Dual functioned BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 for lithium batteries
Ki-Soo
Lee
a,
Seung-Taek
Myung
*b,
Khalil
Amine
c,
Hitoshi
Yashiro
b and
Yang-Kook
Sun
*a
aDepartment of Chemical Engineering, Center for Information and Communication Materials, Hanyang University, Seoul, 133-791, Republic of Korea. E-mail: yksun@hanyang.ac.kr; Fax: +82 2 2282 7329; Tel: +82 2 2220 0524
bDepartment of Chemical Engineering, Iwate University, 4-3-5 Ueda, Morioka, Iwate 020-8551, Japan. E-mail: smyung@iwate-u.ac.jp; Fax: +81 19 621 6345; Tel: +81 19 621 6345
cElectrochemical Technology Program, Chemical Engineering Division, Argonne National Laboratory, 9700 S. Cass Ave., Argonne, IL 60439, USA
First published on 11th February 2009
Abstract
Spinel-type lithium manganese oxides have shown considerable promise for the positive electrode in lithium batteries but have suffered from poor performance at high temperatures (40–60 °C). The electrochemical properties of BiOF-coated spinel Li[Li0.1Al0.05Mn1.85]O4 at elevated temperatures (55 °C) were investigated. BiOF nanoparticles were well coated on spinel Li[Li0.1Al0.05Mn1.85]O4, as confirmed by scanning and transmission electron microscopy. The BiOF-coated spinel Li[Li0.1Al0.05Mn1.85]O4 electrode had excellent capacity retention at 55 °C, maintaining its initial discharge capacity of 96.1% after 100 cycles. This improved cycling performance was ascribed to the presence of the BiOF layer on the spinel particles, which prevented the infiltration of HF generated by the decomposition of electrolytic salt, LiPF6, in the electrolyte. This property resulted in a considerable reduction of manganese dissolution of the BiOF-coated spinel Li[Li0.1Al0.05Mn1.85]O4. Transmission electron microscopy of extensively cycled particles confirmed that the particle surface of the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 remained almost completely undamaged, whereas pristine spinel particles were severely deteriorated. In the present paper, we have tried to understand the role of the BiOF coating layer and why such coatings resulted in better battery performances, using BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. The BiOF coating layer on the surface of Li[Li0.1Al0.05Mn1.85]O4 particles can be defined as follows; the oxyfluoride layer provides a strong protecting layer against HF attack and scavenges HF, thus resulting in reducing HF from the electrolyte. Synergistically, these actions resulted in the significantly improved high temperature cycling performances exhibited by the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4.
Introduction
Lithium secondary batteries are the most important power source for portable equipment on the market today. In addition, these batteries have recently come under consideration as potential power sources for both hybrid and all-electric vehicles, although safety is an important concern for such use. Spinel-type lithium manganese oxides have long been studied as possible positive electrode materials, given that they provide a reliable degree of safety and are cost effective.1,2 However, poor cycling performance at high temperatures (40–60 °C) remains a serious drawback.1–4To overcome the problem of poor cycling at high temperatures and improve electrochemical properties, researchers have explored cation replacement at Mn sites,5–14 surface modifications,15–19 and blending with other materials.20–22 In addition, the electrochemical properties have been improved by surface coating the positive electrode particles with ZnO,23 Al2O3,24 and NiO.25 Further, Myung et al.24–26 reported that, as coating media, amphoteric oxides were transformed to metal fluorides via metal oxyfluorides under an HF environment. In other words, these coating materials were able to scavenge fluorine anions from HF, thus allowing the coating layers to reach the metal fluoride layers.
We recently proposed that a stable AlF3 coating layer on the positive electrode particles would be effective at improving the capacity, capacity retention, rate capability, and thermal stability of lithium batteries.27–30 Based on our discussion above, we expect that a metal oxyfluoride coating would be effective since the oxygen component may allow fluorine anions from HF and fluorine-containing moieties to protect the active materials during cycling.
Komaba et al.5 suggested that Li doping in Mn sites of LiMn2O4 significantly suppresses Mn dissolution during cycling. Further, Myung et al.6 and Sun et al.14 also reported that the partial replacement of Mn by Al improves cycling performance at elevated temperatures. Therefore, we expected that the use of both Li and Al would significantly reduce Mn dissolution and improve high temperature cyclability. Furthermore, surface treatment of spinel compounds with metal oxyfluoride should enhance capacity retention at high temperatures. Thus, in this study, we prepared a surface-modified spinel material, Li[Li0.1Al0.05Mn1.85]O4 coated by bismuth oxyfluoride (BiOF), and evaluated its structural properties and electrochemical performance.
Experimental
To begin, Mn3O4 powder was synthesized via a co-precipitation method previously described.31 The prepared Mn3O4 precursor, which contained an excess of LiOH·H2O and Al(OH)3, was preheated to 500 °C for 5 h and subsequently calcined for 15 h at 850 °C in a furnace under air to form spinel Li1.1Al0.05Mn1.85O4 powder. To prepare the BiOF coating precursor, bismuth nitrate [Bi(NO3)3·5H2O, Kanto] was dissolved in distilled water and nitric acid. Next, ammonium fluoride (NH4F, Aldrich) was dissolved in distilled water, into which the prepared Li1.1Al0.05Mn1.85O4 powder was immersed. Then, the bismuth nitrate solution was slowly added to the mixture simultaneously with the desired amount of NH4OH solution. After continuous mixing for 1 h, the BiOF-coated Li1.1Al0.05Mn1.85O4 powders were filtered and washed several times with distilled water. The resulting powders were dried at 110 °C to remove any residual water and then heated at 400 °C for 5 h under air.The as-precipitated bismuth oxyfluoride powder was characterized by several techniques. The thermal behavior was studied with thermal gravimetry (TG, DTG-60, SHIMADZU Industries) at a heating/cooling rate of 1 °C min−1. The crystalline phase was analyzed with powder X-ray diffraction (XRD, Rigaku, Rint-2000) using Cu Kα radiation. The XRD data were obtained at 2θ = 10° to 110°, with a step size of 0.03°. The collected intensity data from the XRD were analyzed by the Rietveld refinement program Fullprof 2002.32 The morphology of the prepared powders was determined by scanning electron microscopy (SEM, JSM-6340F, JEOL). High-resolution transmission electron microscopy (TEM, JEM2010, JEOL) was also employed to characterize the prepared powders. The chemical compositions of the final powders were determined by atomic absorption spectroscopy (AAS, Vario 6, AnalyticJena).
