DOI:
10.1039/B915978A
(Paper)
J. Mater. Chem., 2009,
19, 8470-8477
Synthesis and characterization of a novel kind of near-infrared electrochromic polymers containing an anthraquinone imide group and ionic moieties†
Received
4th August 2009
, Accepted 17th September 2009
First published on 7th October 2009
Abstract
A novel near-infrared (NIR) electrochromic polyelectrolyte consisting of anthraquinone imide (AQI) pendants and a poly(1-vinylimidazole bromide) main chain was synthesized viaradical polymerization. Its AQI content was varied by copolymerization with 1-vinyl-3-butylimidazole bromide to improve the processability of the polymer as well as the monomer conversion. The electrochemical and electrochromic properties of both homopolymer and copolymers were investigated. All the (co)polymers exhibited two reversible redox states (radical anions and dianions). Spectroelectrochemical analysis in solution showed that the radical anions possessed intense NIR absorptions with λmax values in the range 820 to 830 nm, while the dianions exhibited absorptions between 460 and 560 nm. The homopolymer film displayed an optical attenuation of 3 dB/µm at 810 nm with a switching time of 20 s. Such a polymer can change color in a single film device without any additional electrolytes and showed a rapid response time of 1 s at 810 nm.
Introduction
Electrochromism is broadly defined as a reversible optical change in a material induced by a burst of charge.1 A typical electrochromic device (ECD) is a multilayer cell that contains an electrochromic layer (coloring layer), electrolyte, transparent electrodes and background sheet.2 Usually, two electrochromic materials with complementary optical (cathodic and anodic coloring) properties are used to enhance the optical response of the device.3 The electrolyte layer which separates the two electrochromic materials acts as an ionic conductor between the electrodes as well as a source or sink for ions moving through the electrolyte/electrochrome interfaces during electron transfer.4,5 However, such multilayer configuration is not convenient for the enlargement of the device area, and that the interface defect between layers remarkably affects the cell life time.6 While considerable effort has been expended towards the development of high-performance electrochromic materials7–9 and electrolytes,10–13 less attention has been paid to the simplification of the device structure through material innovation. The realization of practical, long-lived electrochromic devices therefore remains a challenge because of performance limitations that include high cost, small area, and short lifetime.
Carpenter and Conell simplified the device structure by using a single film of Prussian blue (PB) as the only electrochemically active element.6 The device could be reversibly bleached by applying a voltage across the film and the removal of voltage resulted in immediate recoloration. The limitation of the device was that the mobility of ions was dependent on the hydration of PB, which was hard to control. Tsutsumi et al. physically blended coloring material and electrolyte to make a single-film electrochromic display device.12 The coloring response was slow probably due to the incomplete mix of the electrochromic material and the electrolyte. Funahashi and coworkers synthesized a liquid crystal consisting of an imidazolium group as an ion conductive part and a phenylterthiophene moiety, associated with the electronic charge transport, and made a single film device based on the compound.14 The device changed colors when the voltage increased to 5 V. The ion-conductive channel was successfully constructed because of the introduction of an ionic liquid group. However, the cell was manipulated at high temperature.
