Glycine-glutamic-acid-based organogelators and their fluoride anion responsive properties††
Mingjun
Teng
a,
Guichao
Kuang
a,
Xinru
Jia
*a,
Min
Gao
a,
Yan
Li
a and
Yen
Wei
*b
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of the Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing, China. E-mail: xrjia@pku.edu.cn; Fax: +86 10-62751708; Tel: +86 10-62752102
bDepartment of Chemistry, Drexel University, Philadelphia, USA. E-mail: weiyen@drexel.edu; Fax: +1 215 8952 650; Tel: +1 215 8951 265
First published on 23rd June 2009
Abstract
Novel low-molecular-weight organogelators (LMOGs) 1 and 2 derived from glycine-glutamic-acids (GGA) based dipeptide were synthesized. The difference on the chemical structures between 1 and 2 resulted in some interesting variations on their gelling behavior and fluoride anion (F−) responsive properties. Gelator 1 could gel aromatic solvents effectively, while gelator 2 possessed excellent gelation ability in protic solvents. Upon the addition of 0.5 equiv. F−, the toluene gel of 1 transformed into solution due to the disruption of the intermolecular hydrogen-bonding, whereas the alcohol gel of 2 was stable and preserved even by introducing 20 equiv. F−. Interestingly, 2 could be regarded as an efficient receptor for F− in acetonitrile (MeCN) solution. After the addition of F−, the fluorescent emission band of 2 red-shifted from 360 to 420 nm with the emission intensity enhanced drastically; the intensity of the CD signal decreased and almost disappeared after addition of 2 equiv. F−. These changes were due to the conformational change and hydrogen-bonding interaction between urea groups and F−. The results indicated that GGA-based 1 and 2 might represent potential sensor materials for the naked-eye detection of F−.
Introduction
In recent years, considerable efforts have been focused on the design and synthesis of sensor materials.1 As an important kind of soft materials with diverse applications, low-molecular-weight-organogelators (LMOGs) have shown responsive properties under certain external stimuli (temperature, light, pH, solvents, chemicals, and so on).2 Various organogels displaying chemical, photo, metal, proton, mechanical and redox response have been documented in the last few years.3 As it reported, the structures and properties of some supramolecular gels could be modulated by anions. For example, Džolić et al. developed the oxalamide-derived anthraquinone based organogels that turned into solution with specific recognition towards F−.4 Yi et al. studied a switchable fluorescent organogel, whose fluorescent property and gel state could be selectively controlled by F− addition.5 Very recently, Maeda raised a new concept of anion-responsive supramolecular gels, which correlated supramolecular assembly with anion binding.6Due to the close relationship with human body for dental care and the clinical treatment of osteoporosis, the detection of F− in biological systems and the food industry is particularly attractive.7 However, the reports of organogels displaying F− response are still limited. In addition, not all the gelators containing recognition units such as the urea and amide group can selectively respond to F− rather than other anions possibly due to the complication of multiple interactions between gelators and different anions.8,9
Fluorescence, colorimetry and circular dichroism (CD) have been reported as the simple and easy ways for detecting anions because of the high sensitivity and convenience. For example, Nam et al. synthesized a naphthalene derivative with two phenylurea groups at the 1,8-position, which showed a unique new fluorescent emission band upon binding with F−.10 Zhang et al. reported the CD spectra modulation could be realized by the addition of F−.11 Sessler et al. reported a colorimetric anion sensor with two pyrrole N–H groups, and such a receptor could detect the existence of F− by both naked-eye and UV lamp.12 To our knowledge, there is no report of receptor which can recognize F− with combination of both fluorescence and CD spectrum.