Electrochemical testing was performed in 2032 coin type cells. The positive electrodes were fabricated by blending the prepared powders, Super P carbon black, and polyvinylidene fluoride (85 : 7.5 : 7.5) in N-methyl-2-pyrrolidone. The slurry was then cast on aluminium foil and dried at 110 °C for 10 h in a vacuum oven, at which time disks were punched out of the foil. The negative electrode was lithium foil, and the electrolyte was 1 M LiPF6 solution in an ethylene carbonate (EC)-diethyl carbonate (DEC) mixture (3 : 7 ratio by volume, Cheil Industries). The positive and negative electrodes were separated by a porous polypropylene film. All cells were prepared in an Ar-filled dry box. The assembled cells were charged and discharged within a voltage range of 3.0–4.3 V at a constant current density of 1 C (100 mA g−1) and elevated temperature (55 °C). Cycle-life tests were performed at the same voltage and temperature conditions.
For HF titration, the cells that had been cycled at 55 °C (adapting Li metal as the anode) were carefully disassembled, and the resulting contents were washed thoroughly with Li salt-free solvent for one week in a glove box. An aqueous solution of NaOH (Kanto) with Bromothymol Blue (BTB, Aldrich) as an indicator was used for the titration of the used electrolyte.
To confirm the presence of byproducts on the surface of the active materials after extensive cycling, the cycled active materials were examined by time-of-flight secondary ion mass spectroscopy (ToF-SIMS, ULVAC-PHI TFS2000, Perkin Elmer) at 10−9 torr. This instrument was also equipped with a liquid Ga ion source and pulse electron flooding. During the analysis, the targets were bombarded by pulsed 15 keV Ga+ beams. The total collection time was 300 s over a 12 × 12 µm area.
Results and discussion
Material characterization
In an attempt to coat Li[Li0.1Al0.05Mn1.85]O4 with BiOF, we first prepared bismuth oxyfluoride by a co-precipitation method. Fig. 1 shows the TG curves of the as-precipitated bismuth oxyfluoride measured at temperatures up to 900 °C under a flow of dry air. No significant weight loss occurred up to 150 °C, although weight steadily decreased throughout the remaining temperature range. Specifically, the weight loss up to 400 °C was around 1.5 wt%, and 5.5 wt% up to 900 °C. The continuous weight loss was ascribed to the evaporation of some of the elements, such as O2 and/or F2, from the precipitated material. | ||
| Fig. 1 TG curves of the as-co-precipitated BiO0.98F0.51. | ||
The calcination temperature of the as-prepared bismuth oxyfluoride was determined from XRD spectra at temperatures from 400 °C to 750 °C, and the XRD results are shown in Fig. 2. The precipitate exhibited the typical fingerprint peaks associated with BiO0.51F1.98 compounds, having a simple cubic structure of the Fm3m space group (Fig. 2a). The calculated lattice parameters coincided well with the reported values and are listed in Table 1.33–35 Calcination of the precipitated BiO0.51F1.98 to 400 °C in air led to the formation of well-crystallized BiOF (Fig. 2b), which possessed a tetragonal structure as listed in Table 1.34 Heat treatment in air caused evaporation of fluorine but a gain of oxygen via the following reaction:
| 4BiO0.5F2 + O2 → 4BiOF + 2F2↑ | (1) |
| Crystal parameters | ||||||
|---|---|---|---|---|---|---|
| Space group | a/Å | b/Å | c/Å | β/° | Vol/Å3 | |
| BiO0.51F0.98 | Fm3m | 5.8195(5) | — | — | 90 | 197.132 |
| BiOF | P4/nmm | 3.7529(2) | — | 6.2354(4) | 90 | 87.821 |
| Bi2O3 | P21/c | 5.8074(5) | 8.2442(5) | 7.5483(7) | 122.82(1) | 303.700 |
 | ||
| Fig. 2 Powder XRD patterns of (a) the as-co-precipitated BiO0.98F0.51 and of (b) BiO0.98F0.51 calcined at 400 °C, (c) 550 °C, (d) 650 °C, and (e) 750 °C for 5 h in air. | ||
Thus, the weight loss that appeared at approximately 400 °C was most likely due mainly to the evaporation of F from the BiO0.51F1.98, resulting in BiOF. The crystal parameters, as calculated by the least square method, are in good agreement with the literature.34 Heat-treatment at elevated temperatures such as 550 °C and 650 °C resulted in the formation of impurities like bismuth oxide or a mixture of BiOF and bismuth oxide, as shown in Fig. 2c and d. Further, single-phase Bi2O3 appeared at 750 °C (Fig. 2e). Based on these observations, the following reaction is suggested:
| 4BiOF + O2 → 2Bi2O3 + 2F2↑ | (2) |
This reaction suggests that the slow weight loss in the TG curve of Fig. 1 may have stemmed primarily from the evaporation of F, leading to monoclinic α-Bi2O3. The obtained lattice parameters are similar to reported values.35 In summarizing Fig. 1 and Fig. 2, we decided that the optimum calcination temperature for BiOF as a coating medium for the spinel Li[Li0.1Al0.05Mn1.85]O4 was 400 °C in air.
To synthesize Li[Li0.1Al0.05Mn1.85]O4, we first prepared Mn3O4via co-precipitation, mixed it with an excess of LiOH·H2O and Al(OH)3, preheated the mixture to 500 °C for 5 h, and subsequently calcined it at 850 °C for 15 h in air. To confirm the crystal structure of the calcined product, a Rietveld refinement was performed, the results of which are shown in Fig. 3a and Table 2. The chemical composition of the resulting powders obtained from AAS analysis is Li[Li0.1Al0.05Mn1.85]O4. The occupation factors of all elements based on the chemical analyses were invariable, and the sum of the occupation factors of Mn, Al, and Li in the 16d sites was equal to one. As can be deduced from Fig. 3a, the spinel Li[Li0.1Al0.05Mn1.85]O4 had a cubic spinel phase, and the excess amount of Li was well distributed in the 16d manganese site. The calculated lattice parameter, shown in Table 2, was 8.188(1) Å. The reason for this smaller value was ascribed to the reduced amount of the relatively larger Mn3+ (0.65 Å36) by Li+ incorporation and substitution of smaller Al3+ ions for Mn sites in the structure.6,14 Meanwhile, the amount of stable Mn4+ (0.54 Å36) increased relative to the amount of Mn3+, meaning that the lattice constant was smaller than that of the stoichiometrically equivalent LiMn2O4 (8.244 Å).1–4 From the Rietveld refinement results for the XRD data in Fig. 3a and Table 2, the Li+ and Al3+ elements are believed to be well dispersed in the spinel structure.
| Formula | Li[Li0.1Al0.05Mn1.85]O4 | |||||
|---|---|---|---|---|---|---|
| Crystal system | Cubic | |||||
| Space group | Fd3m | |||||
| Atom | Site | x | y | z | g | B/Å2 |
| a M* indicates the elements occupied in 16d site such as Mn, Li, and Al. | ||||||
| Li1 | 8a | 1/8 | 1/8 | 1/8 | 1 | 1.4 |
| Mn | 16d | 1/2 | 1/2 | 1/2 | 0.925 | 1.1 |
| Li2 | 16d | 1/2 | 1/2 | 1/2 | 0.05 | 1.1 |
| Al | 16d | 1/2 | 1/2 | 1/2 | 0.025 | 1.1 |
| O | 32e | 0.263(5) | 0.263(5) | 0.263(5) | 1 | 0.8 |
| R wp/% | 14.8 | |||||
| Cell parameters | a = 8.188(1) Å | |||||
| Unit volume = 548.954 (1) Å3 | ||||||
| Distance | Li–O = 1.9556(2) Å | |||||
| M*–O = 1.9472(1) Å | ||||||
![Rietveld refinement results of XRD data for (a) the pristine and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4.](/image/article/2009/JM/b819224c/b819224c-f3.gif) | ||
| Fig. 3 Rietveld refinement results of XRD data for (a) the pristine and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. | ||
The as-synthesized Li[Li0.1Al0.05Mn1.85]O4 powders were then immersed in bismuth- and fluorine-containing aqueous solution and reacted at 80 °C for 1 h. The obtained powders were dried at 110 °C to remove any residual water and then heated at 400 °C for 5 h in air. The product was also refined with the Rietveld method to determine whether or not the coating medium was introduced into the spinel structure. The corresponding refinement results are shown in Fig. 3b and Table 3. No significant change in the XRD pattern was observed for the coated spinel in Fig. 3b, nor was any change noticed for the resulting crystal parameters (Table 3) compared with the pristine Li[Li0.1Al0.05Mn1.85]O4 shown in Table 2. Thus, we initially thought that the coating medium might not have been introduced into the spinel framework. As confirmed in Fig. 1 and Fig. 2, however, the coated material on the spinel surface was indeed BiOF, although due to the very small amount of coating medium used, we could not find the diffraction peaks of BiOF in Fig. 3b. The amount of deposited BiOF was around 0.25 wt% compared with the spinel Li[Li0.1Al0.05Mn1.85]O4 powders.
| Formula | Li[Li0.1Al0.05Mn1.85]O4 | |||||
|---|---|---|---|---|---|---|
| Crystal system | Cubic | |||||
| Space group | Fd3m | |||||
| Atom | Site | x | y | z | g | B/Å2 |
| a M* indicates the elements occupied in 16d site such as Mn, Li, and Al. | ||||||
| Li1 | 8a | 1/8 | 1/8 | 1/8 | 1 | 1.4 |
| Mn | 16d | 1/2 | 1/2 | 1/2 | 0.925 | 1.1 |
| Li2 | 16d | 1/2 | 1/2 | 1/2 | 0.05 | 1.1 |
| Al | 16d | 1/2 | 1/2 | 1/2 | 0.025 | 1.1 |
| O | 32e | 0.263(4) | 0.263(4) | 0.263(4) | 1 | 0.8 |
| R wp/% | 14.1 | |||||
| Cell parameters | a = 8.188(1) Å | |||||
| Unit volume = 548.965(1) Å3 | ||||||
| Distance | Li–O = 1.9556(2) Å | |||||
| M*–O = 1.9473(1) Å | ||||||
Fig. 4 shows SEM images of the as-synthesized pristine and BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. The pristine particles exhibited a smooth plane and no precipitates (Fig. 4a). Meanwhile, numerous nanometre-sized sediments, which were assumed to be the BiOF compound, were observed on the surface of the Li[Li0.1Al0.05Mn1.85]O4 particles (Fig. 4b). To examine the existence of the coating on the surface of the spinel powders, we performed elemental mappings for Mn, Al, and Bi. Fig. 4c shows the particle morphology of the surface-modified Li[Li0.1Al0.05Mn1.85]O4. The particles are polygonal, and the estimated particle size is 2–3 µm on average. The Mn and Al elements are well distributed in the particles shown in Fig. 4d and e. The weaker mapping traces for the Al was due to the relatively weaker X-ray scattering factor of Al compared with Mn. As seen in the Bi mapping results of Fig. 4f, the Bi ingredient appears to be also uniformly dispersed on the surface of the spinel Li[Li0.1Al0.05Mn1.85]O4. Bright-field TEM images of the pristine Li[Li0.1Al0.05Mn1.85]O4 exhibited a smooth edge line (Fig. 5a), whereas images of the surface-modified particles indicated a thin, smooth coating layer on the spinel particle. The thickness of the coating layer was determined to be approximately 10 nm. In summary, the TG, XRD, SEM, and TEM results led us to believe that the spinel Li[Li0.1Al0.05Mn1.85]O4 oxide was indeed covered by a thin nanocrystalline BiOF coating layer.
![SEM images of (a) pristine and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4; EDX mappings of (c) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, (d) Mn, (e) Al, and (f) Bi (bar indicates 7 µm).](/image/article/2009/JM/b819224c/b819224c-f4.gif) | ||
| Fig. 4 SEM images of (a) pristine and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4; EDX mappings of (c) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, (d) Mn, (e) Al, and (f) Bi (bar indicates 7 µm). | ||
![Bright-field TEM images of (a) pristine and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4.](/image/article/2009/JM/b819224c/b819224c-f5.gif) | ||
| Fig. 5 Bright-field TEM images of (a) pristine and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. | ||
Electrochemical cell performance
Fig. 6 shows the first charge and discharge curves obtained from cells with positive electrodes of Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated spinel oxide. The electrochemical performance was measured at a constant current density of 100 mA g−1 (1-C rate) at 55 °C applied over a voltage range of 3.0–4.3 V versus Li. The initial charge and discharge capacities of the pristine and coated electrodes were quite similar: 102.7 mAh (g-oxide)−1 for the Li[Li0.1Al0.05Mn1.85]O4 and 104.3 mAh (g-oxide)−1 for the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 (Fig. 6a). To confirm the BiOF incorporation into the spinel framework, the discharge curves shown in Fig. 6a were differentiated to produce the results in Fig. 6b. The pristine and the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 had charge and discharge profiles typical of spinel LiMn2O4,1–4 which had two potential plateaus in Fig. 6a. To explain this observation, we considered doping in the oxygen site during the heat treatment at 400 °C. In this case, when the fluorine was substituted for oxygen, the resulting charge and discharge voltages became slightly higher than those in our previous reports.37,38Fig. 6b clearly shows that the voltage plateaus for the charge and discharge of the pristine material were in good agreement with those of the coated Li[Li0.1Al0.05Mn1.85]O4. Myung et al.6 suggested that when a hetero-element is doped in the Mn site of a spinel compound, the resulting redox potential varies due to the change in interaction energies between Li positive ions and Li positive ions in the spinel framework. For example, a partial Al substitution in the Mn site of the spinel LiAlxMn2-xO4 resulted in a slightly higher discharge voltage. For these reasons, we initially believed that Bi and F did not constitute a part of the spinel Li[Li0.1Al0.05Mn1.85]O4 structure, and further, that the BiOF was present only on the surface of the spinel Li[Li0.1Al0.05Mn1.85]O4.![Comparison of the first charge and discharge curves of (a) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C and (b) the corresponding differentiated curves.](/image/article/2009/JM/b819224c/b819224c-f6.gif) | ||
| Fig. 6 Comparison of the first charge and discharge curves of (a) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C and (b) the corresponding differentiated curves. | ||
Fig. 7 shows the cycling performance of the pristine and BiOF-coated electrodes over 100 cycles. The cells were cycled at 100 mA g−1 (1-C rate) over a voltage range of 3.0–4.3 V at 55 °C. The pristine Li[Li0.1Al0.05Mn1.85]O4 exhibited gradual capacity fading upon cycling (Fig. 7a); the capacity retention was about 84.4% over 100 cycles (Fig. 7e). On the other hand, the BiOF-coated electrode exhibited a significantly improved capacity retention (Fig. 7b), maintaining 96.1% of its initial capacity during the 100 cycles (Fig. 7e). For the pristine material, the beginning voltage of discharge was much lower on the 25th cycle than the first (Fig. 7a and c), whereas changes were only barely noticeable for the BiOF-coated sample (see Fig. 7b and d). Furthermore, the voltage profiles at the 100th cycle were fairly close compared with the first cycle for the BiOF-coated electrode, as shown in Fig. 7b and d. We believe that the surface modification of spinel Li[Li0.1Al0.05Mn1.85]O4 by the addition of the BiOF nanolayer gave rise to this significant improvement in high temperature cycling performance.