An all-solid-state device with advantages of mechanical stability, safety, and ease of processing is desired since liquid devices present some inherent drawbacks such as leakage. Ionic liquids have been introduced into the polymer structures to obtain polymer electrolytes for solid state actuators due to their chemical stability, low flammability, negligible vapour pressure, high ionic conductivity, and wide electrochemical window. Pioneering works by Ohno and coworkers demonstrated the preparation of different types of polymeric ionic liquids (PILs), as a way of developing high performance polymer electrolytes.15–19 Mecerreyes and coworkers have demonstrated that the cycle life of electrochromic devices (ECDs) is significantly enhanced (up to 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) when a new class of tailor-made polymer electrolyte based ionic liquids (ILs) and polymeric ionic liquids (PILs) analogues are used in poly(ethylene dioxythiophene) (PEDOT)/electrolyte/PEDOT devices, and this is in contrast to previously used poly(ethylene oxide) electrolytes.20
000 cycles) when a new class of tailor-made polymer electrolyte based ionic liquids (ILs) and polymeric ionic liquids (PILs) analogues are used in poly(ethylene dioxythiophene) (PEDOT)/electrolyte/PEDOT devices, and this is in contrast to previously used poly(ethylene oxide) electrolytes.20
On the other hand, near-infrared (NIR) absorbing EC materials are receiving more attention due to their potential applications in biomedicals, optical communications, camouflage materials in warfare, and thermal control and thermal emission detectors for spacecraft.2,21Conjugated polymers such as polyanilines and polythiophenes, mixed-valence metal complexes, rutheniumsemiquinone complexes, and conjugated aromatic diquinones are known to be NIR electrochromic.9,22 For making a flexible electrochemical device (e.g., electrochromic VOA), both anodically and cathodically NIR-coloring organic materials are preferably needed. However, n-type organic semiconductors such as quinones and imides and n-type polymers such as the polythiophenes containing low band gap electron-withdrawing units are less available and less studied than the p-type semiconductors and polymers for optoelectronic applications.23 Cathodically NIR-coloring organic and polymeric materials are scarce and difficult to obtain, in part due to the relatively poor stability of their charged states (e.g., radical anions of anthraquinone and pentacenediquinone).24
AQIs strongly absorb in the wide range 700–1200 nm upon electrochemical reduction to the state of radical anions, and the maxima absorption is in the NIR region. We have established an efficient method for the synthesis of 6-nitro (and bromo)-substituted anthraquinone imides and demonstrated further functionalization through the nitro displacement and Suzuki coupling reactions. The new anthraquinone imides are redox active and NIR electrochromic in a range 700–1600 nm.25 In this paper, we report the synthesis and electrochromic property of a novel near-infrared (NIR) active polyelectrolyte, which consists of poly(1-vinylimidazole bromide) main chain and anthraquinone imide (AQI) pendants. It is expected that the combination of PILs and AQIs can lead to a high performance NIR electrochromic material with good stability, high ionic conductivity, short switching time, and simplified device structure.
Experimental section
Materials
Azobisisobutyronitrile (AIBN, AR, Wuhan Chemical Co.) was recrystallized from ethanol and dried under vacuum at room temperature. Tetrahydrofuran (THF, AR, Beijing Chemical Co.) was refluxed with sodium and distilled out just before use under nitrogen atmosphere. Dimethyl sulfoxide (DMSO, AR, Beijing Chemical Co.) and N,N-dimethylformamide (DMF, AR, Beijing Chemical Co.) were distilled out from calcium hydride. 6-Aminohexan-1-ol (99%) and 1-vinylimidazole (99%) were purchased from Aldrich and were used as obtained. Anthraquinone-2,3-dicarboxylic acid,211-vinyl-3-butylimidazole bromide (ViBuIm+Br−), and poly(1-vinyl-3-butylimidazole bromide) (PViBuIm+Br−)26 were prepared according to the literature. Other chemicals were purchased from Beijing Chemical Reagents Company and were used as received unless noted otherwise.
Measurements
1H NMR and 13C NMR spectra were obtained on a Bruker ARX400 spectrometer at the ambient temperature with CDCl3 or d6-DMSO as the solvents and tetramethylsilane (TMS) as an internal standard. Mass spectra were measured on a Micromass Finnigan MAT ZAB-HS mass spectrometer. Elemental analyses were performed on an Elementar Vario EL instrument. The number average molecular weights (Mn), weight average molecular weights (Mw), and polydispersity indices (PDI, Mw/Mn) of polymers were estimated by a gel permeation chromatography (GPC) system equipped with a Waters 2410 refractive index detector, a Waters 515 HPLC pump and two Waters styragel columns (HT3 and HT4) using DMF as the eluent at a flow rate of 1 mL/min. The columns were kept at a constant temperature of 35 °C. The obtained data were processed against a series of polystyrene standards. Thermogravimetric analyses (TGA) were conducted on a SDT 2960 TA Instruments at a heating rate of 10 °C/min and an air flow rate of 75 cm3/min. Cyclic voltammograms (CV) were performed on a CHI 630C electrochemical workstation. The solutions were made in CH2Cl2 or DMF or THF containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) and were degassed with nitrogen prior to electrochemical operation. Platinum working and counter electrodes were employed together with a silver pseudoreference electrode. All the electrochemical measurements were referenced to Ag/Ag+. The pseudo-reference was calibrated externally using a 5 mM solution of ferrocene (Fc/Fc+). Optical switching and spectroelectrochemical data were obtained on a Shimadzu UV 3100 UV-visible-NIR spectrophotometer connected to a computer. Spectroelectrochemical experiment of solution was conducted in a 2 mm quartz cuvette, platinum mesh working electrode and platinum counter electrode were employed together with a silver pseudoreference electrode. Spectroelectrochemical experiment of the film was conducted in a 1 cm quartz cutette containing a platinum counter electrode and a silver psedoreference electrode. The ITO glasses coated by thin films of polymer (thickness is about 500 nm) were used as the working electrode.
