Synthesis and properties of NLO chromophores with fine-tuned gradient electronic structures†
Xiaohua
Ma
a,
Fei
Ma
b,
Zhenhua
Zhao
a,
Naiheng
Song
*a and
Jianping
Zhang
*b
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Department of Polymer Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: nsong@pku.edu.cn; Fax: +86-10-62755644; Tel: +86-10-62755644
bDepartment of Chemistry, Renmin University of China, Beijing 100872, People's Republic of China. E-mail: jpzhang@chem.ruc.edu.cn; Tel: +86-10-62516604
First published on 3rd March 2009
Abstract
A novel series of heterocycle-based NLO chromophores based on different combinations of auxiliary donor (i.e., benzene, thiophene and pyrrole) and auxiliary acceptor (i.e., thiazole with different regiochemistries) were designed and synthesized. Due to the different electron-rich and poor nature of the auxiliary donors and acceptors, respectively, the resulting NLO chromophores have systematically varied ground-state electronic structures, as evidenced by the 1H NMR, CV and UV-vis investigations. The nonlinear optical properties of the resulting NLO chromophores were studied by UV-vis spectroscopy, Hyper-Rayleigh scattering (HRS), and semi-empirical computations. All the chromophores have very large molecular hyperpolarizabilities (β1000 nm) in the range of 704–1500 × 10−30 esu (or β0, 318–768 × 10−30 esu), which showed a great sensitivity to the gradient electronic structures. Upon increasing the electron density from benzene to thiophene and to pyrrole, substantial increases in β0 were observed; significantly larger β0 values were also observed for NLO chromophores based on “matched” thiazole (C2 is connected to the acceptor) than those based on “un-matched” thiazole (C5 is connected to the acceptor). TGA investigations showed good thermal stability for the resulting NLO chromophores. However, with the increase of electron density of the auxiliary donor, a decrease in thermal and photochemical stability was observed. It is interesting to note that NLO chromophores based on triarylamine as the donor and thiazole as the auxiliary acceptor exhibited not only high thermal stability but also very large β0 values.
Introduction
Organic second-order nonlinear optical (NLO) polymers with large optical nonlinearities have potential applications in ultra-fast electro-optic (e-o) devices.1 Recently, breakthroughs in materials development have been made to afford NLO polymers with extremely large e-o coefficients (e.g., r33 > 100 pm/V).2–4 However, for practical e-o applications, NLO polymers with good comprehensive materials properties are still to be developed. One critical challenge towards highly efficient NLO polymers has been to develop NLO chromophores with simultaneously very large molecular first hyperpolarizability (β), good optical transparency, and excellent thermal and photochemical stabilities. Despite the large number of NLO chromophores that have been developed in the past two decades, only a few of them exhibit promising properties for e-o device applications.2–6 Thus, to further enhance the NLO properties and to explore the full potential of NLO chromophores, rational structural designs based on advanced understanding of the structure-property relationships are highly desired.According to the bond-length alternation model,5 maximal β of D–π–A NLO chromophores can be realized for certain degrees of ground-state polarization. Therefore, much effort has been devoted to the design and synthesis of NLO chromophores with different combinations of the electron donor, the π-bridge, and the electron acceptor, aiming at the optimization of the ground-state electronic structure. Currently, the state-of-the-art NLO chromophores are the so-called “FTC” and “CLD” type of chromophores developed by Dalton and co-workers, which coupled dialkylamino donor with a strong electron acceptor (i.e., 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran, TCF) to yield very large molecular optical nonlinearities (µβ > 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × 10−48 esu).6 However, maximal β values for these chromophores remain largely unrealized, especially when considering the astonishingly large µβ values (e.g., µβ = −488000 × 10−48 esu) achieved for zwitterionic chromophores by Marks and co-workers.4,7
000 × 10−48 esu).6 However, maximal β values for these chromophores remain largely unrealized, especially when considering the astonishingly large µβ values (e.g., µβ = −488000 × 10−48 esu) achieved for zwitterionic chromophores by Marks and co-workers.4,7
To achieve even larger β values by using stronger electron donor and acceptor represents a challenging task due to the limited accessibility to such groups at the current stage. A strategy to alleviate such problem could be through the optimization of the π-bridge.8–10 Theoretical and experimental studies have shown that, upon introduction of electron-rich heterocycle to the donor end of the π-bridge as auxiliary donor and electron-poor heterocycle to the acceptor end as auxiliary acceptor, the donor and acceptor strength will be enhanced.8–13 Substantial increases in β values can also be expected for chromophores with such gradient electronic structures.
Among the many aromatic heterocycles, pyrrole is known to have very high electron density and low aromaticity and can serve as an efficient auxiliary donor to yield large molecular optical nonlinearities.8 However, experimental studies on NLO chromophores containing pyrole as the auxiliary donor were not known until our very recent report on the first synthesis of piperidin-1-ylpyrrole and methyl 4-methoxyphenylpyrrole-based NLO chromophores.14 Later on, Davies et al. reported the synthesis and properties of a diarylaminopyrrole-based NLO chromophore.15 Although these chromophores showed promising properties, the potential of pyrrole in enhancing the donor strength remains largely unexplored. On the other hand, the electron-poor five-membered heterocycle thiazole has recently drawn great attention due to its good potential in increasing the β values when used as the auxiliary acceptor.10–13 Due to the inherent dipolar nature, thiazole can be incorporated into NLO chromophores in either “matched” (i.e., the electron-poor C2 is connected to the acceptor) or “un-matched” (i.e., the electron-rich C5 is connected to the acceptor) configurations. Although both connections are expected to give enhanced β values, the former can be more efficient, as indicated by the theoretical computations.10,11 Up to now, experimental investigations on thiazole-based NLO chromophores are still limited; those containing “matched” thiazole at the acceptor end of the π-bridge are not yet known.
Towards an advanced understanding of structure-property relationship and to achieve highly efficient NLO chromophores, we have herein designed and prepared a novel series of heterocycle-based NLO chromophores with fine-tuned gradient electronic structures. These chromophores have the same tricyanovinyl electron acceptor and the same length of π-conjugation, but contain different aromatic rings with varying electron densities (i.e., pyrrole > thiophene > benzene)8,10 as the auxiliary donor and 1,3-heteroaromatic thiazole with different regiochemistries as the auxiliary acceptor. Examination of NLO chromophores with such a complete combination of auxiliary donor and acceptor should allow us to gain more insight on the potential contribution of π-bridge to β values and offer guidelines for rational structure design toward highly efficient NLO chromophores. In this study, modifications on the amino donor part were also made by using different substitutions (e.g., dialkyl, aryl alkyl, and diaryl substitutions) to fine tune the donor ability. Triarylamine donor15,16 that have recently been successfully used in NLO chromophores to afford large β values and high thermal stability was for the first time coupled with thiazole as auxiliary acceptor in this study to yield novel NLO chromophores with good combination of properties.
Experimentals
Materials
1-Methyl-5-(piperidin-1-yl)pyrrole-2-carbaldehyde,145-(piperidin-1-yl) thiophene-2-carbaldehyde,142-chloromethyl thiophene,172-(chloromethyl) thiazole,182-chloro-5-(chloromethyl)thiazole,19thiazole-5-carbaldehyde,204-methoxy-N-(4-methoxyphenyl)-N-phenylbenzenamine,214-(bis(4-methoxy phenyl)amino)benzaldehyde22 and tetracyanoethene23 were prepared referring to literature methods. n-Butyl lithium (n-BuLi) (Acros Organics), 1-iodo-4-methoxybenzene (Linzi Tianyuan Welfare Chemical Factory), potassium t-butyloxide (t-BuOK) (Alfa Aesar Corp.), tetrabutylammonium perchlorate (Bu4NClO4, 99%) (Fluka), 2,2′-bipyridine (Sinopharm Chemcial Reagent Co. Ltd) and thiazol-2-amine (Beijing Ouhe Technology Co., Ltd) were purchased and used as received. Phosphorous oxide trichloride (POCl3), trimethylsilane chloride (TMSCl), tetrachloromethane (CCl4), thionyl chloride, sulfuryl dichloride and aniline were purchased from Beijing Chemical Reagent Ltd and distilled before use. N,N-Dimethylformamide (DMF) and tetrahydrofuran were distilled over calcium hydride and metal sodium, respectively, and stored over molecular sieves (3 Å) under nitrogen. All other chemicals were purchased from Beijing Chemical Reagent Ltd and used as received.Instruments
1H and 13C NMR spectra were recorded on a Varian 300 MHz or a Bruker ARX400 spectrometer using deuterated chloroform (CDCl3) or dimethylsulfoxide (DMSO-d6) as the solvent. Chemical shifts were reported in ppm scale with tetramethylsilane as the internal standard. Infrared (IR) spectroscopies were measured on a Nicolet Magna 750 Fourier transform infrared spectrometer. UV-Vis absorption spectra were recorded with a DU 640 UV-vis spectrometer. Thermogravimetric analysis (TGA) was performed in nitrogen on a TA TGA-DSC Q600 thermogravimetric analyzer with a heating rate of 20 °C/min. Melting points were measured with a SGW-X-4 microscopic melting point instrument. High and low resolution mass spectroscopy was recorded on a ZAB-HS mass spectrometer. Elemental analysis was performed on an Elementar Vario EL instrument (Elementar Analysensysteme GmbH). Cyclic voltammetry (CV) was performed on a CHI 600C cyclic voltametric analyzer in a solution of tetrabutylammonium perchlorate (0.1 M) in acetonitrile at a scanning rate of 200 mV/s. Before each measurement, the cell was deoxygenated with argon for 20 min. The working electrode was a glassy carbon disk (2 mm diameter, freshly polished) for voltammetry. A platinum stick (1 mm thick) was used as the counter-electrode and AgCl/Ag the reference electrode. All potentials are reported versus the AgCl/Ag electrode. Geometric optimization and computation of static molecular first hyperpolarizabilities were carried out with the MOPAC program (2007) in AM1 method.Hyper-Rayleigh scattering (HRS) measurements
HRS measurements of the NLO chromophores were performed at the excitation wavelength of 1000 nm. A third harmonic wave at 355 nm with a pulse duartion of 7 ns (repetition rate, 10 Hz) was supplied by a Q-switched Nd3+:YAG laser, and was used to drive an optical parametric osciallator (MOPO-SL/MOPO-PO, Spectra-Physics) to deliver the light pulses at 1000 nm. The laser beam was passed through a Pelin–Broca prism and other steering optics, and focused into the sample cell with a lens (f = 200 mm). The pulse energy measured in front of the sample cell was 2 mJ/pulse. The HRS signal collected with a camera lens (f/1.4) was further focused via a lens (f = 150 mm) onto the entrance slit of a triplet spectrometer (Trivista, SP2500i, Princeton Instruments/Acton), and was detected by an intensified CCD detector (ICCD PI-MAX, Princeton Instruments/Acton) operated with a gate width of 10 ns. For each HRS measurement the sample was exposed to 1000 laser shots, and the accumulated harmonic signal at 500 nm was extracted, via spectral deconvolution, from the background fluorescence induced by two-photon absorption. The intensity of HRS signal was calibrated with the Beer–Lambert law for the correction of the chromophore self-absorption. To determine the β value of a chromophore, p-nitroaniline (p-NA) in dimethylsulfoxide (DMSO) was used as an external standard, whose β value at 1000 nm (34.5 × 10−30 esu) was calculated based on the known β value at 1064 nm (28.8 × 10−30 esu)24 within the framework of the two-level model. The concentration of p-NA in DMSO was in the range of 1 × 10−3–5 × 10−1 mol/L. All of the sample solutions in chloroform were prepared in the concentration range of 1 × 10−5–1 × 10−4 mol/L.Preparation of intermediates
Diethyl thiophen-2-ylmethyl phosphonate
A solution of 2-chloromethyl thiophene (3.00 g, 22.7 mmol) in triethyl phosphite (7.50 g, 45.2 mmol) was heated to 130 °C under nitrogen and stirred for 36 h. The excess amount of triethyl phosphite was removed under vacuum (∼0.5 mmHg) to leave a sticky yellow liquid product (5.0 g, 94% yield): 1H NMR (300 MHz, CDCl3): δ 7.17–7.20 (1H, m), 6.95–7.01 (2H, m), 3.95–4.10 (4H, m), 3.3 (1H, d, J = 22 Hz), 1.28 (6H, t, J = 7.2 Hz, 7.2 Hz); 13C NMR (75 MHz, CDCl3): δ 132.4, 132.2, 127.3, 127.2, 126.9, 126.8, 124.6, 124.5, 62.3, 62.4, 28.8, 26.9, 16.3, 16.2.Diethyl thiazol-2-ylmethylphosphonate
A mixture of 2-(chloromethyl) thiazole (4.00 g, 30.0 mmol) and triethyl phosphite (9.96 g, 60.0 mmol) was heated at 130 °C under nitrogen for 48 h. The excessive amount of triethyl phosphite was removed under vacuum (∼0.5 mmHg) to give a yellow viscous liquid product (6.80 g, 96% yield): 1H NMR (300 MHz, CDCl3): δ 7.73 (1H, d, J = 3.4 Hz), 7.29–7.32 (1H, m), 4.05–4.20 (4H, m), 3.70 (2H, d, J = 21.2 Hz), 1.30 (6H, t, J = 7.0 Hz, 7.0Hz).[(2-Chlorothiazol-5-yl)methyl]triphenylphosphonium chloride