Herein, we describe the synthesis of two F− receptors 1 and 2 and investigate their difference in response towards F− as a consequence of structural change. Both compounds have the same glycine-glutamic-acids (GGA) dipeptide moiety, with 1 containing urea group at the center and 2 using biphenyl group as the linker (Scheme 1). Particularly, the compound 2 was designed with three features: (1) 2,2′-diaminobiphenyl bearing two amino groups may enable a facile recognition of F− due to the suitable separation distance between the two amino groups; (2) the changeable dihedral angle between biphenyl moiety and distance of two urea groups may endow the molecule with optical activity after combining with F−; (3) GGA moiety as the gelation auxiliary would enhance non-covalent forces such as π–π stacking and hydrogen-bonding interactions.
 | ||
| Scheme 1 Synthetic route of compounds 1, 2 and 3. Reagents and conditions: (a) triphosgen, Et3N, CH2Cl2, ambient temperature, 12 h; (b) Fe, HCl, reflux, 48 h; (c) triphosgen, pyridine, CH2Cl2, ambient temperature, 12 h; (d) triphosgen, pyridine, propyl amine, ambient temperature, 12 h. | ||
In the gel preparation, we found that 1 could gel aromatic solvents whereas 2 could gel alcohols solvents. The gel formation of 2 was modulated by heating-ultrasound circle rather than common heating-cooling circle. For 1, naked-eye detection of F− could be achieved through the gel–sol transition. In contrast, 2 showed unique properties for detecting F−via the changes from both fluorescence and CD spectrum in acetonitrile (MeCN) solution. These results demonstrate that the minor change of the structure does have a great impact on their gelation properties and the F− responsive behavior.
Experimental
Characterization techniques
1H NMR spectra were recorded on Brucker 300 MHz spectrometers at room temperature using CDCl3 and d6-dimethyl sulfoxide as solvents and tetramethylsilane as an internal standard. Mass spectrum (MS) was recorded on a ZAB-HS (Micromass Co.) mass spectrometer. ESI high resolution mass-spectra (HRMS) were acquired on a Bruker Apex IV FTMS mass spectrometer. UV-vis spectra were acquired on a Varian CARY 1E UV-vis spectrophotometer at 298 K. Fluorescence excitation and emission measurements were carried out at 298 K using a Hitachi F-4500 fluorescence spectrophotometer. FT-IR spectra were obtained using a Bruker VECTOR22 IR spectrometer. Powder wide-angle X-ray diffraction (WAXD) patterns of xerogels were obtained using a Bruker D8 Discover diffractometer with GADDS as a 2D detector. Air scattering was subtracted from the sample patterns. Diffraction patterns were recorded in a transmission mode at room temperature employing Cu Kα radiation. (Small-angle X-ray scattering) SAXS measurements were performed on equipment with a SAXSess camera (Anton-Paar, Graz Austria) which is connected with an X-ray generator (Philips) operating at 40 kV and 50 Ma employing Cu Kα radiation (λ = 0.154 nm). The 1D scattering function (log I (q)) was obtained by integrating the 2D scattering pattern which was recorded on an imaging-plate detector (Perkin Elmer) using SAXSQuant software (Anton-Paar, Graz Austria). Gel was naturally dried for two days at room temperature before WAXD experiment CD spectra of the gels and solutions were recorded on a Jasco J-810 spectropolarimeter equipped with a Julabo F25 thermostatic apparatus. The samples were dropped into or prepared in a quartz cuvette with a path length of 1 cm, respectively. Transmission electron microscopy (TEM) was applied by a JEM-100 CXII microscope, operating at an acceleration voltage of 100 kV without any staining. Sonication (SY-3100 D) was conducted in water baths with sonication power 150 W.Materials
2,2′-dinitrobisphenyl and triphosgene were purchased from Acros Company; pyridine and the other chemicals were purchased from Beijing Chemicals Corporation and used without further purification unless otherwise specified. The GGA moiety and the focal Boc-deprotected compound were synthesized according to our previous report.13Synthesis and characterization.