Synthesis
N-6-Hydroxyhexyl-anthraquinone-2,3-dicarboxylic imide (4).
Anthraquinone-2,3-dicarboxylic anhydride (0.94 g, 3.4 mmol) and DMF (15 mL) were added into a 50 mL three necked flask. Under an argon atmosphere, 6-aminohexan-1-ol (0.36 g, 3.4 mmol) was added. After being stirred for 1 h at room temperature, the mixture was heated to reflux for 12 h. After cooling to room temperature, the solution was poured into water. The solids were collected by filtration, washed with water, and dried in air. The product was purified by silica gel chromatography (methanol/dichloromethane: 1/10 (v/v) as eluent) to afford a light yellow powder (0.76 g) in 60% yield. 1H NMR (400 MHz, CDCl3, δ ppm): 8.79 (s, 2H, Ar), 8.38 (dd, 2H, J = 3.3 and 5.8 Hz, Ar), 7.89 (dd, 2H, J = 3.4 and 5.8 Hz, Ar), 3.78 (t, 2H, J = 7.2 Hz, –N–CH2), 3.65 (t, 2H, J = 6.5 Hz, –CH2–O), 1.75 (m, 2H, –CH2), 1.58 (m, 2H, –CH2), 1.43 (m, 4H, –CH2). 13C NMR (100 MHz, CDCl3, δ ppm): 25.19, 26.54, 28.39, 32.51, 38.54, 62.73, 122.52, 127.73, 133.05, 134.90, 135.88, 138.01, 166.61, 181.54. MS (ESI, m/z) for C22H19NO5: 377 (M+). Anal.Calcd for C22H19NO5: C, 70.02; H, 5.07; N, 3.71. Found: C, 70.17; H, 5.15; N, 3.77.
N-6-Bromohexylanthraquinone-2,3-dicarboxylic imide (3).
To a 50 mL three necked flask, 0.5 g (1.3 mmol) of 4 and 15 mL of CHCl3, 1 mL of PBr3 and 3.2 g (2.0 mmol) bromine were added under argon. The mixture was heated to reflux for 12 h. After cooling to room temperature, the solution was washed with water followed by drying over anhydrous Na2SO4 and evaporated to dryness. The product was further purified by silica gel chromatography (dichloromethane as eluent) to afford a light yellow powder (0.38 g) in 65% yield. 1H NMR (400 MHz, CDCl3, δ ppm): 8.78 (s, 2H, Ar), 8.37 (dd, 2H, J = 3.4 and 5.4 Hz, Ar), 7.89 (dd, 2H, J = 3.3 and 5.5 Hz, Ar), 3.77 (t, 2H, J = 7.2 Hz, –N–CH2), 3.40 (t, 2H, J = 6.7 Hz, –CH2–Br), 1.87 (m, 2H, –CH2), 1.75 (m, 2H, –CH2), 1.51 (m, 2H, –CH2), 1.40 (m, 2H, –CH2). 13C NMR (125 MHz, CDCl3, δ ppm): 26.18, 27.81, 28.42, 32.69, 33.78, 38.66, 122.69, 127.69, 133.88, 135.19, 136.00, 138.17, 166.73, 181.68. MS (ESI, m/z) for C22H18BrNO4: 437 (MH+). Anal.Calcd for C22H18BrNO4: C, 60.01; H, 4.12; N, 3.18. Found: C, 60.05; H, 4.11; N, 3.06.
1-Vinyl-3-[6-(1,3,5,10-tetraoxonaphtho[2.3-l]isoindolin-2-yl)hexyl]imidazole bromide (2).
A mixture of 3 (2.0 g, 4.5 mmol) and 1-vinylimidazole (2.0 g, 22 mmol) was heated for 12 h in an oil bath at 70 °C with vigorous stirring. Then the mixture was washed three times with ethyl acetate. The residue was collected by filtration and purified by silica gel chromatography (methanol/dichloromethane: 1/5 (v/v) as eluent) to afford a yellow powder (1.7 g) in 70% yield. 1H NMR (400 MHz, d6-DMSO): δ 9.67 (s, 1H, –N–CH), 8.28 (s, 2H, Ar), 8.26 (m, 1H, –N–CH), 8.15 (dd, 2H, J = 3.4 and 5.5 Hz, Ar), 7.99 (s, 1H, –N–CH) 7.95 (dd, 2H, J = 3.3 and 5.5 Hz, Ar), 7.33 (dd, 1H, J = 8.8 and 15.6 Hz, –N–CH), 6.00 (dd, 1H, J = 1.9 and 15.6 Hz, = CH), 5.42 (dd, 1H, J = 1.8 and 8.6 Hz, = CH), 4.23 (t, 2H, J = 6.9 Hz, –N–CH2), 3.60 (t, 2H, J = 7.1 Hz, –N–CH2), 1.85 (t, 2H, J = 6.4 Hz, –CH2), 1.64 (t, 2H, J = 6.3 Hz, –CH2), 1.35 (t, 2H, –CH2). 13C NMR (100 MHz, d6-DMSO, δ ppm): 25.18, 25.78, 27.71, 29.09, 38.00, 49.24, 108.70, 119.29, 120.74, 123.38, 127.10, 129.02, 132.71, 135.21, 135.50, 137.57, 166.44, 181.24. MS (MALDI-TOF, m/z) for C27H24N3O4+:454. Anal.Calcd for C27H24BrN3O4: C, 60.68; H, 4.53; N, 7.86. Found: C, 61.01; H, 4.70; N, 7.96.
Homopolymerization
.
A mixture of 2 (0.30 g, 0.56 mmol), AIBN(4.6 mg, 0.028 mmol) and DMF (1.7 g) was introduced into a polymerization tube. After three freeze/thaw cycles, the tube was sealed under vacuum and placed into a preheated oil bath set at 60 °C. After 36 h, the tube was opened and the reaction mixture was diluted with DMSO. The polymer was precipitated and washed by ethyl ether. To remove unreacted monomer, the crude product was dissolved and extracted in methanol for one week. The residue was dried under vacuum at room temperature to afford a dark yellow powder (48 mg) in a yield of 16%.
Copolymerization
.
A mixture of 2 (0.030 g, 0.056 mmol), ViEtIm+Br− (0.18 g, 0.50 mmol), AIBN (4.6 mg, 0.028 mmol) and DMF (1.7 g) was introduced into a polymerization tube. After three freeze/thaw cycles, the tube was sealed under vacuum and placed into a preheated oil bath set at 60 °C. After 36 h, the tube was opened and the reaction mixture was diluted with DMF. The polymer was precipitated and washed by ethyl ether. To remove unreacted monomer, the crude product was dissolved and dialysed in methanol for a week. The methanol was changed three times every day. The solution was concentrated with a rotatory evaporator. The residue was dried under vacuum at room temperature to afford a light yellow powder (0.20 g) in a yield of 97%.
Preparation of the polymer film on ITO for optical attenuation studies.
Thin films of the polymers on ITO glass were prepared viaspin casting from the polymer solution (5 wt%) in DMF. The film on ITO was dried at room temperature for 6 h and then thermally cured under nitrogen at 120 °C for 12 h.
Fabrication of a prototype EC device.
The sandwich-type test devices were assembled using two ITO glass-plates coated with the EC materials and pressing them together. The device was assembled under atmospheric condition. After the preparation of the polymer ECD, it was left to dry in a vacuum at 120 °C for 24 h.
Results and discussion
Synthesis of monomer
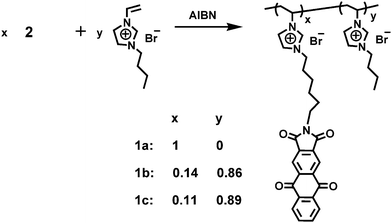
The monomer 1-vinyl-3-[6-(1,3,5,10-tetraoxonaphtho[2.3-l]isoindolin-2-yl)hexyl]imidazole bromide (2) was prepared via a multistep synthetic route as illustrated in Scheme 1. Imidization of anthraquinone-2,3-dicarboxylic anhydride with 6-aminohexan-1-ol followed by bromination with liquid bromine and PBr3 yielded N-6-bromohexylanthraquinone-2,3-dicarboxylic imide (3), which reacted with 1-vinylimidazole to produce the monomer 2. The structures of all the intermediates and monomers were confirmed by NMR, mass spectrometry, and elemental analysis.
![Synthesis of 1-vinyl-3-[6-(1,3,5,10-tetraoxonaphtho[2.3-l]isoindolin-2-yl)hexyl]imidazole bromide (2).](/image/article/2009/JM/b915978a/b915978a-s1.gif) |
| | Scheme 1 Synthesis of 1-vinyl-3-[6-(1,3,5,10-tetraoxonaphtho[2.3-l]isoindolin-2-yl)hexyl]imidazole bromide (2). | |
 |
| | Scheme 2 Preparation of the (co)polymers. | |
To tune the properties of 1a, 2 was copolymerized with ViEtIm+Br−. A similar procedure to the homopolymerization was used. As a result, two new copolymers 1b and 1c were obtained (Scheme 2). The compositions of the copolymers were determined from UV-vis absorptions of AQI according to the correlation curve calibrated using the model compound 3 and poly(ViEtIm+Br−) (Fig. S1, S2 and S3, ESI†) and agreed well with the feed ratio of the two monomers (Table 1). The polymerization yield increased to 97% through the addition of ViEtIm+Br−. This is similar to the quinizarin copolymers synthesized by Ahn and coworkers.28 The copolymers dissolved in not only NMP, DMAc, DMF, and DMSO but also methanol and water. The excellent solubility of the copolymers made them potential candidates for practical applications by spin or dip-coating processes. Furthermore, these copolymers were soluble in the commercially available ionic liquid 1-methyl-3-butylimidazole bromide (MeBuIm+Br−) due to their similar chemical structures. Therefore, these polymers could be the matrix of choice for obtaining interesting gels composed of an ionic liquid and a polymer that are relatively scarce and potentially useful in a number of applications.17,29,301b had a molecular weight of 9.1 × 104 Dalton with a PDI of 1.29 by GPC analysis and 1c had a molecular weight of 12.1 × 104 Dalton with a PDI of 1.25. The decomposition temperatures of 1b and 1c were both around 200 °C in N2.
Table 1 Characterization of AQI-containing polymer
|
Polymer
|
AQI in feed (x:y)a |
AQI in polymer (x:y)b |
Mn (× 10−4)c |
Mw/Mn |
Yield (%) |
Td (°C)d |
|
Feed molar ratio of AQI.
The molar ratio of AQI in polymer as calculated from the UV-vis absorption of each polymer in DMSO.
From GPC measurements in DMF using polystyrene standards.
The onset temperature for 5% weight loss in N2.
|
|
1a
|
100% (0:1) |
— |
10 |
1.18 |
16 |
310 |
|
1b
|
16.7% (1:5) |
13.8% (0.8:5) |
9.1 |
1.29 |
30 |
200 |
|
1c
|
9.1% (1:10) |
10.7% (1.2:10) |
12.1 |
1.25 |
97 |
200 |
Electrochemical and spectroelectrochemical studies in solution
The electrochemical properties of 2 and 1a–c were examined using cyclic voltammetry (CV) in DMF solutions containing 0.1 M TBAP. Reduction potentials were measured relative to the internal standard ferrocene/ferricenium (Fc/Fc+). The electrochemical data are summarized in Table 2. For all the monomers and polymers, two reduction waves were revealed, indicating the formation of stable radical anions and dianions respectively.25 Multi-sweep experiments showed a negligible change after 30 cycles of scanning, suggesting a good reversibility of the process. The typical cyclic voltammograms for monomer 2 and polymer 1a are shown in Fig. 2. There are two chemically reversible redox couples at E1/2 values of −1.15 and −1.62 V for monomer 2 and −1.09 and −1.62 V for polymer 1a in the reductive scan. For 1b and 1c, the E1/2 values are −0.95 and −1.52 V, −1.34 and −1.77 V, respectively. (Fig. S6 and S7, ESI†) It suggested that the electrochemical property of AQI groups was retainable after being grafted to polymer backbones.