A solution of 2-chloro-5-(chloromethyl)thiazole (3.00 g, 17.9 mmol) and triphenylphosphine (11.7 g, 44.6 mmol) in chloroform (30 mL) was stirred at refluxing temperature for 48 h before filtration to collect the solid product, which was washed with acetone (10 mL) twice to give a white powdered product (6.20 g, 90% yield): 1H NMR (300 MHz, CDCl3): δ 7.86–7.94 (6H, m), 7.76–7.82 (3H, m), 7.63–7.68 (6H, m), 7.25 (1H, d, J = 4.2 Hz), 5.70 (2H, d, J = 14.1 Hz).Thiazol-5-ylmethanol
To a solution of thiazole-5-carbaldehyde (0.226 g, 2.00 mmol) in methanol (10 mL), was added powdered sodium borohydride (0.08 g, 1.00 mmol) in portions. The resulting solution was stirred at room temperature for 1 h. After removal of the solvent by rotary-evaporation, the liquid residue was purified by silica-gel column chromatography (eluent: ethanol/ethyl acetate/ligroin = 1/1/4, v/v/v, Rf = 0.31) to give a colorless oily product (0.17 g, 75% yield): 1H NMR (300 MHz, CDCl3): δ 8.75 (1H, s), 7.74 (1H, s), 4.90 (2H, d, J = 4.8 Hz), 3.1 (1H, t, J = 5.4 Hz).5-(Chloromethyl)thiazole
A mixture solution of thiazol-5-ylmethanol (11.5 g, 100 mmol) and thionyl chloride (2.36 g, 200 mmol) in chloroform was refluxed at 80 °C for 2 h. It was then poured into water (150 mL) and extracted with ethyl acetate (3 × 50 mL). The organic extracts were combined, washed with saturated aqueous sodium carbonate solution (30 mL), and dried over anhydrous magnesium sulfate. After removal of the solvent by rotary-evaporation, a yellow liquid product was obtained, which was used for the next step of synthesis without further purification (3.5 g, 31% yield): 1H NMR (300 MHz, CDCl3): δ 8.80 (1H, s), 7.80 (1H, s), 4.91 (2H, s).Diethyl thiazol-5-ylmethylphosphonate
A mixture of 5-(chloromethyl)thiazole (2.00 g, 15.1 mmol) in triethyl phosphite (10.0 mL) was refluxed at 130 °C for 36 h. The excessive amount of triethyl phosphite was removed under high vacuum (∼0.5 mmHg) to give a light-yellow viscous liquid product (2.0 g, 57% yield): 1H NMR (300 MHz, CDCl3): δ 8.75 (1H, d, J = 1.2 Hz), 7.55 (1H, d, J = 1.2 Hz), 3.83 (4H, d, J = 7.2 Hz), 3.36 (2H, d, J = 21 Hz), 1.28 (6H, t, J = 7.2 Hz).Preparation of NLO chromophores
(E)-2-{2-[1-methyl-5-(piperidin-1-yl)-1H-pyrrol-2-yl] vinyl} thiazole (P-Ti2)
A solution of 1-methyl-5-(piperidin-1-yl)-1H-pyrrole-2-carbaldehyde (1.93 g, 10.0 mmol) and diethyl thiazol-2-ylmethylphosphonate (4.70 g, 20.0 mmol) in anhydrous THF (60.0 mL) was prepared, purged with nitrogen, and cooled with a dry ice/acetone mixture to −78 °C. To the solution was added dropwise a solution of potassium t-butyloxide (2.24 g, 20.0 mmol) in anhydrous tetrahydrofuran (60.0 mL). After the addition, the reaction solution was warmed gradually up to room temperature in 3 h and then poured into distilled water (150 mL). The mixture solution was extracted in a separatory funnel with ethyl acetate (3 × 30 mL). The organic extracts were combined, washed with distilled water (2 × 30 mL), and dried over anhydrous magnesium sulfate. After removal of the solvent, the solid residue was purified by silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/4, v/v, Rf = 0.5) and then recrystallized from ligroin to give yellow sheet crystals (2.32 g, 85% yield): mp. 107–108 °C; 1H NMR (300 MHz, CDCl3): δ 7.70 (1H, d, J = 3.3 Hz), 7.30 (1H, d, J = 16.2 Hz), 7.13 (1H, d, J = 3.3 Hz), 6.97 (1H, d, J = 15.9 Hz), 6.51 (2H, d, J = 3.9 Hz), 5.71 (2H, d, J = 3.9 Hz), 3.56 (3H, s), 2.83 (4H, t, J = 5.1 Hz, 5.4 Hz), 1.53–1.70 (6H, m); 13C NMR (75 MHz, CDCl3): δ 168.3, 146.1, 143.0, 126.1, 123.4, 116.3, 114.5, 108.9, 96.3, 53.8, 29.8, 26.0, 24.0; anal. calcd for C15H19N3S: C, 65.90; H, 7.00; N, 15.37; found: C, 65.90; H, 6.93; N, 15.43.(E)-2-(2-{2-[1-methyl-5-(piperidin-1-yl)-1H-pyrrol-2-yl]vinyl}thiazol-5-yl) ethene-1,1,2-tricarbonitrile (P-Ti2-TCV)
To a solution of P-Ti2 (0.273 g, 1.00 mmol) in anhydrous tetrahydrofuran (3 mL) at −78 °C under nitrogen, was added n-butyl lithium (1.6 M in hexane, 0.94 mL, 1.5 mmol). The resulting solution was allowed to warm gradually up to −10 °C in 2 h and then cooled again to −78 °C. A solution of tetracyanoethene (0.192 g, 1.50 mmol) in tetrahydrofuran (5 mL) was added dropwise to the solution. After the reaction solution was stirred at −78 °C for 10 min, hydrochloric acid (1 M, 1 mL) was dropped in. The solution was warmed up to room temperature and poured into distilled water (30 mL). The mixture was extracted in a separatory funnel with dichloromethane (5 × 30 mL). The organic extracts were combined, washed with distilled water (50 mL) twice, dried over anhydrous magnesium sulfate, and rotary-evaporated to give a dark green solid residue, which was purified by silica-gel column chromatography (eluent: ethyl acetate/ligroin, 1/1, v/v, Rf = 0.6) to yield a green solid product (0.060 g, 16% yield); 1H NMR (400 MHz, CDCl3): δ 8.43 (1H, s), 7.69 (1H, d, J = 14.8 Hz), 6.96 (1H, d, J = 4.4 Hz), 6.83 (1H, d, J = 14.8 Hz), 5.92 (1H, d, J = 4.4 Hz), 3.60 (3H, s), 3.01 (4H, t, J = 4.8 Hz), 1.71–1.75 (4H, m), 1.63–1.64 (4H, m); 13C NMR (75 MHz, CDCl3): 178.2, 157.9, 154.5, 130.1, 128.6, 127.8, 119.7, 113.4, 113.0, 112.6, 110.2, 101.9, 52.8, 31.2, 25.7, 23.9; FT-IR (KBr, cm−1): 2935, 2855, 2192, 1570, 1508, 1439, 1409, 1352, 1226, 1127, 1100, 1026; HRMS for C20H18N6S (m/z): 374.1314 (calcd), 374.1311 (found).(E)-N-(4-Methoxyphenyl)-N,1-dimethyl-5-[2-(thiazol-2-yl)vinyl]-1H-pyrrol- 2-amine (MP-Ti2)
To a solution of 5-(N-4-methoxyphenyl N-methyl amino)-1-methyl-1H-pyrrole-2-carbaldehyde (1.22 g, 5.00 mmol) and diethyl thiazol-2-ylmethylphosphonate (2.56 g, 10.0 mmo) in anhydrous tetrahydrofuran (60.0 mL) at −78 °C under nitrogen, was added dropwise a solution of potassium t-butyloxide (1.12 g, 10.0 mmol) in anhydrous tetrahydrofuran (30.0 mL). After the addition, the solution was allowed to gradually warm up to room temperature and then poured into distilled water (100 mL). The mixture was extracted with ethyl acetate (3 × 30 mL). The organic extracts were combined, washed with distilled water (30 mL) twice, and dried over anhydrous magnesium sulfate. After removal of the solvent, the liquid residue was purified by a silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/8, v/v, Rf = 0.3) to give a sticky liquid product (0.15 g, 93% yield): 1H NMR (300 MHz, CDCl3): δ 7.76 (1H, d, J = 3.3 Hz), 7.39 (1H, d, J = 15.9 Hz), 7.18 (1H, d, J = 3.3 Hz), 7.03 (1H, d, J = 15.9 Hz), 6.82 (2H, d, J = 9.0 Hz), 6.58–6.63 (3H, m), 5.96 (1H, d, J = 4.2 Hz), 3.76 (3H, s), 3.41(3H, s), 3.23 (3H, s); 13C NMR (75 MHz, CDCl3): δ 167.9, 152.9, 143.4, 143.0, 140.9, 126.9, 123.1, 116.8, 115.6, 114.6, 108.6, 103.0, 55.7, 40.8, 29.8.(E)-2-[2-(2-{5-[(4-methoxyphenyl)(methyl)amino]-1-methyl-1H-pyrrol-2-yl}vinyl)thiazol-5-yl]ethene-1,1,2-tricarbonitrile (MP-Ti2-TCV)
To a solution of MP-Ti2 (0. 900 g, 2.77 mmol) in anhydrous tetrahydrofuran (20 mL) at −78 °C under nitrogen, was added n-butyl lithium (1.6 M in hexane, 2.08 mL, 3.33 mmol). The resulting solution was allowed to warm gradually up to 0 °C in 0.5 h and then cooled again to −78 °C, to which a solution of tetracyanoethene (0.531 g, 4.15 mmol) in anhydrous tetrahydrofuran (10.0 mL) was added by syringe over 5 min. After the resulting reaction solution was stirred at −78 °C for additional 10 min, hydrochloric acid (1 M, 1 mL, 1.00 mmol) was dropped in. The solution was warmed up to room temperature, poured into distilled water (30 mL), and extracted with dichloromethane (3 × 30 mL). The extracts were combined, washed with distilled water, and dried over anhydrous magnesium sulfate. After rotary-evaporation to remove the solvent, the residue was run through a silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/1, v/v, Rf = 0.55) to give the pure green solid product (0. 10 g, 8.5% yield); 1H NMR (400 MHz, CDCl3): δ 8.44 (1H, s), 7.80 (1H, d, J = 15.2 Hz), 7.08 (1H, d, J = 4.4 Hz), 6.81–6.89 (5H, m), 6.16 (1H, d, J = 4.4 Hz), 3.80 (3H, s), 3.34 (3H, s), 3.33 (3H, s); 13C NMR (75 MHz, CDCl3): δ; 117.9, 157.7, 155.3, 148.6, 141.5, 129.6, 128.2, 128.1, 127.5, 120.2, 118.3, 114.9, 113.1, 112.8, 112.5, 111.1, 104.9, 55.6, 42.1, 31.1. FT-IR (KBr, cm−1): 2948, 2208, 1595, 1505, 1462, 1396, 1337, 1233, 1172, 1037; HRMS for C23H18N6OS (m/z): 426.1263 (calcd), 426.1276 (found).(E)-2-{2-[5-(piperidin-1-yl)thiophen-2-yl]vinyl}thiazole (T-Ti2)
To a solution of 5-(piperidin-1-yl)thiophene-2-carbaldehyde (1.95 g, 10.0 mmol) and diethyl thiazol-2-ylmethylphosphonate (4.70 g, 20.0 mmol) in anhydrous tetrahydrofuran (80.0 mL) at −78 °C under nitrogen, was added dropwise a solution of potassium t-butyloxide (2.24 g, 20.0 mmol) in anhydous tetrahydrofuran (60.0 mL). After the addition, the solution was gradually warmed up to room temperature over 1 h and then poured into distilled water (150 mL). The mixture was extracted with ethyl acetate (3 × 50 mL). The organic extracts were combined, washed with distilled water (2 × 30 mL), and dried over magnesium sulfate. After removal of the solvent by rotary-evaporation, the solid residue was purified by a silica-gel chromatography (eluent: ethyl acetate/ligroin = 1/4, v/v, Rf = 0.7) and recrystallized from ligroin to give yellow needle-like crystals (2.07 g, 75% yield): mp. 111–112 °C; 1H NMR (300 MHz, CDCl3): δ 7.70 (1H, d, J = 3.3 Hz), 7.42 (1H, d, J = 15.6 Hz), 7.11 (1H, d, J = 3.3 Hz), 6.86 (2H, d, J = 3.9 Hz), 6.75 (1H, d, J = 15.6 Hz), 5.94 (2H, d, J = 3.9 Hz), 3.19 (4H, m), 1.67–1.74 (4H, m), 1.58–1.63 (2H, m); 13C NMR (75 MHz, CDCl3): δ 167.8, 160.8, 143.0, 130.4, 128.4, 126.0, 116.5, 114.9, 103.7, 51.5, 25.0, 24.0; Anal. Calcd. for C14H16N2S2: C, 60.83; H, 5.83, N, 10.13; Found: C, 60.73; H, 5.81; N, 10.10.(E)-2-(2-{2-[5-(piperidin-1-yl)thiophen-2-yl]vinyl}thiazol-5-yl)ethene-1,1,2- tricarbonitrile (T-Ti2-TCV)
To a stirred solution of T-Ti2 (0.273 g, 0.989 mmol) in anhydrous tetrahydrofuran (3.00 mL) at −78 °C under nitrogen, n-butyl lithium (1.6 M in hexane, 0.940 mL, 1.50 mmol) was added in drops. After the addition, the solution was allowed to warm up to −10 °C over 2 h and then cooled again to −78 °C, to which a solution of tetracyanoethene (0.192 g, 1.50 mmol) in anhydrous tetrahydrofuran (5 mL) was added dropwise. After stirring for 5 min, distilled water (3.00 mL) was added and the resulting solution was warmed up to room temperature. After removal of the solvent under vacuum, the residue was washed thoroughly with diethyl ether (2 × 30 mL) and dichloromethane until the washings were colorless. The dichlormethane washings were combined, dried over anhydrous magnesium sulfate, and rotary-evaporated to give a solid residue that was recrystallized from chloroform to yield golden colored crystals (0.150 g, 40% yield): 1H NMR (300 MHz, CDCl3): δ 8.43 (1H, s), 7.89 (1H, d, J = 15.4 Hz), 7.27 (1H, d, J = 4.5 Hz), 6.56 (1H, d, J = 14.4 Hz), 6.14 (1H, d, J = 4.5 Hz), 3.44 (4H, t, J = 4.8 Hz), 1.73–1.70 (6H, m); FT-IR (KBr, cm−1):2942, 2859, 2209, 1581, 1496, 1445, 1411, 1382, 1324, 1234, 1172, 1070, 1011, 943; HRMS for C19H15N5S2 (m/z): 377.0769 (calcd), 377.0769 (found).(E)-2-chloro-5-{2-[5-(piperidin-1-yl)thiophen-2-yl]vinyl} thiazole (T-Ti5)
To a solution of (2-chlorothiazol-5-yl)methyl triphenyl phosphonium chloride (3.56 g, 8.90 mmol) in toluene (160 mL), was added potassium t-butyloxide (1.79 g, 17.8 mmol). The solution was stirred at room temperature for 2.5 h, followed by the addition of 5-(piperidin-1-yl) thiophene-2-carbaldehyde (1.56 g, 8.00 mmol) in toluene (20 mL) in drops. The reaction solution was stirred at room temperature for another 6 h before being poured into distilled water (200 mL). The mixture was extracted in a separatory funnel with ethyl acetate (3 × 30 mL). The organic extracts were combined, washed with distilled water, dried over anhydrous magnesium sulfate, and rotary-evaporated to give a yellow solid residue that was purified by silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/10, v/v, Rf = 0.33) and recrystallized from ligroin to yield yellow needle-like crystals (1.30 g, 54% yield): mp: 113–114 °C; 1H NMR (300 MHz, CDCl3): δ 7.32 (1H, s), 6.78 (1H, d, J = 15.6 Hz), 6.76 (1H, d, J = 3.9 Hz), 6.50 (1H, d, J = 15.6 Hz), 5.91 (1H, d, J = 3.9 Hz), 3.18 (3H, t, J = 5.4 Hz), 1.56–1.74 (6H, m); 13C NMR (75 MHz, CDCl3): δ 160.0, 152.7, 147.7, 140.9, 137.6, 128.8, 126.4, 111.4, 103.7, 51.6, 25.1, 23.6. Anal. calcd for C14H15ClN2S2: C, 54.09; H, 4.86; N, 9.01; found: C, 54.06; H, 4.90; N, 8.96.(E)-2-(5-{2-[5-(Piperidin-1-yl)thiophen-2-yl]vinyl}thiazol-2-yl)ethene-1,1,2–tricarbonitrile (T-Ti5-TCV)
To a solution of T-Ti5 (0.100 g, 0.323 mmol) in anhydrous tetrahydrofuran (10.0 mL) at −78 °C under nitrogen, was added dropwise a solution of t-butyl lithium (1.5 M in pentane, 0.25 mL, 0.375 mmol). The reaction solution was stirred at −78 °C for 1 h before tetracyanoethene (0.042 g, 0.328 mmol) in anhydrous tetrahydrofuran (10.0 mL) was added in drops. The color of the solution turned to dark green right away. The solution was stirred at −78 °C for another 5 min and then poured into distilled water (30.0 mL). Hydrochloric acid (2 M) was added to the mixture solution to adjust the pH value to 5, followed by extraction with dichloromethane (3 × 20 mL). The organic extracts were combined, washed with distilled water, dried over anhydrous magnesium sulfate, and rotary-evaporated to give a dark green solid that was purified by recrystallization from ethyl acetate (0.03 g, 27% yield): 1H NMR (300 MHz, CDCl3): δ 7.99 (1H, s), 7.32 (1H, d, J = 15 Hz), 7.13 (1H, d, J = 4.5 Hz), 6.54 (1H, d, J = 15 Hz), 6.09 (1H, d, J = 4.5 Hz), 3.38–3.41 (4H, m), 1.54–1.72 (6H, m); FT-IR (KBr, cm−1): 2944, 2856, 2210, 1584, 1499, 1446, 1386, 1347, 1247, 1609, 1012, 919; HRMS for C19H15N5S2 (m/z): 377.0769 (calcd), 377.0770 (found).(E)-N,N-Dimethyl-4-[2-(thiazol-2-yl)vinyl]benzenamine (B-Ti2)
To a solution of 4-(dimethylamino)benzaldehyde (1.49 g, 10.0 mmol) and diethyl thiazol-2-ylmethylphosphonate (4.70 g, 20.0 mmol) in anhydrous tetrahydrofuran (100 mL) at −78 °C under nitrogen, was added dropwise potassium t-butyloxide (2.24 g, 20.0 mmol) in anhydrous tetrahydrofuran. The solution was allowed to warm up to room temperature in 0.5 h and then poured into distilled water (100 mL). The resulting mixture was extracted with ethyl acetate (3 × 30 mL). The organic extracts were combined, washed with distilled water, dried over anhydrous magnesium sulfate, and rotary-evaporated to remove most of the solvent. After a flash column chromatography on silica-gel (eluent: ethyl acetate/ligroin = 1/4, v/v, Rf = 0.5), the resulting yellow solid was further purified by recrystallization from ligroin to give yellow sheet-like crystals (2.07 g, 90% yield): mp. 123–124 °C; 1H NMR (300 MHz, CDCl3): δ 7.74 (1H, d, J = 3.3 Hz), 7.43 (2H, d, J = 6.9Hz), 7.36 (2H, d, J = 16.2 Hz), 7.10–7.16 (2H, m), 6.69 (2H, d, J = 6.9Hz), 2.99 (3H, s); 13C NMR (75 MHz, CDCl3): δ 168.2, 150.7, 143.0, 134.8, 128.3, 123.6, 116.8, 116.7, 112.0, 40.2.(E)-2-{2-[4-(Dimethylamino)styryl]thiazol-5-yl}ethene-1,1,2-tricarbonitrile (B-Ti2-TCV)
To a solution of B-Ti2 (0.230 g, 1.00 mmol) in anhydrous tetrahydrofuran (3.00 mL) at −78 °C under nitrogen, was added n-buthyl lithium (1.6 M in hexane, 0.94 mL, 1.50 mmol). The resulting solution was stirred at −78 °C for 1 h before tetracyanoethene (0.192 g, 1.50 mmol) in anhydrous tetrahydrofuran (4 mL) was added. The reaction solution was stirred at −78 °C for another 10 min and then poured into distilled water (3 mL). The mixture was dried under vacuum with a cold trap and the solid residue was washed with diethyl ether (30 mL) twice and extracted with dichloromethane (30 mL) three times. The dichloromethane extracts were combined, dried over anhydrous magnesium sulfate, and rotary-evaporated to give a dark green residue that was purified by recrystallization from chloroform to yield the dark green solid product (0.371 g, 75% yield): 1H NMR (300 MHz, CDCl3): δ 8.55 (1H, s), 7.80 (1H, d, J = 15.6 Hz), 7.54 (2H, d, J = 9.0 Hz), 7.13 (1H, d, J = 15.6 Hz), 6.73 (2H, d, J = 9.0 Hz), 3.11 (6H, s); FT-IR (KBr, cm−1): 2899, 2212 (–CN), 1595, 1510, 1414, 1366, 1232, 1223, 1157, 941, 832; HRMS for C18H13N5S (m/z): 331.0892 (calcd), 331.0896 (found); anal. calcd for C18H13N5: C, 65.24; H, 3.95; N, 21.13; found: C, 64.15; H, 4.85; N,22.44.(E)-N,N-dimethyl-4-[2-(thiophen-2-yl)vinyl]benzenamine: (B-T)
To a solution of 4-(dimethylamino)benzaldehyde (1.49 g, 10.0 mmol) and diethyl thiophen-2-ylmethylphosphonate (4.68 g, 20.0 mmol) in anhydrous tetrahydrofuran (60 mL) at −78 °C under nitrogen, was added dropwise potassium t-butyloxide (2.24 g, 20.0 mmol) in anhydrous tetrahydrofuran (60 mL). After the addition, the solution was gradually warmed up to room temperature and then poured into distilled water (100 mL). The resulting mixture was extracted with ethyl acetate (3 × 50 mL). The organic extracts were combined, washed with distilled water (2 × 30 mL), and dried over magnesium sulfate. After removal of the solvent by rotary-evaporation, the solid residue was purified by a silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/4, v/v, Rf = 0.7) and recrystallized from ligroin to give yellow sheet crystals (1.90 g, 85% yield): mp. 137–139 °C; 1H NMR (300 MHz, CDCl3): δ 7.36 (2H, d, J = 8.7Hz), 7.11–12 (1H, m), 7.05 (1H, d, J =15.9 Hz), 6.96–7.01 (2H, m), 6.87 (1H, d, J = 15.9 Hz), 6.71 (2H, d, J = 8.7 Hz); 13C NMR (100 MHz, CDCl3): δ 150.0, 143.9, 128.6, 127.4, 127.36, 127.32, 124.5, 122.9, 117.6, 112.4, 40.4.(E)-2-{5-[4-(dimethylamino)styryl]thiophen-2-yl}ethene-1,1,2-tricarbonitrile (B-T-TCV)
The synthesis referred to the literature method25: to a solution of B-T (0.460 g, 20.0 mmol) in anhydrous DMF (15 mL), tetracyanoethene (0.312 g, 24.0 mmol) in anhydrous DMF (10 mL) was added dropwise. The solution was stirred for 24 h before being poured into distilled water (100 mL). The solid product was collected by filtration and recrystallized from ethyl acetate to give a dark green crystalline product (0.396 g, 60% yield): 1H NMR (200 MHz, CDCl3): δ 7.94 (1H, d, J = 4.4 Hz), 7.48 (2H, d, J = 9.0 Hz), 7.29 (1H, d, J = 15.8 Hz), 7.18 (1H, d, J = 4.2Hz), 7.08 (1H, d, J = 15.8 Hz), 6.82 (2H, d, J = 8.4 Hz), 3.10 (6H, s); FT-IR (KBr, cm−1): 3086, 2804, 2207, 1583, 1513, 1411, 1367, 1292, 1163, 1070, 995, 942, 857, 717; HRMS for C19H14N4S (m/z): 330.0939 (calcd), 330.0935 (found).(E)-4-methoxy-N-(4-methoxyphenyl)-N-{4-[2-(thiazol-2-yl)vinyl]phenyl} benzenamine (TPA-Ti2)
To a solution of 4-[bis(4-methoxyphenyl)amino]benzaldehyde (1.00 g, 3.00 mmol) and diethyl thiazol-2-ylmethylphosphonate (1.06 g, 4.50 mmol) in anhydrous tetrahydrofuran (30 mL) at −78 °C under nitrogen, was added dropwise potassium t-butyloxide (0.672 g, 6.00 mmol) in anhydrous tetrahydrofuran. After the addition, the solution was gradually warmed up to room temperature and then poured into distilled water (100 mL). The mixture was extracted with ethyl acetate (3 × 30 mL). The organic extracts were combined, washed with distilled water (2 × 30 mL), dried over anhydrous magnesium sulfate, and rotary-evaporated to remove the solvent. The solid residue was purified by a silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/4, v/v, Rf = 0.5) to give a light yellow solid product (1.19 g, 96% yield): mp. 88–89 °C; 1H NMR (300 MHz, CDCl3): δ 7.76 (1H, d, J = 3.3 Hz), 7.32–7.37 (3H, m), 7.07–7.13 (4H, m), 3.85 (6H, s); 13C NMR (75 MHz, CDCl3): 167.8, 156.2, 149.4, 143.2, 140.1, 134.2, 127.9, 127.3, 127.0, 119.4, 118.3, 117.2, 114.72, 55.4; FT-IR (KBr, cm−1): 2954, 1595, 1505, 1327, 1296, 1245, 1178, 1134, 1104, 1031, 968, 832, 719, 573, 533.(E)-2-(2-{4-[bis(4-methoxyphenyl)amino]styryl}thiazol-5-yl)ethene-1,1,2-tricarbonitrile (TPA-Ti2-TCV)
To a stirring solution of TPA-Ti2 (0.345 g, 0.833 mmol) in anhydrous tetrahydrofuran (10 mL) at −78 °C under nitrogen, was added n-butyl lithium (1.6 M in hexane, 0.573 ml, 0.917 mmol). The solution was allowed to warmed to room temperature in 3 h and then cooled again down to −78 °C. Tetracyanoethane (0.160 g, 1.30 mmol) in anhydrous tetrahydrofuran (5 mL) was added dropwise to the solution. The reaction solution was stirred for another 5 min in cold bath, warmed gradually up to room temperature, and then poured into distilled water (60.0 mL). The mixture was extracted with ethyl acetate (3 × 30 mL). The organic extracts were combined, washed with distilled water, dried over anhydrous magnesium sulfate, and rotary-evaporated to remove the solvent. The solid residue was purified by a silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/1, v/v, Rf = 0.6) to give green crystalline products (0.368 g, 86% yield): mp. 235–237 °C; 1H NMR (400 MHz, CDCl3): δ 8.56 (1H, s), 7.76 (1H, d, J = 15.6 Hz), 7.41 (2H, d, J = 8.7 Hz), 7.11–7.14 (5H, m), 6.86–6.92 (6H, m), 3.83 (6H, s); 13C NMR (75 MHz, CDCl3): δ 178.3, 157.2, 157.0, 152.0, 143.0, 138.8, 130.3, 128.1, 127.8, 125.8, 118.0, 115.3, 115.0, 112.2, 112.1, 111.8, 55.5; FT-IR (KBr, cm−1): 3038, 2833, 2219, 1581, 15022, 1435, 1391, 1327, 1285, 1240, 1161, 1032, 968, 824; HRMS for C30H21N5O2S (m/z): 515.1416 (calcd), 515.1416 (found).(E)-4-methoxy-N-(4-methoxyphenyl)-N-{4-[2-(thiazol-5-yl)vinyl]phenyl}benzenamine (TPA-Ti5)
To a solution of 4-[bis(4-methoxyphenyl)amino]benzaldehyde (0.333 g, 1.00 mmol) and diethyl thiazol-5-ylmethyl phosphonate (0.523 g, 1.20 mmol) in anhydrous tetrahydrofuran (20 mL) at −78 °C under nitrogen, was added dropwise potassium t-butyloxide (1.34 g, 1.20 mmol) in anhydrous tetrahydrofuran (20 mL). After the addition, the solution was warmed gradually up to room temperature and then poured into distilled water (100 mL). The mixture was extracted with ethyl acetate (3 × 50 mL). The organic extracts were combined, washed with distilled water (2 × 30 mL), dried over anhydrous magnesium sulfate, and rotary-evaporated to remove the solvent. The solid residue was purified by a silica-gel column chromatography (eluent: ethyl acetate/ligroin = 1/4, v/v, Rf = 0.5) to give a yellow solid product (0.362 g, 88% yield): 1H NMR (300 MHz, CDCl3): δ 8.61 (1H, s), 7.79 (1H, s), 7.26 (2H, d, J = 8.7Hz), 7.07–7.10 (5H, m), 6.84–6.90 (7H, m), 3.80 (6H, s); 13C NMR (75 MHz, CDCl3): δ 156.1, 150.5, 148.8, 140.8, 140.4, 131.9, 128.2, 127.2, 126.8, 119.9, 1115.8, 115.0, 114.7, 55.4.(E)-2-(5-{4-[bis(4-methoxyphenyl)amino]styryl}thiazol-2-yl)ethene-1,1,2-tricarbonitrile (TPA-Ti5-TCV)
To a nitrogen-purged, stirring solution of TPA-Ti5 (0.200 g, 0.480 mmol) in anhydrous tetrahydrofuran (10.0 mL) at −78 °C, was added n-butyl lithium (1.6 M in hexane, 0.350 mL, 0.560 mmol) through a syringe. The solution was stirred at −78 °C for 2 h before tetracyanoethene (0.094 g, 0.730 mmol) in anhydrous tetrahydrofuran (5 mL) was added dropwise. The solution was stirred at −78 °C for another 5 min and then poured into distilled water (100 mL). The mixture was extracted with ethyl acetate (50 mL) three times. The organic extracts were combined, washed with distilled water (2 × 20 mL), dried over anhydrous magnesium sulfate, and rotary-evaporated to concentrate the solution to about 20 mL. The resulting golden crystals were collected by filtration and washed with small amount of ethyl acetate (0.05 g, 20% yield): mp. 228–239 °C; 1H NMR (400 MHz, CDCl3): δ 8.13 (1H, s), 7.33 (2H, d, J = 8.8 Hz), 7.04–7.23 (6H, m), 6.84–6.89 (6H, m), 3.82 (6H, s); 13C NMR (100 MHz, CDCl3): 157.0, 152.1, 151.1, 145.6, 145.4, 139.7, 139.1, 130.8, 129.1, 127.6, 120.1, 118.4, 114.9, 112.4, 112.3, 111.9, 111.7, 55.4; FT-IR (KBr, cm−1): 3069, 2834, 2217, 1583, 1502, 1458, 1384, 1285, 1242, 1152, 1034, 958; HRMS-ESI for C30H21N5O2S (m/z): 515.1416 (Calcd.), 515.1420 (Found).Results and discussion
Synthesis
The heterocycle-based gradient NLO chromophores were synthesized according to Scheme 1. All the chromophores have the same tricyanovinyl electron acceptor and the same length of π-conjugation, but contain different aromatic rings (i.e., pyrrole, thiophene and benzene) as the auxiliary electron donor and 1,3-heteroaromatic thiazole with different regiochemistries as the auxiliary electron acceptor.10,11 Different amino donors (i.e., dialkyl, aryl alkyl, and diaryl substituted amines) were also employed in the study to tune the donor strength. Although the diethyl analogues of B-Ti2-TCV and B-T-TCV are known in the literature,12 these chromophores were prepared and characterized for a systematic comparison between the structures and optical nonlinearities. | ||
| Scheme 1 Synthesis of NLO chromophores with different gradient electronic structures. | ||
The syntheses of key pyrrole intermediates, e.g., 1-methyl-5-(piperidin-1-yl)-1H-pyrrole-2-carbaldehyde and 5-(N-4-methoxyphenyl N-methyl amino)-1-methyl-1H-pyrrole-2-carbaldehyde, were previously reported.14 Other intermediates, i.e., the dialkylamino- and bis(4-methoxyphenyl)amino-substituted aryl aldehydes and the thiazole-based Horner–Wadsworth–Emmons (HWE) reagents, were prepared referring to the literature methods.17–24 The coupling reactions between the aldehydes and HWE reagents (Scheme 1) were carried out at −78 °C using potassium t-butyloxide as a base to afford the chromophore precursors with exclusively the (E)-alkene configuration at high yields (e.g., 75–96%). Lithiation of the chromophore precursors at the 2- or 5-position of thiazole with n- or t-butyl lithium, followed by a nucleophilic substitution reaction with tetracyanoethene, yielded the desired NLO chromophores. The purity and the chemical structures of the precursors and the chromophores were demonstrated by NMR spectroscopy, elemental analysis and high-resolution mass spectroscopy.
Electronic structure
The different electronic structures of the chromophores were first analyzed by 1H NMR spectroscopy and cyclic voltammetry. As shown in Table 1, the protons (i.e., H-1 and H-2, Scheme 1) on the auxiliary donors appear at the highest fields for pyrrole (i.e., P-Ti2-TCV), followed by thiophene (i.e., T-Ti2-TCV) and benzene (i.e., B-Ti2-TCV) in sequence, indicating the varying electron density in the order of pyrrole > thiophene > benzene, which agrees with the calculated results.8,10 Substitution of the alicyclic donor (e.g., piperidine for P-Ti2-TCV) with a semi-aromatic amine (e.g., methyl 4-methoxyphenyl amine) leads to a decreased electron density of the pyrrole in MP-Ti2-TCV, as evidenced by the pronouced shifts of H-1 and H-2 to lower fields. Similarly, the use of diarylamine as the donor (e.g., TPA-Ti2-TCV) results in a decrease in electron density on the auxiliary-donor benzene in relative to that of B-Ti2-TCV. The regiochemistry of thiazole, i.e., either C2 (“matched” dipole vectors between thiazole and the chromophore) or C5 (“un-matched”) of thiazole is connected to the acceptor, affects only slightly the electron density of the auxiliary donors (e.g., T-Ti2-TCVvs.T-Ti5-TCV, and TPA-Ti2-TCVvs.TPA-Ti5-TCV).| NLO chromophores | δ/ppm a | E ox /Vb | E red/Vc | ΔE(CV)/eVd | |
|---|---|---|---|---|---|
| H-1 | H-2 | ||||
| a Chemical shifts of protons (H1 and H2, Scheme 1) on auxiliary donor. b Anodic peak potential vs.Ag/Ag+ reference electrode for irreversible oxidation; scan rate 0.2 V s−1. c Half potential for reversible redox processes. d ΔE = |EHOMO − ELUMO| = e(Eox, onset − Ered, onset). | |||||
| P-Ti2-TCV | 5.94 | 6.97 | 0.42 | −0.42 | 0.64 |
| MP-Ti2-TCV | 6.16 | 7.08 | 0.49 | −0.36 | 0.64 |
| T-Ti2-TCV | 6.15 | 7.27 | 0.59 | −0.43 | 0.85 |
| T-Ti5-TCV | 6.09 | 7.13 | 0.51 | −0.27 | 0.55 |
| B-Ti2-TCV | 6.74 | 7.52 | 0.83 | −0.37 | 0.98 |
| B-T-TCV | 6.85 | 7.50 | 0.91 | −0.33 | 0.88 |
| TPA-Ti2-TCV | 6.87 | 7.42 | 0.73c | −0.36 | 0.93 |
| TPA-Ti5-TCV | 6.86 | 7.27 | 0.70c | −0.23 | 0.76 |
Table 1 displays the redox potentials of the NLO chromophores measured in Bu4NClO4/MeCN (0.1 M) by cyclic voltammetry. As expected, the increase of electron density of auxiliary donor from benzene to thiophene and to pyrrole led to a decrease in oxidation potential (Eox) in the same order, e.g. from 0.83 V for B-Ti2-TCV to 0.59 V for T-Ti2-TCV and to 0.42 V for P-Ti2-TCV. Since oxidation potential is directly related to the ionization potential or the tendency of losing electron, it is expected that chromophores with electron-rich auxiliary donors would have an efficient intramolecular charge tranfer (CT) from the donor to the acceptor upon excitation. It is worth noting that, of all the chromophores, only triarylamine (TPA)-based ones (i.e., TPA-Ti2-TCV and TPA-Ti5-TCV), exhibited a fully reversible oxidation process, as shown in Fig. 1. Such a good reversibility in oxidation may contribute to a good thermal and photochemical stability. Recently, thermally stable TPA-based NLO chromophores have drawn some attention.15,16 In contrast to the oxidation, all the chromophores exhibited a reversible reduction process at low potentials of −0.23–−0.43 V, indicating a high electron affinity of the chromophores. The regiochemistry of thiazole affect greatly on Ered,1/2. Chromophores with “matched” thiazole (i.e., T-Ti5-TCV and TPA-Ti5-TCV) showed much lower Ered,1/2 than their “un-matched” isomers (i.e., T-Ti2-TCV and TPA-Ti2-TCV, respectively), suggesting a stronger electron-accepting ability of the former.