Results and discussion
Gelation properties of 1 and 2
The synthesis procedure is shown in the Scheme 1. Gelator 1 was obtained from the reaction of Gly-Glu peptides and triphosgene in CH2Cl2 at ambient temperature for 12 h. Gelator 2 was synthesized involving two steps: 2,2′-diaminobiphenyl was obtained by reducing 2,2′-dinitrobiphenyl with Fe in HCl aqueous solution at 110 °C, and then the Gly-Glu peptides reacted with it in the presence of triphosgene at room temperature.A typical gel preparation procedure was as follows: A specific weight of gelator was dissolved in an organic solvent or mixed solvents to get a homogeneous solution upon gentle heating. After cooling to room temperature (T ≈ 25 °C), the solvent was immobilized without any flow, it could be considered as a gel. Another method for preparation of a gel was applied by simple ultrasound.14 The gelation properties of compounds 1 and 2 in various solvents were evaluated. As shown in Table 1, the gelation ability and efficiency of 1 and 2 changed dramatically resulting from the structural variation. 1 could gel aromatic solvents at a relatively low concentration, while 2 gelled different kinds of protic solvents such as ethanol and isopropyl alcohol. The gelation capability and efficiency of 1 is greater than that of 2, which is supported by the fact that 1 displayed lower critical gel concentration and higher gel–sol transition temperature (Tgel) in toluene at the same concentration (Tgel of 1 is 55 °C while Tgel of 2 is 40 °C, c = 2 × 10−2 M). These results may be due to the steric hindrance of 2, in which the bulky biphenyl group would hinder the interactions of the gelators to some extent. An unexpected phenomena of 2 was that an opaque organogel could form in the mixed solvents (toluene/alcohols = 1:4, v/v) only when the mixture suffered a brief sonification. In contrast, precipitate was obtained in the same mixed solvents after heating–cooling cycle without ultrasound, suggesting the indispensable role of sonification in the preparation of gel from 2. Compared to heat-induced gelation, ultrasound-induced gelation is easy handled and the sample could maintain either solution or gel state at room temperature, while the heat-induced gelation sample can only maintain gel state at room temperature. All the gels were very stable and maintained for several months at room temperature.
| Solvents | 1 | 2 |
|---|---|---|
| a S, P, and G denotes solution, precipitate, gelation, respectively. Numbers in parentheses indicate the critical gel concentration (CGC, mg/mL). | ||
| EtOH | P | G(20) |
| iPrOH | P | G(20) |
| Benzene | G(10) | S |
| Toluene | G(12) | S |
| o-Xylene/CHCl3 (10:1, v/v) | G(24) | S |
| CHCl3 | S | S |
| CHCl2 | S | S |
| THF | S | S |
| Cyclohexane | P | P |
| Ethyl ether | P | S |
| 1,4-dioxane | S | S |
| DMSO | S | S |
| DMF | S | S |
To obtain the visual images of the gel phase assemblies from 1 and 2, transmission electron microscopy (TEM) measurement was performed. The samples were prepared by drying the gel on a grid through evaporating the solvent slowly at room temperature under vacuum. As shown in Fig. 1, both gelators developed well-defined three-dimensional network structures. The gel morphology of 1 prepared in toluene displayed entangled tape-like assemblies of width ranging from 50 to 300 nm and length of several micrometres (Fig. 1a). The gel of 2 prepared in ethanol showed intertwined fibers with larger aspect ratio (length over width). Also, polarized optical microscopy (POM) was used to get insight into the images of the pristine gels. Compared to ethanol gel of 2 with fused and strong birefringent fibers, toluene gel of 1 displayed a less birefringent texture, suggesting that 2 displayed a higher degree of anisotropic molecular arrangement.
 | ||
| Fig. 1 TEM images of (a) 1 xerogel made in toluene (12 mg/mL) and (b) 2 xerogel (20 mg/mL) made in ethanol. Insets show the POM images of corresponding pristine gels (scale bars = 200 µm and 300 µm, respectively). | ||
It is well-known that the hydrogen-bonding and aromatic π–π stacking are the major driving forces responsible for the self-assembly of organogelators in organic solvents.15–18 The hydrogen-bonding interaction among the amide and urea groups was easily proved by the FT-IR measurements.19,20 As shown in Fig. 2, comparison of FT-IR spectroscopy of 1 in toluene gel state and in the solution demonstrated that the characteristic peaks of amide I, II bands at 1622 and 1543 cm−1 in the gel state shifted to 1641 and 1521 cm−1 in the sol state, respectively, and the peak corresponding to the N–H stretching vibration shifted from 3371 to 3448 cm−1, indicating the presence of urea–urea, urea–amide or amide–amide hydrogen bonds. Due to the ability of 2 in gelling protic-aromatic mixed solvents and some alcohols that strongly competed for hydrogen-bonding formation, we assumed that the hydrogen-bonding and aromatic π–π stacking may both contribute to the gel formation.21–24