Table 2 Electrochemistry of the monomer and polymers
| Code |
In solutiona λ(nm) |
In film λ(nm) |
In solutionb (V) |
In film(V) |
| Anion |
Dianion |
Anion |
Dianion |
E°red1 |
E°red2 |
E°red1 |
E°red2 |
|
c = 5 × 10−3 mol/L in DMF/TBAP (0.1 M).
c = 10−3 mol/L in DMF/TBAP (0.1 M) vs.Fc/Fc+ at 100 mV/s.
|
|
2
|
830 |
540 |
— |
— |
−1.15 |
−1.62 |
— |
— |
|
1a
|
830 |
560 |
810 |
— |
−1.09 |
−1.62 |
−1.01 |
−1.50 |
|
1b
|
820 |
530 |
810 |
540 |
−0.95 |
−1.52 |
−1.04 |
−1.52 |
|
1c
|
820 |
460 |
810 |
540 |
−1.34 |
−1.77 |
−1.50 |
−1.95 |
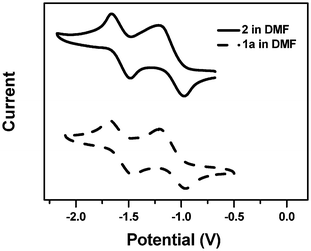 |
| | Fig. 2
Cyclic voltammograms of (a) monomer 2 and (b) polymer 1a in DMF (10−3 M) containing TBAP (0.1 M) at room temperature, scan rate of 100 mV/s, Pt disk working electrode, potentials vs.Ag/AgCl. | |
The electrochromic properties of 2 and 1a–c were analyzed in DMF solution using an optical transparent thin layer cell (OTTLE). All these monomer and polymers exhibited similar electrochromic properties, and the typical electrochromic transmittance spectra of polyamide 1a are shown in Fig. 3. The UV-vis absorption of the neutral state showed a maximum peak around 330 nm with a small shoulder around 420 nm. Upon reduction, the radical anion of 1a shows a maximum peak (λmax) at 830 nm, while 1b and 1c have their λmax both at 820 nm. These NIR absorptions can be attributed to π*–π* (SOMO → LUMO) transitions of the radical anions of the imides.25 Further reduction to dianions resulted in the disappearance of the NIR absorptions and the appearance of new absorption bands at 560 nm for 1a, 530 nm for 1b and 460 nm for 1c. (Fig. S8 and S10, ESI†).
 |
| | Fig. 3
UV-vis-NIR spectra of polymer 1a (5 × 10−3 M containing 0.1 M TBAP) in DMF in its neutral, anionic, and dianionic states. | |
Electrochemical and spectroelectrochemical studies of polymer films
Thin films of the polymers on ITO glass were prepared by spin casting from the polymer solution (5 wt%) in DMF and investigated by cyclic voltammetry in anhydrous THF containing 0.1 M TBAP as an electrolyte under a nitrogen atmosphere. The electrochemical behavior of the films were similar to those in solution. (Fig. S6 and S7, ESI†).
Electrochromism of the polymer thin films was examined by an OTTLE coupled with UV-vis spectroscopy. The electrode preparations and solution conditions were identical to those used in cyclic voltammetry. All the polymer films exhibited similar electrochromic properties, and a typical electrochromic absorbance spectrum of polymer 1a is shown in Fig. 4. Upon electrochemical reduction, the NIR-absorbing radical anionic state was readily reached. The UV-vis absorption peak of polymer 1a shifts from 830 in solution to 810 nm in the solid-state, implying π–π interaction between the AQI groups. For 1b and 1c, the peaks shift from 820 to 810 nm. The smaller shift should result from the addition of copolymer units which retard the π–π interaction. The dianionic states of AQI groups in polymers all exhibited absorption at 540 nm. The absorption of 1b and 1c in the film was weak due to the low content of the AQI group (Fig. S9 and S11, ESI†). As a result, the polymer 1a was chosen for the following characterization.
 |
| | Fig. 4
UV-vis-NIR spectra of polymer 1a in film in its neutral, anionic, and dianionic states (THF containing 0.1 M TBAP). | |
Optical attenuation studies
The studies on dynamic attenuation and optical switching were performed using a quartz cuvette cell, which contained an ITO plate coated with a layer of cured polymer 1a with a thickness of about 500 nm, a silver reference electrode, and a platinum counter electrode. Anhydrous THF containing 0.1 M TBAP was used as the electrolyte solution.31
The optical attenuation can be tuned as a function of the applied voltage and the optical attenuation is calculated as
| | | Optical attenuation = 10(log (Tb/Tc)) | (1) |
Tb is the transmittance in the bleached state and
Tc is the transmittance in the colored state. Optical attenuation increased dramatically with increasing applied potential as shown in
Fig. 5. It reached a maximum at a potential of −2 V. In the more negative range, optical attenuation gradually decreased due to the partial reduction of the
radical anion state to the dianion state. Therefore, the
polymer film coated on the ITO electrode was applied with potentials stepping between its oxidized (+0.4 V
vs.silver
electrode) and reduced states (−2 V
vs.silver
electrode). As shown in
Fig. 6, the film displayed an optical attenuation of 1.5 dB at 810 nm with a switching time of 20 s. Accordingly, its attenuation power is 3 dB/µm in thickness. The attenuation at 540 nm can be seen in the Fig. S12 (ESI
†).
