. Richmond, M. Stremler, C. Y. Chung and J. P. Wikswo, Lab Chip, 2005, 5, 611 RSC.
- M. Yang, J. Yang, C.-W. Li and J. Zhao, Lab Chip, 2002, 2, 158 RSC.
- C. W. Frevert, G. Boggy, T. M. Keenan and A. Folch, Lab Chip, 2006, 6, 849–856 RSC.
- B. G. Chung, F. Lin and N. L. Jeon, Lab Chip, 2006, 6, 764–768 RSC.
- T. M. Keenan, C.-H. Hsu and A. Folch, Appl. Phys. Lett., 2006, 89, 114103 CrossRef.
- J.-Y. Cheng, C.-W. Wei, K.-H. Hsu and T.-H. Young, Sens. Actuators B, 2004, 99, 186 CrossRef.
- A. Bancaud, G. Wagner, K. D. Dorfman and J.-L. Viovy, Anal. Chem., 2005, 77, 833–839 CrossRef CAS.
- C.-H. Lee, H.-Y. Mong and W.-C. Lin, Opt. Lett., 2002, 27, 1773–1775 Search PubMed.
- T.-H. Hsu, W.-Y. Liao, P.-C. Yang, C.-C. Wang, J.-L. Xiao and C.-H. Lee, Opt. Express, 2007, 15, 76–82 CrossRef.
- M. A. A. Neil, R. Juskaitis and T. Wilson, Opt. Lett., 1997, 22, 1905–1907.
- J.-Y. Shih, S.-C. Yang, T.-M. Hong, A. Yuan, J. J. W. Chen, C.-J. Yu, Y.-L. Chang, Y.-C. Lee, K. Peck, C.-W. Wu and P.-C. Yang, J. Natl. Cancer Inst., 2001, 93, 1392–1400 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |