Ultra-fine porous SnO2 nanopowder prepared via a molten salt process: a highly efficient anode material for lithium-ion batteries†
Zai Ping
Guo
*ab,
Guo Dong
Du
b,
Yanna
Nuli
c,
Mohd Faiz
Hassan
b and
Hua Kun
Liu
b
aSchool of Mechanical, Materials & Mechatronics Engineering, University of Wollongong, Australia NSW 2522. E-mail: zguo@uow.edu.au; Fax: +61 2 4221 5731; Tel: +61 2 4221 5225
bInstitute for Superconducting & Electronic Materials, University of Wollongong, Australia NSW 2522
cDepartment of Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, PR China
First published on 27th March 2009
Abstract
Ultra-fine porous SnO2nanoparticles for lithium ion batteries were prepared by a simple, easily scaled-up molten salt method at 300 °C. The structure and morphology were confirmed by X-ray diffraction and transmission electron microscopy. The as-prepared SnO2 had a tetragonal rutile structure with crystal sizes around 5 nm. The electrochemical performance was tested compared with commercial nanopowder and previously reported nanowires. The as-prepared nanoparticles delivered a significantly higher discharge capacity and better cycle retention. The nanoparticle electrode delivered a reversible capacity of 410 mAh g−1 after 100 cycles. Even at high rates, the electrode operated at a good fraction of its capacity. The excellent electrochemical performance of the ultra-fine porous SnO2 can be attributed to the ultra-fine crystallites (which tend to decrease the absolute volume changes) and the porous structure (which promotes liquid electrolyte diffusion into the bulk materials and acts as a buffer zone to absorb the volume changes).
Introduction
Large efforts are presently devoted to developing advanced materials to substitute for the currently used graphite anodes in lithium-ion batteries. Among them, tin-based oxides have attracted much attention, owing to their higher theoretical specific capacity (781 mAh g−1 for SnO2) compared with graphite electrode (372 mAh g−1).1–3 However, tin-based materials have a disadvantage, in that they undergo significant volume expansion and contraction, greater than 100%, during lithium intercalation and de-intercalation.4,5 This causes cracking and crumbling, resulting in “dead volume”, which is electrically disconnected from the bulk material or the current collector. The mechanically inactive “dead volume” results in subsequent degradation of electrode performance during cycling. Special structures have been designed to stabilize the morphology of electrodes in order to alleviate the volume changes and mechanical stress, such as Sn-metal alloy6–10 or tin-based composite.11–13 In such cases, another inactive or less active composition served as a supporting and conducting matrix to buffer the volume change. Another approach was to make porous structures or nanostructures, such as nanowires,14–16nanotubes,17,18nanorods,19nanoparticles,20,21 or hollow structures.22–24 When the size was decreased, the absolute volume change and cracking would decrease. These reports showed improved electrochemical performance for the SnO2-based materials, but they either used templates or involved tedious synthetic procedures, which handicapped them in terms of large-scale application. An alternative relatively simple synthetic technique was the hydrothermal method,25,26 but it was not easy to control the real pressure and thus the batch to batch reproducibility. Furthermore, this method made it difficult to obtain large batch production.In our previous work, we reported the effect of morphology on the electrochemical performance of SnO2 anodes27 and found that SnO2nanowires exhibit better electrochemical performance than nanotubes and nanopowder with average crystal sizes of less than 30 nm, due to the higher electrical conductivity of the nanowires. However, the synthesis process for nanowires was expensive and complicated, which made it unsuitable for real industrial applications. Recently, it was reported that a porous framework was an ideal structure to buffer the volume changes of SnO2 during charge/discharge.15 Most types of porous SnO2 have been prepared through hard templating or soft templating synthesis, which were expensive and not easy to control. In this work, we have developed a simple, easily scaled-up method, the molten salt method, to synthesize ultra-fine porous SnO2nanostructures. The as-prepared ultra-fine porous SnO2 nanomaterials show excellent electrochemical performance, better than some reported to date in previous publications.14,20–22,28–32 Molten salts used as the reaction solvent have been reported for the preparation of cathode materials elsewhere.33–35 The molten salt method can result in very fine morphology due to the large viscosity and dielectric behaviour of the eutectic system. Furthermore, this method is easy to achieve and scale up, the treatment temperature is low, and the raw molten salt materials are recyclable, so it is economical and promising for industrial application. The molten salt method could also be used to produce other metal oxides, such as nanosized magnetic or electrode materials.
Experimental
Material preparation
Porous SnO2 nanopowder was synthesized by a molten salt method. 100 mmol of lithium nitrate (Aldrich), 100 mmol of lithium hydroxide monohydrate (Aldrich), and 10 mmol of tin(II) chloride dehydrate (Analar-BDH) were mixed in an agate mortar to obtain a homogeneous mixture. Then, 50 mmol hydrogen peroxide (Sigma-Aldrich) was added into the mixture, which was stirred for 24 h. The mixture was then given a heat-treatment at 120 °C for 4 h in a vacuum oven, followed by further heat-treatment in air at 300 °C for 3 h in a muffle furnace. After cooling down, the SnO2 was separated from the eutectic mixture by washing with a large amount of de-ionized water and by centrifugation, and then the product was dried under vacuum at 60 °C for 24 h.Material characterization
Thermogravimetric analysis (TGA) was carried out with Setaram 92 equipment at a heating rate of 10 °C min−1 under air. X-Ray diffraction (XRD) patterns were obtained using a Philips PW1730 diffractometer with CuKα radiation and a graphite monochromator. Transmission electron microscope (TEM) investigations were performed using a JEOL 2011 200 keV analytical electron microscope. TEM samples were prepared by deposition of ground particles onto lacey carbon support films.The electrochemical tests were carried out via CR2032 coin-type cells. The electrodes were prepared by mixing as-prepared nano-SnO2 and commercial SnO2 as active materials with 10 wt% carbon black (Super P, MMM, Belgium) and 10 wt% polyvinylidene fluoride (PVDF) dissolved in N-methyl-2-pyrrolidinone (NMP) to form a homogeneous slurry, which was then spread onto copper foil. The coated electrodes were dried in a vacuum oven at 120 °C for 4 h. Coin cells were assembled in an argon-filled glove box (Mbraun, Unilab, Germany) by stacking a porous polypropylene separator containing liquid electrolyte between the composite electrode and a lithium foil counter electrode. The electrolyte used was 1 M LiPF6 in a 50 : 50 (v/v) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC), provided by MERCK KgaA, Germany. The cells were galvanostatically charged and discharged in different voltage ranges at different current densities. All the electrochemical tests were carried out at room temperature (25 °C).
Results and discussion
The overall synthetic procedure is described in Scheme 1. The molten salt method shows an accelerated reaction rate and controllable particle morphology, because the salt melt acts as a strong solvent and exhibits a high ionic diffusion rate.36 Furthermore, the melted salts with their low melting point are helpful in hindering the growth of SnO2 particles. When cooled down, the SnO2 particles were surrounded by the salts, which were then washed away by de-ionized water, leaving pores behind, and thus generating ultra-fine porous nanostructures. | ||
| Scheme 1 Schematic model of synthetic procedure: (a) mixed raw materials; (b) solid molten salts and SnO2; (c) porous and nanosized SnO2. | ||
Temperature is a crucial parameter for crystal growth, so the temperature option is the first consideration. Fig. 1 shows the TGA result on the mixed raw materials. No weight losses above 250 °C can be observed in the curve. Since low temperatures are typically favourable for the synthesis of nanostructures and porous structures, 300 °C was chosen in the experiment to ensure that the reaction occurred in a liquid phase.