![Continuous charge and discharge curves of (a) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and (b) Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C; corresponding differentiated curves for discharge of (c) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and (d) Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4; (e) cyclability of Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C.](/image/article/2009/JM/b819224c/b819224c-f7.gif) | ||
| Fig. 7 Continuous charge and discharge curves of (a) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and (b) Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C; corresponding differentiated curves for discharge of (c) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and (d) Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4; (e) cyclability of Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C. | ||
Post-cycling analyses
Fig. 8 shows the XRD patterns of the extensively cycled pristine and BiOF-coated electrodes (Fig. 7). An Al current collector and graphite were used as the inner standard to calibrate the diffraction patterns. The original cubic spinel phase was maintained throughout cycling for both electrodes; however, the diffraction peaks for the pristine Li[Li0.1Al0.05Mn1.85]O4 were shifted more towards a higher angle (2θ) compared with those of the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. The lattice parameters were calculated from the XRD patterns using a least squares method, the results of which are given in Table 4. The lattice parameters for the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 were almost identical to those of the original material prior to cycling, while the lattice parameters decreased slightly for the pristine Li[Li0.1Al0.05Mn1.85]O4. Myung et al.6,25 previously reported that such variation of lattice parameters after cycling is due to gradual dissolution of manganese from the active material. Based on this information and our data, we took the smaller change in the lattice parameters, meaning that less manganese dissolution occurred for the BiOF-coated material.| Lattice parameter/Å | ||
|---|---|---|
| Before cycling | After cycling | |
| Pristine Li[Li0.1Al0.05Mn1.85]O4 | 8.188(1) | 8.147(6) |
| BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 | 8.188(1) | 8.182(3) |
![XRD patterns of extensively cycled electrodes at 55 °C for (a) pristine Li[Li0.1Al0.05Mn1.85]O4 and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4.](/image/article/2009/JM/b819224c/b819224c-f8.gif) | ||
| Fig. 8 XRD patterns of extensively cycled electrodes at 55 °C for (a) pristine Li[Li0.1Al0.05Mn1.85]O4 and (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. | ||
To elucidate the reason behind the good cycling performance of the BiOF-coated electrode, we stored the fully charged Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells at 55 °C for 4 weeks, and measured the open-circuit voltages (OCVs) every 24 h. Both cells exhibited a gradual decrease in their OCVs over the length of the storage (Fig. 9); however, the OCVs of the BiOF-coated electrode were higher relative to those of the pristine sample. Specifically, the OCV after 4 weeks was 4.135 V for the Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cell, whereas it was 4.088 V for the pristine sample.
![The change of open-circuit voltages for the fully charged (to 4.3 V) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells during 4 weeks at 55 °C.](/image/article/2009/JM/b819224c/b819224c-f9.gif) | ||
| Fig. 9 The change of open-circuit voltages for the fully charged (to 4.3 V) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells during 4 weeks at 55 °C. | ||
The continuous lowering of the OCVs was thought to have been driven by the decomposition of the electrolytic salt at higher voltages, resulting in HF generation. To test this idea, we performed an HF titration of the analyzed electrolytes stored with the fully charged pristine or BiOF-coated electrodes for 4 weeks at 55 °C. Indeed, as shown in Fig. 10, the amount of HF increased with time, and after 4 weeks, the HF content for the electrolyte with the pristine Li[Li0.1Al0.05Mn1.85]O4 was around 400 ppm, while that of the electrolyte stored with BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 was approximately 150 ppm. This result indicates that the coating of the active material effectively suppressed HF propagation.
![HF titration results for the electrolyte of the fully charged (to 4.3 V) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells during 4 weeks at 55 °C.](/image/article/2009/JM/b819224c/b819224c-f10.gif) | ||
| Fig. 10 HF titration results for the electrolyte of the fully charged (to 4.3 V) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells during 4 weeks at 55 °C. | ||
Another possible explanation for the lowered OCV is the disproportionate dissolution of Mn from the active material. Thus, the concentration of dissolved Mn in the electrolyte used in Fig. 10 was also measured by AAS, and similar tendencies were observed (Fig. 11). Namely, a smaller amount of Mn was dissolved for the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. Specifically, the dissolved amount of Mn for the Li[Li0.1Al0.05Mn1.85]O4 was approximately 63 ppm during the first week, while the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 exhibited greatly reduced Mn dissolution in the electrolyte, having a concentration of about 27 ppm. After 4 weeks, the dissolved Mn concentrations for the pristine and coated material were 306 ppm and 92 ppm, respectively.