 |
| | Fig. 5 Optical attenuation of film of polymer 1a at 810 nm on ITO glass as a function of applied potential with a switching time of 20 s and a stepping potential (0.4 V vs.silver electrode). | |
 |
| | Fig. 6 Changes in optical attenuation of film (500 nm) of 1a at 810 nm over time with stepping potentials between −2 V and +0.4 V. | |
To probe the effect of the switching time on the redox process and optical attenuation, optical attenuation of the polymer 1a was measured at various switching times between −2 and +0.4 V versussilver electrode. Fig. 7 depicts the optical attenuation as a function of switching time. It exhibited a gradual increase in attenuation with longer switching time.
 |
| | Fig. 7 Dependence of optical attenuation on switching time for film (500 nm) of 1a on ITO glass at 810 nm with stepping potentials between −2 and +0.4 V. | |
Prototype EC device
The cell was assembled in the air with a configuration ITO||polymer 1a||ITO. Construction of the single film ECDs was easy because of the good adhesion and good solubility of the polymer. As we expected, the device exhibited electrochromism without an electrolyte layer. Fig. 8 shows the vis-NIR spectra of the device under polarization of −2 V (colored form) and 0 V (bleached form), respectively. This potential range is sufficient to cause a visually perceptible optical contrast (dark purple to light yellow). The color change should also be attributed to the formation of radical anions and dianions of 1a. The device required one second at −2 V for switching transmittance at 810 nm and one second for bleaching. Such a device demonstrated a rapid response in the NIR region to the applied potentials. The fast optical response of the device indicates fast kinetics of charge transfer. The presence of the dopant anion on the polymer chain was deemed to potentially initiate faster switching within the polymer film. When an electric field is applied to the device, the negative ions would be accumulated on the surface of the electrode forming an electrical double layer. Subsequently, electrical charges should be injected from the electrode into the AQI group of the molecules and transported between them, leading to electrochromism.
However, the optical contrast of the device is low compared to that observed in thin films dipped in electrolyte solutions. The device reached an attenuation of 0.48 dB (0.96 dB/µm) at 810 nm and an increase in the switching time (up to 5 s) did not further expand the attenuation range (Fig. 9). The reason is probably that an efficient electrochromic cell requires two electrochromic materials with complementary optical (cathodic and anodic coloring) properties to enhance the optical response. When a potential is applied to the working electrode of the single film device, the negatively biased region of the film undergoes reduction, with an accompanying color change. The electrochromic material near the counter-electrode remains unchanged. To improve the performance of the device, an anodically-NIR coloring material should be introduced to the system. This part of work is underway and will be reported separately.
 |
| | Fig. 9 Changes in optical attenuation of single film device of 1a at 810 nm over time with stepping potentials between −2 V and 0 V. | |
Conclusions
A novel class of NIR electrochromic polymers containing AQI and ionic moieties are synthesized and characterized for the first time. When electrochemically reduced to a radical anionic state, the polymers exhibit intense NIR absorptions in both solution and film. Owing to the ionic moieties in the polymer, a single-layer NIR electrochromic device without electrolyte layer was demonstrated. The device exhibits a rapid response time of one second. To achieve a larger dynamic range, one needs an AQI-containing polymer as a cathodically NIR-coloring layer and an anodically NIR-coloring polymer such as some p-type polythiophenes and mixed-valence ruthenium complex materials. Above all, this new kind of polymer can be applied to various simple electrochromic devices without an electrolyte layer, which is much easier to prepare and could afford comparable performance with complex devices.
Acknowledgements
The financial support of the National Natural Science Foundation of China through the Key Program (No. 20834001), the National Science Fund for Distinguished Young Scholars (No. 20325415), and the Research Fund for Doctoral Program of Higher Education of MOE (No. 20060001029) are greatly appreciated.
Notes and references
- G. Sonmez, H. B. Sonmez, C. K. F. Shen, R. W. Jost, Y. Rubin and F. Wudl, Macromolecules, 2005, 38, 669 CrossRef CAS.