 | ||
| Fig. 1 TGA curve of raw material consisting of SnCl2·2H2O mixed with LiOH and LiNO3 (molar ratio 1 : 1). | ||
The structure of the as-prepared SnO2 nanopowder was characterized using X-ray diffraction and compared with a commercial powder (with average crystal size of 61 nm), as shown in Fig. 2. All reflections are in good agreement with a tetragonal rutile structure (JCPDS 41-1445), belonging to the space groupP42/mnm (136). The highly broadened peaks indicate that the as-prepared powder is composed of SnO2 crystallites with very small size. The calculated mean crystallite size of the SnO2, estimated from Scherrer's formula, is 3.6 nm, based on the (110), (101), and (211) peaks.
 | ||
| Fig. 2 X-Ray diffraction patterns for commercial and as prepared SnO2. | ||
The morphology images are displayed in Fig. 3. The commercial powder was characterized by SEM (Fig. 3(a) and (b)), while the as-prepared SnO2 was characterized by high resolution TEM (Fig. 3(c) and (d)). The TEM images show that the crystal sizes of the as-prepared SnO2 are around 5 nm, and lattice fringes are present with d spacing values of 0.318 nm, corresponding to the (110) and (220) planes of SnO2. There are also a great many pores among the nanoparticles, and the average pore width calculated from Brunauer–Emmett–Teller (BET) measurements is 2.015 nm. These pores provide void space to buffer volume expansion during lithium insertion.
 | ||
| Fig. 3 Scanning electron microscope (SEM) images (a, b) of commercial SnO2 and transmission electron microscope (TEM) images (c, d) of the as-prepared nano-SnO2. The scale bars are 1 µm, 0.5 µm, 20 nm and 5 nm, respectively. | ||
The electrochemical performance of the as-prepared ultra-fine porous SnO2 was characterized by charge/discharge cycling over different voltage ranges and compared with the performance of commercial SnO2nanoparticles and of SnO2nanowires prepared in our laboratory. Fig. 4 shows the first and the second charge/discharge curves of the SnO2 anodes. The as-prepared SnO2 nanopowder delivers a reversible capacity of 660 mAh g−1 and 645 mAh g−1 in the first and second cycles, respectively, while the commercial sample shows only 372 mAh g−1 and 287 mAh g−1, respectively. The initial Coulombic efficiency of the SnO2 nanopowder is 44.2%, which is much higher than that of the commercial powder (37.8%).
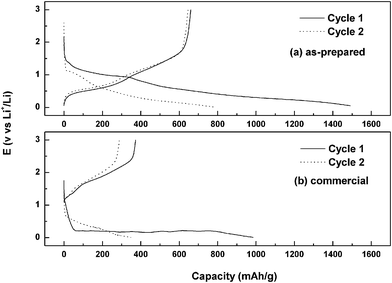 | ||
| Fig. 4 First and second cycles of charge and discharge curves for as-prepared (a) and commercial (b) SnO2. | ||
Fig. 5(a) and (b) compare the cycling performance of the as-prepared nanoparticles with that of SnO2nanowires in the voltage range of 0.05–1.5 V. The as-prepared ultra-fine porous nanopowder shows a higher capacity and better cycling performance. The nanopowder electrode delivers a reversible capacity of 410 mAh g−1 after 100 cycles, which is significantly better than that of the SnO2nanowires. Curves (c) and (d), respectively, show the cycling performance of the as-prepared and the commercial SnO2 in the voltage range of 0.01–3 V. Both the materials show a decrease in discharge capacity with time. However, obviously, the as-prepared ultra-fine porous nanopowder shows a much higher discharge capacity than the commercial powder.