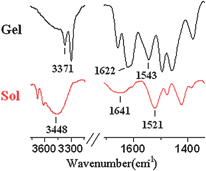 | ||
| Fig. 2 Infrared spectra of solution and gel from 1 in toluene. | ||
Detailed microstructures of the gel were elucidated by wide angle X-ray diffraction (WAXD) and small angle X-ray scattering (SAXS). From the WAXD pattern of the xerogel 1 formed in toluene, a series of sharp peaks was observed, demonstrating a typical feature of crystalline-like order structure. We have tried different gel preparation ways that showed the identical results. The WAXD pattern of 1 exhibited a strong peak at 2θ = 4.06° (d-spacing of 2.22 nm), which was consistent with the size of the molecule (Fig. S2–3, ESI†). Although it was hard to interpret the WAXD pattern into a specific molecular packing pattern, the observed diffraction pattern suggested the ordered arrangements of 1 in the gel state to some extent. On the other hand, the SAXS pattern of xerogel 2 formed in the mixed solvents (toluene/alcohols = 1:4, v/v) displayed three peaks in the low angle region (Fig. 3). Their scatter vector ratios were 1: 3: 4, which could be indexed as (100), (300) and (400) diffractions. The result implied the existence of a lamellar packing pattern of 2 with the d spacing of 4.20 nm. Considering that molecular length of 2 was 2.29 nm (Fig. S4, ESI†), the d spacing was larger than the molecular length but smaller than twice of it. A bilayer interdigitated packing of 2 was proposed (Fig. S5, ESI†).
 | ||
| Fig. 3 SAXS pattern of 2 xerogel made from toluene and alcohols (1:4, v/v). | ||
Fluoride anion responsive properties
The most remarkable feature of the synthesized compounds was their different response towards F− with 1 in gel state and 2 in solution, resulting from the minor structural difference. The anion binding property of 1 in the gel state was investigated by adding certain amounts of different anions to the toluene gel. Interestingly, the addition of tetrabutylammonium fluoride (TBAF) resulted in a rapid transition from a translucent gel to a homogenous solution (Fig. 4a). The solution could not turn back into a stable gel regardless of heating or ultrasound. However, the gel state of 1 could be preserved after adding the same amount of other halide anions (Cl−, Br−, I−). The results indicate that the compound 1 possesses a high selectivity towards F− with naked-eye sensing simply by gel–sol transition. | ||
| Fig. 4 (a) Photographs of 1 organogel formed in toluene (12 mg/mL) and the collapsing gel by adding 1 equiv. TBAF to the gel. (b) Photographs of 2 organogel formed in toluene/alcohols (1:4, v/v) (20 mg/mL) and the retained gel by adding 20 equiv. TBAF. | ||
In order to further understand the interaction between 1 and F−, the 1H NMR spectra of 1 were recorded by adding different amounts of F−. As shown in Fig. S6, ESI† with 0.5 equiv. F− addition, the proton signal assigned to the urea group disappeared and the amide N–H signal shifted downfield from 7.41 to 7.95 ppm. Subsequently increasing the concentration of F− led to a more obvious downfield shift and finally to the disappearance of the amide N–H signal. This implied that the amide groups and urea groups either interacted with F− through hydrogen-bonding or were deprotonated by excess amounts of F−. Thus, it is reasonable to assume that the gel–sol transition of 1 originated from the disruption of hydrogen-bonding interaction between the urea and amide groups in the presence of F−.25,26 CD spectroscopy was also recorded after the addition of different anions (Fig. S7, ESI†). We found that the band of CD signal at 220 nm assigned to CONH group27 was enhanced and red-shifted to 226 nm after the introduction of F−. However, the addition of other anions such as Cl− and AcO− only resulted in a neglectable change in CD spectrum. It is possible that the strong interaction between CONH groups and F− was responsible for the change of CD spectra. In addition, the F− responsive property of 1 in sol state was also examined by UV-vis and fluorescent spectrometry. However, no obvious absorption and fluorescent change were observed (Fig. S8, ESI†).