- B. V. Bergeron, K. C. White, J. L. Boehme, A. H. Gelb and P. B. Joshi, J. Phys. Chem. C, 2008, 112, 832 CrossRef CAS.
- W. A. Gazotti Jr., G. Casalbore-Miceli, A. Geri and M.-A. de Paoli, Adv. Mater., 1998, 10, 60 CrossRef.
-
T. A. Skotheim, R. L. Elsenbaumer, and J. R. Reynolds, Handbook of Conducting Polymers, New York, 1998 Search PubMed.
-
P. M. S. Monk, R. Mortimer, and D. R. Rosseinsky, Electrochromism:Fundamentals and Applications,VCH, Weinheim, Germany, 1995 Search PubMed.
- M. K. Carpenter and R. S. Conell, Appl. Phys. Lett., 1989, 55, 2245 CrossRef CAS.
- L. L. Miller, C. J. Zhong and P. Kasai, J. Am. Chem. Soc., 1993, 115, 5982 CrossRef CAS.
- G. S. Liou and C. W. Chang, Macromolecules, 2008, 41, 1667 CrossRef CAS.
- M. Deepa, S. Bhandari, M. Arora and R. Kant, Macromol. Chem. Phys., 2008, 209, 137 CrossRef CAS.
- A. Lewandowski and A. Swiderska, Solid State Ionics, 2004, 169, 21 CrossRef CAS.
- A. Lewandowski and A. Swiderska, Solid State Ionics, 2003, 161, 243 CrossRef CAS.
- H. Tsutsumi, Y. Nakagawa, K. Miyazaki, M. Morita and Y. Matsuda, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1725 CrossRef CAS.
- D. M. Tigelaar, M. A. B. Meador and W. R. Bennett, Macromolecules, 2007, 40, 4159 CrossRef CAS.
- S. Yazaki, M. Funahashi and T. Kato, J. Am. Chem. Soc., 2008, 130, 13206 CrossRef CAS.
- H. Ohno, Electrochim. Acta, 2001, 46, 1407 CrossRef CAS.
- T. Mizumo and H. Ohno, Polymer, 2004, 45, 861 CrossRef CAS.
- W. Ogihara, J. Sun, M. Forsyth, D. R. MacFarlane, M. Yoshizawa and H. Ohno, Electrochim. Acta, 2004, 49, 1797 CrossRef CAS.
- S. Washiro, M. Yoshizawa, H. Nakajima and H. Ohno, Polymer, 2004, 45, 1577 CrossRef CAS.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2004, 50, 255 CrossRef CAS.
- R. Marcilla, F. Alcaide, H. Sardon, J. A. Pomposo, C. Pozo-Gonzalo and D. Mecerreyes, Electrochem. Commun., 2006, 8, 482 CrossRef CAS.
- Y. He and Y. Zhao, J. Phys. Chem. C, 2008, 112, 61 CrossRef CAS.
-
P. M. S. Monk, R. J. Mortimer, and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, 2007 Search PubMed.
- E. K. Todd, S. Wang, X. Wan and Z. Y. Wang, Tetrahedron Lett., 2005, 46, 587 CrossRef CAS.
- S. Wang, E. K. Todd, M. Birau, J. Zhang, X. Wan and Z. Y. Wang, Chem. Mater., 2005, 17, 6388 CrossRef CAS.
- W. Qiao, J. Zheng, Y. Wang, Y. Zheng, N. Song, X. Wan and Z. Y. Wang, Org. Lett., 2008, 10, 641 CrossRef CAS.
- R. Marcilla, J. A. Blazquez, J. Rodriguez, J. A. Pomposo and D. Mecerreyes, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 208 CrossRef CAS.
-
G. Odian, Principles of Polymerization, John Wiley and Sons, New Jersey, 4th edn, 2004 Search PubMed.
- C.-W. Lee, Z. Yuan, K.-D. Ahn and S.-H. Lee, Chem. Mater., 2002, 14, 4572 CrossRef CAS.
- M. A. B. H. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976 CrossRef CAS.
- B. Singh and S. S. Sekhon, J. Phys. Chem. B, 2005, 109, 16539 CrossRef CAS.
- Y. Qi and Z. Y. Wang, Macromolecules, 2003, 36, 3146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Figures S1–S12. See DOI: 10.1039/b915978a |
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 