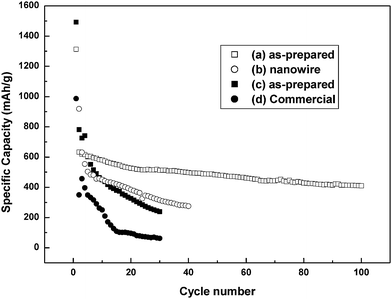 | ||
| Fig. 5 Cycling performance with different voltage profiles: (a, b) between 0.05 and 1.5 V; (c, d) between 0.01 and 3 V. | ||
In order to fully estimate the electrochemical performance of the as-prepared sample, the cycling data at various charge/discharge rates (0.1, 0.5, 1, 2, 5 and 10 C) are shown in Fig. 6. The charge capacity at the 5 C rate is 57% of the capacity of 0.1 C; even as high as the 10 C rate, it still shows 43% of the capacity of 0.1 C. By returning to 1 C, the electrode delivers a capacity of about 500 mAh g−1. We tried to compare with reported results, but only a few papers did high rate testing. The results are shown in the supporting information (Table S1).† Obviously, our results are comparable. Considering the simple synthetic process and template free, the molten salt method is very promising for large-scale application. The good cycling response is well supported by the TEM images of cycled SnO2 (Fig. 7), which show that the crystal sizes are from 5–10 nm in size, very similar to the sizes in the as-prepared sample, and with pores among the particles. In further work, transition metal oxides or carbon will be incorporated to improve the conductivity and buffer volume change.
 | ||
| Fig. 6 Capacity delivered upon cycling at different rates between 0.05 and 1.5 V in coin-type half-cells. | ||
 | ||
| Fig. 7 (a) TEM images of the SnO2electrode after 20 cycles; (b) a high resolution image of (a). | ||
The promising electrochemical performance of the as-prepared SnO2nanoparticles could be attributed to: (1) the pores, which provides further buffering against the local volume change during the Li–Sn alloying/de-alloying reactions; (2) the ability of the pores to promote liquid electrolyte diffusion into the bulk of the anode and provide fast transport channels for the Li ions; (3) the lower absolute volume changes during the Li–Sn alloying/de-alloying reactions due to the nanosized oxide crystals (around 5 nm); and (4) the shorter diffusion length for Li insertion due to the ultra-fine nanosized particles, with benefits in retaining the structural stability, thereby leading to good cycling performance.
Conclusions
In this paper, a simple molten salt method was applied to synthesize ultra-fine porous SnO2 nanopowder. The nanosized crystals (around 5 nm) delivered a higher capacity and more stable cyclability compared to SnO2nanowires. The reversible capacity was 410 mAh g−1 after 100 cycles in the voltage range of 0.05–1.5 V; even at high charge/discharge rates, the performance was comparable to the reported results. This template or surfactant free method is an efficient way to produce ultra-fine porous nanopowder and shows promise for industrial use.Acknowledgements
Financial support provided by the Australian Research Council (ARC) through an ARC Discovery project (DP0878611) is gratefully acknowledged. Moreover, the authors would like to thank Dr Tania Silver at the University of Wollongong for critical reading of the manuscript.References
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395 CrossRef CAS.
- I. A. Coutney and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2045 CAS.
- N. Li, C. R. Martin and B. Scrosati, Electrochem. Solid-State Lett., 2000, 3, 316 CrossRef CAS.
- J. Yang, Y. Takeda, N. Imanishi and O. Yamamoto, J. Electrochem. Soc., 1999, 146, 4009 CrossRef CAS.
- L. Wang, S. Kitamura, T. Sonoda, K. Obata, S. Tanase and T. Sakai, J. Electrochem. Soc., 2003, 150, A1346 CrossRef CAS.
- J. Wolfenstine, S. Campos, D. Foster, J. Read and W. Behl, J. Power Sources, 2002, 109, 230 CrossRef CAS.
- H. Shin and M. Liu, Adv. Funct. Mater., 2005, 15, 582 CrossRef CAS.
- X. Cheng and P. Shi, J. Alloys Compd., 2005, 391, 241 CrossRef CAS.
- J. Hassoun, S. Panero, P. Simon, P.-L. Taberna and B. Scrosati, Adv. Mater., 2007, 19, 1632 CrossRef CAS.
- L. Shi, H. Li, Z. Wang, X. Huang and L. Chen, J. Mater. Chem., 2001, 11, 1502 RSC.
- O. Mao and J.-R. Dahn, J. Electrochem. Soc., 1999, 146, 423 CrossRef CAS.
- F. Chen, Z. Shi and M. Liu, Chem. Commun., 2000, 2095 RSC.