In sharp contrast, it was interesting to find that the ethanol organogel of 2 retained without any change even by adding 20 equiv. TBAF. In order to exclude the diffusion factor, we heated 2 in the mixture of ethanol and TBAF until it became a homogeneous solution, for which could enhance the interaction between F− and 2. To our surprise, a stable gel was obtained as the solution was cooled to room temperature under ultrasound. It was likely that the F− in a polar protic solvent behaved as a less efficient competitor to deprotonate the N–H groups.4 We tried to use toluene/alcohols mix-solvents to weaken the interaction between alcohols and F−. However, the gel still retained stable after addition of 20 equiv. TBAF (Fig. 4b). FT-IR spectra of 2 in gel state showed that the amide I, II bands were nearly identical before and after F− addition (Fig. S9, ESI†), indicating that hydrogen-bonding interaction was not significantly influenced by the addition of F−. In addition, we have also investigated the impact of the size of cations on the gelation property of 2. After adding three kinds of cations with different sizes separately, we found that the size of cations had neglectable effect on the gel behavior (Fig. S10, ESI†).
The most interesting property of 2 was the selective response towards F− in MeCN solution. The experiment was carried out by adding certain amounts of tetrabutylammonium salt of the envisaged anion (F−, Cl−, Br−, I −, and AcO−) into the solution of 2. Of all the anions tested, 2 combined selectively with F− over other anions in MeCN. It is likely that F− possesses the suitable size to the core of two urea groups of 2, which resulted in the strongest ability to combine with the N–H groups.
UV-vis spectrometry was performed to evaluate the recognition ability of 2 towards F− in MeCN. As shown in Fig. S4a, ESI† during the addition of different amounts of TBAF to the solution of 2, the absorption band at 250 nm decreased greatly, and a new band at 305 nm appeared and increased concomitantly. In sharp contrast, the UV-vis spectra showed neglectable change after addition of other target anions (Fig. S11, ESI†), which strongly indicated the specific recognition of 2 towards F−.
The fluorescent spectra were obtained with the excitation of 2 at 280 nm. As shown in Fig. 5, upon the addition of F−, the fluorescence spectra changed drastically. A unique new peak emerged and developed substantially at 420 nm with a shoulder band at 400 nm, while the intensity of the original emission band at 360 nm decreased concomitantly. Notably, a bathochromic shift up to 60 nm was generated after addition of 5 equiv. F−. Moreover, the excitation spectra monitored at 400 nm were red-shifted from 280 nm to 300 nm after F− addition, indicating the formation of a new complex (Fig. S12, ESI†). However, addition of other anions (Cl−, Br−, I−, and AcO−) only caused neglectable variation in fluorescent spectra (Δλ < 15 nm), demonstrating the pronounced selective recognition of 2 towards F−. It is assumed that the conjugation and electronic communication between N–H and biphenyl group might be affected by F−,28 which is responsible for the red-shift of the fluorescent emission.
 | ||
| Fig. 5 Emission spectra of 2 at the concentration of 1 × 10−4 M in MeCN with different amounts of (a) F−; (b) different kinds of anions: F−, Cl−, Br−, I− and AcO−. λex = 280 nm. | ||
It was worth noting that the emission band of 2 varied from UV to visible region by adding F−, which made it an efficient F− sensor with naked-eye detection upon illumination at 254 nm under common UV light. As shown in Fig. 6, a strong blue emission induced by addition of F− was observed in the solution of 2. In contrast, addition of other anions such as Cl−, Br−, I−, and AcO− did not result in noticeable fluorescent emission at the same excited wavelength. Fig. S13, ESI† showed the intensity ratios of 420 to 360 nm upon addition of different anions. The result clearly suggested the high selectivity of 2 towards F−.
 | ||
| Fig. 6 Photographs of free 2 in MeCN solution and 2 by adding F−, Cl−, Br−, I−, and AcO− under illumination at 254 nm. | ||
CD measurement was also performed to probe the F− responsive property of 2. As shown in Fig. 7a, upon addition of F−, the intensity of CD signal at 240 nm, which was assigned to the absorbance of the two urea groups, decreased gradually. According to the report of Sakurai,29 it might be assumed in our case that the distance between the two urea groups and the dihedral angle of the biphenyl unit were altered in the presence of F−. On the other hand, addition of other anions only led to a slight change in the CD signals (Fig. 7b), denoting that the binding between those anions and sensor 2 was negligible. This feature allows us to distinguish F− from others through the signal intensity of CD spectrum.