![Mn dissolution for the electrolyte of the fully charged (to 4.3 V) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells during 4 weeks at 55 °C.](/image/article/2009/JM/b819224c/b819224c-f11.gif) | ||
| Fig. 11 Mn dissolution for the electrolyte of the fully charged (to 4.3 V) Li/pristine Li[Li0.1Al0.05Mn1.85]O4 and Li/BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 cells during 4 weeks at 55 °C. | ||
If decomposition of the active material was a result of an attack by the produced HF, byproducts would inevitably have been formed. To confirm the presence of such byproducts, ToF-SIMS was carried out for the pristine and BiOF-coated electrodes after the extensive cycling at 55 °C, and the corresponding results are presented in Fig. 12. Though quantitative analysis is difficult with ToF-SIMS, this tool has a high sensitivity for detecting ions qualitatively for the purpose of surface analysis. In addition, Ga+ ion sputtering was carried out for 3 s to remove the air-contaminated layer during sampling. Fig. 12a and b show LiF+ fragments for the pristine and the BiOF-coated electrodes. The decomposition of the electrolytic salt, in the form of LiPF6, was also clearly evident, as LiF+ fragments generate strong and easily recognizable peaks. To explain this result, Aurbach et al.39 and Edström et al.40 have suggested the following reactions:
| LiPF6 → LiF↓ + PF5 | (3) |
| PF5 + H2O → POF3 + 2HF | (4) |
| 2POF3 + 3Li2O → 6LiF↓ + P2O5↓ (or LixPOFy) | (5) |
![ToF-SIMS results of extensively cycled electrodes at 55 °C: (a) Li2F+ fragment for the pristine Li[Li0.1Al0.05Mn1.85]O4, (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, (c) MnO+ and MnF+ fragments for the pristine Li[Li0.1Al0.05Mn1.85]O4, (d) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, (e) BiO+ and BiF+ fragments for the pristine Li[Li0.1Al0.05Mn1.85]O4, and (f) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4.](/image/article/2009/JM/b819224c/b819224c-f12.gif) | ||
| Fig. 12 ToF-SIMS results of extensively cycled electrodes at 55 °C: (a) Li2F+ fragment for the pristine Li[Li0.1Al0.05Mn1.85]O4, (b) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, (c) MnO+ and MnF+ fragments for the pristine Li[Li0.1Al0.05Mn1.85]O4, (d) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, (e) BiO+ and BiF+ fragments for the pristine Li[Li0.1Al0.05Mn1.85]O4, and (f) BiOF-coated Li[Li0.1Al0.05Mn1.85]O4. | ||
According to reaction 3, in which the decomposition of the electrolytic salt is accelerated at elevated temperatures, there is a simultaneous induction in the amount of insulating LiF compound deposited on the electrode surface. The decomposed byproduct, PF5, reacts with the water molecules contained in the electrolyte (30–50 ppm) even in the fresh state, resulting in the formation of HF. This reaction is also increased at elevated temperatures. The insulating LiF compound would then have been deposited on the outermost surface of those electrodes. From our results, making a quantitative comparison would be difficult, but we inferred that the amount of LiF present on the electrode surface increased for the pristine Li[Li0.1Al0.05Mn1.85]O4 electrodes because the total count for the material was much higher compared to that of the BiOF-coated electrodes. These results help to explain why the operation voltages upon cycling and OCVs at the deeply charged states for the pristine Li[Li0.1Al0.05Mn1.85]O4 are lower in Fig. 7 and 9.
The formed HF was also able to attack the surface of the active materials. Thus, the disproportionate dissolution of manganese (2Mn3+ → Mn2+ + Mn4+)3 is possible, and the following byproducts formed as suggested by Edström et al.:40
| 2LiMn2O4 + 2HF + Li+ + e− → BiOF-coated 3λ-MnO2 + MnO + 2LiF + H2O | (6) |
As can be seen in Fig. 12c and d, the MnO fragments result from the disproportionate dissolution of the spinel compounds. The total counts for the MnO+ fragments appeared to be stronger for the pristine material, which confirmed that larger amounts of Mn were dissolved. This finding is in good agreement with the results of Fig. 11. The observed MnF+ fragments can be ascribed to the following reaction:
| MnO + 2HF → MnF2 + H2O | (7) |
It is likely that the amount of insulating MnF2 formed with the pristine Li[Li0.1Al0.05Mn1.85]O4 was larger than that of BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 (Fig. 12c and d) because of the increased decomposition of active materials present in the latter. As a consequence, capacity fading occurred much faster for the pristine oxide (Fig. 7). Further, less HF and dissolved Mn were found in the electrolyte for the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, as shown in Fig. 10 and Fig. 11.
Reaction of HF with the active material resulted in the formation of MnO, LiF, MnF2, and water molecules. Indeed, as the degradation of the active material progressed, the amount of newly generated water molecules increased. Similarly, these byproducts continuously decomposed the electrolytic salts, and the degradation process simultaneously continued further, leading to even more capacity fading. For the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, BiO+ and BiF+ fragments continued to remain on the surface of the active materials; these fragments were not seen in the case of the pristine material (Fig. 12e).
The BiOF coating located at the outermost surface of the Li[Li0.1Al0.05Mn1.85]O4 was thought to act as a protecting layer against HF attack during cycling. Our results indicate that the high capacity retention was indeed due to the coating layer, which successfully protected the active material from degradation into the electrolyte following HF attack. According to the reactions listed above, less deterioration of the active material meant less generation of HF, as those decomposition reactions were always accompanied by the formation of water molecules, which in turn facilitated the decomposition of electrolytic salts. Degeneration of the spinel active materials progressed in a similar manner. Using ToF-SIMS, we previously reported that amphoteric Al2O3 and NiO nanoparticles are able to scavenge the acidic HF species from the electrolyte.24,25 Similarly, the BiOF layer may have functioned to scavenge HF, and thus the propagated amount of HF was significantly reduced for the BiOF-coated material, as shown in Fig. 10. XRD patterns were obtained for BiOF powders treated with HF solution (49% solution, J.T. Baker) for 1 week at 55 °C. As indicated by Fig. 13, the well-developed BiOF reflections are almost diminished compared with untreated powder. Meanwhile, BiF3 appears quite strong after the HF treatment. This result indicates that the BiOF reacts with HF and finally transforms to BiF3, as follows:
| BiOF + 2HF → BiF3 + H2O | (8) |
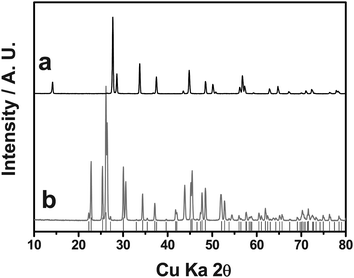 | ||
| Fig. 13 XRD patterns of (a) BiOF and (b) HF-treated BiOF. Grey vertical bars indicate Bragg peak positions of BiF3.41 | ||
We previously suggested that the protection of active material by an AlF3 coating improves the capacity and capacity retention during cycling.27–30 Thus, the role of the BiOF coating layer on the surface of Li[Li0.1Al0.05Mn1.85]O4 particles can be defined as follows: the oxyfluoride layer serves as a strong protecting layer against HF attack and scavenges HF, thus reducing HF from the electrolyte. Together, these actions resulted in the significantly improved high temperature cycling performance exhibited by cells with the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 electrode.