- Y. Wang, J.-Y. Lee and T.-C. Deivaraj, J. Electrochem. Soc., 2004, 151, A1804 CrossRef CAS.
- M.-S. Park, G.-X. Wang, Y.-M. Kang, D. Wexler, S.-X. Dou and H.-K. Liu, Angew. Chem., Int. Ed., 2007, 46, 750 CrossRef CAS.
- H. Kim and J. Cho, J. Mater. Chem., 2008, 18, 771 RSC.
- N. Zhao, G. Wang, Y. Huang, B. Wang, B. Yao and Y. Wu, Chem. Mater., 2008, 20, 2612 CrossRef CAS.
- N. Du, H. Zhang, B.-D. Chen, X.-Y. Ma and D.-R. Yang, Chem. Commun., 2008, 3028 RSC.
- Y. Wang, J.-Y. Lee and H.-C. Zeng, Chem. Mater., 2005, 17, 3899 CrossRef CAS.
- D.-F. Zhang, L.-D. Sun, J.-L. Yin and C.-H. Yan, Adv. Mater., 2003, 12, 1022 CrossRef.
- S. H. Ng, D. I. Dos Santos, S. Y. Chew, D. Wexler, J. Wang, S. X. Dou and H. K. Liu, Electrochem. Commun., 2007, 9, 915 CrossRef CAS.
- V. Subramanian, K. I. Gnanasekar and B. Rambabu, Solid State Ionics, 2004, 175, 181 CrossRef CAS.
- S. Han, B. Jang, T. Kim, S. M. Oh and T. Hyeon, Adv. Funct. Mater., 2005, 15, 1845 CrossRef CAS.
- X. W. Lou, Y. Wang, C. L. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325 CrossRef CAS.
- Y. Wang, F. Su, J. Y. Lee and X. S. Zhao, Chem. Mater., 2006, 18, 1347 CrossRef CAS.
- N. Baik, G. Sakai, N. Miura and N. Yamazoe, J. Am. Ceram. Soc., 2000, 83, 2983 CrossRef CAS.
- C. Kim, M. Noh, M. Choi, J. Cho and B. Park, Chem. Mater., 2005, 17, 3297 CrossRef CAS.
- M.-S. Park, Y.-M. Kang, G.-X. Wang, S.-X. Dou and H.-K. Liu, Adv. Funct. Mater., 2008, 18, 455 CrossRef CAS.
- J. Fan, T. Wang, C. Yu, B. Tu, Z. Jiang and D. Zhao, Adv. Mater., 2004, 16, 1432 CrossRef CAS.
- L. Yuan, K. Konstantinov, G. X. Wang, H. K. Liu and S. X. Dou, J. Power Sources, 2005, 146, 180 CrossRef CAS.
- Y. Wang and J. Y. Lee, J. Power Sources, 2005, 144, 220 CrossRef CAS.
- L. Yuan, Z. P. Guo, K. Konstantinov, H. K. Liu and S. X. Dou, J. Power Sources, 2006, 159, 345 CrossRef CAS.
- Y. C. Chen, J. M. Chen, Y. H. Huang, Y. R. Lee and H. C. Shih, Surf. Coat. Technol., 2007, 202, 1313 CrossRef CAS.
- C. H. Han, Y.-S. Hong, C. M. Park and K. Kim, J. Power Sources, 2001, 92, 95 CrossRef CAS.
- H. Y. Liang, X. P. Qiu, S. C. Zhang, Z. Q. He, W. T. Zhu and L. Q. Chen, Electrochem. Commun., 2004, 6, 505 CrossRef CAS.
- X. Wang, J. M. Song, L. S. Gao, H. G. Zheng, M. G. Ji and Z. D. Zhang, Solid State Commun., 2004, 132, 783 CrossRef CAS.
- W. Tang, X. Yang, Z. Liu, S. Kasaishi and K. Ooi, J. Mater. Chem., 2002, 12, 2991 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of the high rate performance of the SnO2 in this work with those reported elsewhere. See DOI: 10.1039/b821519g |
| This journal is © The Royal Society of Chemistry 2009 |