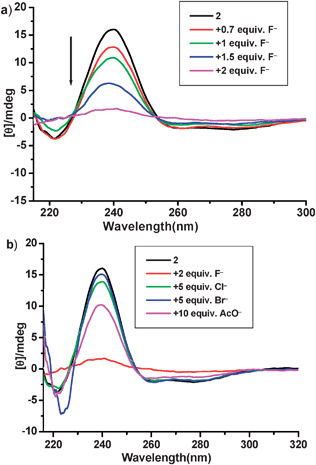 | ||
| Fig. 7 (a) CD spectra of 2 in MeCN at 1 × 10−4 M with different amounts of F−; (b) CD spectra after addition of other anions such as Cl−, Br−, and AcO−. | ||
Since 2 possessed several NHs competing for the anions and multiple equilibriums existed, it was difficult to calculate the association constants between urea group and F− precisely. Therefore, compound 3 with bisurea unit was synthesized and used as a model compound to examine the combination with F−. The titration curve was well fitted to a 1:1 binding isotherm, suggesting the formation of the complex [3·F−] (Fig. S14, ESI†).
The possible explanation of selective response toward F− might be as follows: to reduce the steric hindrance and repulsion interaction, the biphenyl group preferred to keep a nonplanar arrangement and two urea groups were located far from each other as presented in Scheme 2. The addition of F− would destroy the preferred conformation and cause the rotation of biphenyl group to achieve a greater planar complex.30 Therefore, it was understandable that 2 and F− formed a more rigid complex in which F− was located at the core generated by urea groups, which increased the intensity of fluorescent emission.

Additionally, the F− selective recognition is rationalized based on the size and basicity of the anions. With the smallest size, F− matches best to the core formed by bisurea groups when compared to other anions. In addition, the highest electronegative of the F− makes it feasible to interact with urea groups strongly and modulate their distance, which results in the great variation in fluorescent and CD spectra. Moreover, the appropriate distance of urea groups endows 2 as a proper receptor for F−, thus promoting the recognition selectivity.
Conclusion
In conclusion, the gelators 1 and 2 were prepared both with the same GGA moieties and different central groups. The difference on the central structures between 1 and 2 resulted in great variation in gelation property. Both 1 and 2 displayed selective recognition of F− over other anions. Upon addition of F−, toluene gel of 1 transformed to solution because of the disruption of intermolecular hydrogen bonds, while 2 in MeCN solution underwent conformational alteration with strong blue emission under common UV light. The compound 2 is a rare receptor recognizing F− by both fluorescent and CD spectra.Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (NSFC No. 20774003) and National Basic Research Program of China (973 Program, 2007CB935800-2007CB935801) to X.R.J. We thank Dr J. X. Cui for helpful discussions on CD measurement.References
- (a) S. Yagai, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2005, 11, 4054 CrossRef CAS; (b) M. Z. Alam, T. Yoshioka, T. Ogata, T. Nonaka and S. Kurihara, Chem. – Eur. J., 2007, 13, 2641 CrossRef.
- K. S. Moon, H. J. Kim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2007, 46, 6807 CrossRef CAS; N. Koumura, M. Kudo and N. Tamaoki, Langmuir, 2004, 20, 9897 CrossRef CAS; S. I. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592 CrossRef CAS; J. L. Pozzo, G. M. Clavier and J. P. Desvergne, J. Mater. Chem., 1998, 8, 2575 RSC; W. G. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663 CrossRef CAS; W. Deng, H. Yamaguchi, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2007, 46, 5144 CrossRef CAS.
- T. Ishi-i and S. Shinkai, Top. Curr. Chem., 2005, 258, 119 CAS; A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS; A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC.
- Z. Džolić, M. Cametti, A. D. Cort, L. Mandolini and M. Žinić, Chem. Commun., 2007, 3535 RSC.
- H. Yang, T. Yi, Z. G. Zhou, Y. F. Zhou, J. C. Wu, M. Xu, F. Y. Li and C. H. Huang, Langmuir, 2007, 23, 8224 CrossRef CAS.
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274 CrossRef.
- V. Amendola, D. Esteban-Gmez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343 CrossRef CAS; P. A. Gale, Acc. Chem. Res., 2006, 39, 465 CrossRef CAS.
- M. Yamanaka, T. Nakamura, T. Nakagawa and H. Itagaki, Tetrahedron Lett., 2007, 48, 8990 CrossRef CAS.