Fig. 14a shows a bright-field TEM image of the pristine Li[Li0.1Al0.05Mn1.85]O4 particles after 100 electrochemical cycles. Without the BiOF coating, the particle surface was severely damaged, as indicated by the particle edges no longer being visible in Fig. 14a. As the particle surface deteriorated due to the dissolution of Mn, an amorphous phase covering the entire particle surface built up. A magnified image of another particle (Fig. 14b) shows that the particle surface was eroded by acid attack and replaced by an amorphous phase (the particle edge is indicated by the arrow in Fig. 14b). The [113] zone from the high-resolution TEM in Fig. 14c clearly demonstrated the structural damage on the surface of the Li[Li0.1Al0.05Mn1.85]O4 particle during cycling at 55 °C. The lattice fringes became increasingly discontinuous towards the particle edge, and the particle surface was extremely rough due to the acid attack. Further, the particle surface was covered by a thick layer of an amorphous phase, the presence of which likely hindered the transport of Li through the electrode, raising surface resistivity and leading to a gradual degradation of the electrode during cycling.
![Bright-field TEM images of (a) the pristine Li[Li0.1Al0.05Mn1.85]O4 particles after 100 electrochemical cycles, (b) magnified image of another particle, and (c) high-resolution TEM images (inset: electron diffraction pattern at [113] zone axis).](/image/article/2009/JM/b819224c/b819224c-f14.gif) | ||
| Fig. 14 Bright-field TEM images of (a) the pristine Li[Li0.1Al0.05Mn1.85]O4 particles after 100 electrochemical cycles, (b) magnified image of another particle, and (c) high-resolution TEM images (inset: electron diffraction pattern at [113] zone axis). | ||
In contrast, the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 particles exhibited a flat, smooth particle surface after 100 cycles, as can be seen from the bright-field TEM image in Fig. 15a. Unlike the uncoated sample, no secondary phase was detected on the particle after 100 cycles, and the flat surface of the pristine material prior to cycling was maintained throughout. Fig. 15b shows a BiOF particle remaining on the Li[Li0.1Al0.05Mn1.85]O4 particle. Energy-dispersive X-ray spectroscopy and high-resolution TEM images confirmed that the particles, indicated by an arrow in Fig. 15b, were indeed composed of BiOF. Fig. 15b demonstrates that a protective BiOF coating remained on the particle surface throughout cycling. The [112] zone high-resolution TEM image in Fig. 15c clearly indicates that the particle structure was remarkably well preserved during the 100 cycles. Compared to Fig. 14c, the lattice fringes were straight and extended right to the particle edge, indicating that the crystallinity of Li[Li0.1Al0.05Mn1.85]O4 was well-maintained on the particle surface. While a minimal amorphous layer was found on the coated Li[Li0.1Al0.05Mn1.85]O4 particle, the TEM results sufficiently demonstrated the beneficial effect of the BiOF coating in protecting the structure of the Li[Li0.1Al0.05Mn1.85]O4 electrode, especially near the particle surface during electrochemical cycling.
![Bright-field TEM images of (a) the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 particles after 100 electrochemical cycles, (b) magnified image of another particle, and (c) high-resolution TEM images (inset: electron diffraction pattern at [112] zone axis).](/image/article/2009/JM/b819224c/b819224c-f15.gif) | ||
| Fig. 15 Bright-field TEM images of (a) the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 particles after 100 electrochemical cycles, (b) magnified image of another particle, and (c) high-resolution TEM images (inset: electron diffraction pattern at [112] zone axis). | ||
Conclusion
Spinel Li[Li0.1Al0.05Mn1.85]O4 was synthesized by a co-precipitation method and coated by BiOF in the presence of bismuth nitrate and ammonium fluoride. XRD revealed that the prepared Li[Li0.1Al0.05Mn1.85]O4 and BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 materials had a single-phase spinel structure with Fd3m space group. Analysis by SEM and TEM showed the fine dispersion of the BiOF coating over the pristine Li[Li0.1Al0.05Mn1.85]O4 particles. From the TEM images for the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4, an average coating layer 10 nm in thickness was determined. The BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 electrode exhibited excellent cycling performance in the voltage range of 3.0–4.3 V at elevated temperatures (55 °C). The capacity retention of the BiOF-coated Li[Li0.1Al0.05Mn1.85]O4 electrode after 100 cycles was 96.1%, while that of the pristine material was only 84.4% compared with the initial discharge capacity. These improved properties were ascribed to the coating effect of BiOF, which was thought to exert its positive effects by acting as a scavenger of HF and as a protecting layer against HF attack, thus preventing its migration into the electrolyte. Consistent with this hypothesis, the decreased amount of HF during cycling resulted in a significant decrease of manganese dissolution from the spinel Li[Li0.1Al0.05Mn1.85]O4 material. Based on the TEM images of the cycled electrodes, the BiOF coating layer was confirmed to be the material responsible for protecting against HF infiltration into the electrolyte.Acknowledgements
This work was supported by the Division of Advanced Batteries in NGE Program.References
- R. J. Gummow, A. de Kock and M. M. Thackeray, Solid State Ionics, 1994, 69, 59 CrossRef CAS.
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769 CAS.
- D. H. Jang, Y. J. Shin and S. M. Oh, J. Electrochem. Soc., 1996, 143, 2204 CAS.
- Y. Xia, Y. Zhou and M. Yoshio, J. Electrochem. Soc., 1997, 144, 2593 CAS.
- S. Komaba, N. Kumagai, T. Sasaki and Y. Miki, Electrochemistry, 2001, 69, 784 CAS.
- S.-T. Myung, S. Komaba and N. Kumagai, J. Electrochem. Soc., 2001, 148, A482 CrossRef CAS.
- B. Banov, Y. Todorov, A. Trifonova, A. Momchilov and V. Manev, J. Power Sources, 1997, 68, 578 CrossRef CAS.
- K. Amine, H. Tukamoto, H. Yasuda and Y. Fujita, J. Electrochem. Soc., 1996, 143, 1607 CAS.
- K. Amine, H. Tukamoto, H. Yasuda and Y. Fujita, J. Power Sources, 1997, 68, 604 CrossRef CAS.
- C. Sigala, A. Vebaere, J. L. Mansot, D. Guyomard, Y. Piffard and T. Tournoux, J. Solid State Chem., 1997, 132, 372 CrossRef CAS.
- L. Hernan, J. Morales, L. Sanchez and J. Santos, Solid State Ionics, 1999, 118, 179 CrossRef CAS.
- M. Hosoya, H. Ikuta, T. Uchida and M. Wakihara, J. Electrochem. Soc., 1997, 144, L52 CrossRef CAS.
- A. Amatucci, A. Du Pasquier, A. Blyr, T. Zheng and J. M. Tarascon, Electrochimica Acta, 1999, 45, 255 CrossRef CAS.
- Y.-K. Sun, Y.-S. Jeon and H.-J. Lee, Electrochem. Solid-state Lett., 2000, 3, 7 CrossRef CAS.
- J. Cho, T.-J. Kim, Y.-J. Kim and B. Park, Chem. Commun., 2001, 1074 RSC.
- Y.-K. Sun, K.-J. Hong, Jai Prakash and K. Amine, Electrochem. Commun., 2002, 4, 344 CrossRef CAS.
- J. S. Gnanaraj, V. G. Pol, A. Gedanken and D. Aurbach, Electrochem. Commun., 2003, 11, 940 CrossRef.
- Y.-M. Lin, H.-C. Wu, Y.-C. Yen, Z.-Z. Guo, M.-H. Yang, H.-M. Chen, H.-S. Sheu and N.-L. Wu, J. Electrochem. Soc., 2005, 152, A1526 CrossRef CAS.
- J.-M. Han, S.-T. Myung and Y.-K. Sun, J. Electrochem. Soc., 2006, 153, A1290 CrossRef CAS.
- T. Numata, C. Amemiya, T. Kumeuchi, M. Shirakata and M. Yonezawa, J. Power Sources, 2001, 97–98, 358 CrossRef CAS.
- Z. F. Ma, X. Q. Yang, X. Z. Liao, X. Sun and J. McBreen, Electrochem. Commun., 2001, 3, 425 CrossRef CAS.
- S.-T. Myung, M. H. Cho, H. T. Hong, T. H. Kang and C.-S. Kim, J. Power Sources, 2005, 146, 222 CrossRef CAS.
- Y.-K. Sun, K.-J. Hong and Jai Prakash, J. Electrochem. Soc., 2003, 150, A970 CrossRef CAS.
- S.-T. Myung, K. Hosoya, S. Komaba, H. Yashiro, Y.-K. Sun and N. Kumagai, Electrochim. Acta, 2006, 51, 5912 CrossRef CAS.
- S.-T. Myung, K. Izumi, S. Komaba, Y.-K. Sun, H. Yashiro and N. Kumagai, Chem. Mater., 2005, 17, 3695 CrossRef CAS.
- S.-T. Myung, K. Izumi, S. Komaba, H. Yashiro, H. J. Bang, Y.-K. Sun and N. Kumagai, J. Phys. Chem. C, 2007, 111, 4061 CrossRef CAS.
- Y.-K. Sun, J.-M. Han, S.-T. Myung, S.-W. Lee and K. Amine, Electrochem. Commun., 2006, 8, 821 CrossRef CAS.
- B.-C. Park, H.-B. Kim, S.-T. Myung, K. Amine, I. Belharouak, S.-M. Lee and Y.-K. Sun, J. Power Sources, 2008, 178, 826 CrossRef CAS.
- H.-B. Kim, B.-C. Park, S.-T. Myung, K. Amine, Jai Prakash and Y.-K. Sun, J. Power Sources, 2008, 179, 347 CrossRef CAS.
- Y.-K. Sun, S.-W. Cho, S.-T. Myung, K. Amine and Jai Prakash, Electrochim. Acta, 2007, 53, 1013 CrossRef CAS.
- M.-H. Lee, Y.-J. Kang, S.-T. Myung and Y.-K. Sun, Electrochim. Acta, 2004, 50, 939 CrossRef CAS.
- T. Roisnel and J. Rodriguez-Carjaval, Fullprof Manual, Institut Laue-Langevin, Grenoble, 2002 Search PubMed.
- Joint Committee on Powder Diffraction Standards, File No., 24–0147.
- Joint Committee on Powder Diffraction Standards, File No., 14–0699.
- Joint Committee on Powder Diffraction Standards, File No., 73–1595.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 756.
- G.-H. Kim, S.-T. Myung, H. J. Bang, J. Prakash and Y.-K. Sun, Electrochem. Solid-State Lett., 2004, 7, A477 CrossRef CAS.
- G.-H. Kim, J.-H. Kim, S.-T. Myung, C. S. Yoon and Y.-K. Sun, J. Electrochem. Soc., 2005, 152, A1707 CrossRef CAS.
- D. Aurbach, J. Electrochem. Soc., 1989, 136, 906 CrossRef CAS.
- K. Edström, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397 CrossRef.
- Joint Committee on Powder Diffraction Standards, File No. 70–2407.
| This journal is © The Royal Society of Chemistry 2009 |