- T. H. Kim, M. S. Choi, B.-H. Sohn, S.-Y. Park, W.-S. Lyoo and T. S. Lee, Chem. Commun., 2008, 2364 RSC.
- E. J. Cho, J. W. Moon, S. W. Ko, J. Y. Lee, S. K. Kim, J. Yoon and K. C. Nam, J. Am. Chem. Soc., 2003, 125, 12376 CrossRef CAS.
- C. Wang, D. Zhang and D. Zhu, Langmuir, 2007, 23, 1478 CrossRef CAS.
- C. B. Black, B. Andrioletti, C. Try. Andrew, C. Ruiperez and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 10438 CrossRef CAS.
- W. S. Li, X. R. Jia, B. B. Wang, Y. Ji and Y. Wei, Tetrahedron, 2007, 63, 8794 CrossRef CAS; W. S. Li, Y. Ji, X. R. Jia, B. B. Wang and Y. Wei, Acta Polym. Sin., 2006, 712 Search PubMed.
- K. Isozaki, H. Takaya and T. Naota, Angew. Chem., Int. Ed., 2007, 46, 2855 CrossRef CAS; Y. B. Wang, C. L. Zhan, H. B. Fu, X. Li, X. H. Sheng, Y. S. Zhao, D. B. Xiao, Y. Ma, J. S. Ma and J. N. Yao, Langmuir, 2008, 24, 7635 CrossRef CAS.
- M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, S. J. Shirai and K. Hanabusa, Langmuir, 2003, 19, 8622 CrossRef CAS.
- S. J. George and A. Ajayaghosh, J. Am. Chem. Soc., 2001, 123, 5148 CrossRef CAS.
- T. Ishi-I, T. Hirayama, K. Murakami, H. Tashiro, T. Thiemann, K. Kubo, A. Mori, S. Yamasaki and T. Akao, Langmuir, 2005, 21, 1261 CrossRef CAS.
- J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759 CrossRef CAS.
- Y. Kamikawa and T. Kato, Langmuir, 2007, 23, 274 CrossRef CAS.
- Y. F. Zhou, M. Xu, T. Yi, S. Z. Xiao, Z. G. Zhou, F. Y. Li and C. H. Huang, Langmuir, 2007, 23, 202 CrossRef CAS.
- Y. Ji, Y. F. Luo, X. R. Jia, E. Q. Chen, Y. Huang, C. Ye, B. B. Wang, Q. F. Zhou and Y. Wei, Angew. Chem., Int. Ed., 2005, 44, 6025 CrossRef CAS.
- Y. Ji, G. C. Kuang, X. R. Jia, E. Q. Chen, B. B. Wang, W. S. Li, Y. Wei and L. Jiang, Chem. Commun., 2007, 4233 RSC.
- G. C. Kuang, Y. Ji, X. R. Jia, Y. Li, E. Q. Chen and Y. Wei, Chem. Mater., 2008, 20, 4173 CrossRef CAS.
- G. C. Kuang, Y. Ji, X. R. Jia, E. Q. Chen, M. Gao, J. Yeh and Y. Wei, Chem. Mater., 2009, 29, 456 CrossRef.
- T. H. Kim, M. S. Choi, B.-H. Sohn, S. Y. Park, W. S. Lyoo and T. S. Lee, Chem. Commun., 2007, 2364 Search PubMed.
- R. Varghese, S. J. George and A. Ajayaghosh, Chem. Commun., 2005, 593 RSC.
- C. S. Love, A. R. Hirst, D. K. VictorChechikSmith, I. Ashworth and Colin Brennan, Langmuir, 2004, 20, 6580 CrossRef CAS.
- S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Org. Biomol. Chem., 2003, 1, 741 RSC.
- Y. Jeong, K. Hanabusa, H. Masunaga, I. Akiba, K. Miyashi, S. Sakurai and K. Sakurai, Langmuir, 2005, 21, 586 CrossRef CAS.
- D. H. Lee, J. H. Im, H. Y. Lee and J. I. Hong, Tetrahedron Lett., 2002, 43, 9637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–14 including ESI HRMS spectra, NMR spectra and CD spectra. See DOI: 10.1039/b904263f |
| This journal is © The Royal Society of Chemistry 2009 |