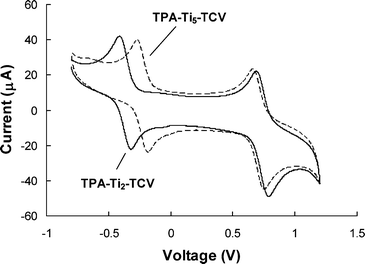 | ||
| Fig. 1 Cyclic voltammetry of TPA-Ti2-TCV and TPA-Ti5-TCV in acetonitrile containing tetrabutylammonium perchlorate (0.1 M) as the supporting electrolyte. | ||
Optical properties
The resulting NLO chromophores showed moderate to good solubility in common organic solvents such as choroform, tetrahydrofuran and DMF. Good solubility in non-polar solvents such as dioxane and toluene was also observed for pyrrole- and TPA-based chromophores. The UV-vis absorption maxima (λmax) and the extinction coefficients (ε) of the NLO chromophores in chloroform are summarized Table 2. All the chromophores exhibited broad and intense CT absorptions in the red spectral region (λmax = 668–840 nm) with onset absorptions in the range of 820–1030 nm (Fig. 2). A strong sensitivity of the linear spectra to both the auxiliary donor and the auxiliary acceptor was revealed. The significant bathochromic shift of λmax to lower energy observed from B-Ti2-TCV to TPA-Ti2-TCV to P-Ti2-TCV and to T-Ti2-TCV suggests an increasing ground-state polarization for these chromophores and a donor strength sequence in the order of Me2NPh < (MeOPh)2NPh < 2-piperidin-1-ylpyrrole < piperidin-1-ylthiophene. On the other hand, the introduction of “matched” thiazole as the auxiliary acceptor red-shifted significantly the λmax (e.g., 66 nm for TPA-Ti5-TCV and 90 nm for T-Ti5-TCV) in relative to the “un-matched” isomers, suggesting a further shifted ground-state toward charge separation. The observed low optical bandgaps of “matched” chromophores agrees with the electrochemical measurement results (Table 1).| NLO chromophores | λ a/nm | logε b | Δλdioxane-CHCl3c/nm | β1000d/10−30 esu | β0, HRSe/10−30 esu | β0, Calcdf/10−30 esu | Tdg/°C |
|---|---|---|---|---|---|---|---|
| a Maximal CT absorption wavelength measured in chloroform. b Logarithm of extinction coefficient in chloroform. c Differential CT wavelength between dioxane and chloroform. d Molecular hyperpolarizability (β) measured by Hyper-Rayleigh scattering experiment at 1000 nm with p-nitroaniline as an external standard. Intensity of second harmonic light was calibrated with the Beer–Lambert law. e Static β values calculated within the two-level model by using the formula β = β0 × (1/λmax)4/{[(1/λmax2) − (4/λ2)] × [(1/λmax2) − (1/λ2)]}. f Static β values calculated with the MOPAC program in AM1 method. g On-set temperature for weight loss determined by TGA in nitrogen at a heating rate of 20 °C/min. | |||||||
| P-Ti2-TCV | 731 | 4.73 | 71 | 976 | 516 | 189.5 | 218 |
| MP-Ti2-TCV | 752 | 4.68 | 74 | 923 | 486 | 129.1 | 182 |
| T-Ti2-TCV | 750 | 4.85 | 70 | 888 | 485 | 182.2 | 272 |
| T-Ti5-TCV | 840 | 4.90 | 117 | 1073 | 576 | 202.7 | 224 |
| B-Ti2-TCV | 668 | 4.95 | 57 | 882 | 383 | 176.7 | 263 |
| B-T-TCV | 678 | 4.71 | 54 | 704 | 318 | 144.6 | 288 |
| TPA-Ti2-TCV | 684 | 4.75 | 55 | 1501 | 698 | 189.9 | 257 |
| TPA-Ti5-TCV | 750 | 5.02 | 79 | 1406 | 768 | 222.3 | 274 |
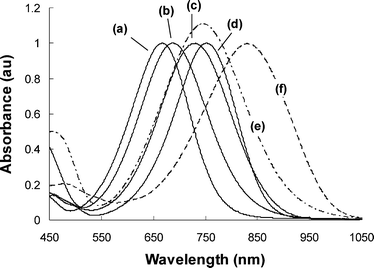 | ||
| Fig. 2 UV-vis absorption spectra of (a) B-Ti2-TCV, (b) TPA-Ti2-TCV, (c) P-Ti2-TCV, (d) T-Ti2-TCV, (e) TPA-Ti5-TCV, and (f) T-Ti5-TCV in chloroform. | ||
All the NLO chromophores exhibited large positive solvatochromism (Δλ = 54–117 nm) from dioxane to chloroform, suggesting the large optical nonlinearities of these chromophores (Table 1).26 In comparison, the benzene-based chromophores have a smaller Δλ (54–79 nm) than their pyrrole and thiophene analogues (Δλ = 70–117 nm). Meanwhile, the “matched” chromophores (i.e., T-Ti5-TCV and TPA-Ti5-TCV) showed significantly larger Δλ than their “un-matched” isomers (i.e., T-Ti2-TCV and TPA-Ti2-TCV), suggesting larger β values of the “matched” chromophores.
The molecular first hyperpolarizabilities (β) of the NLO chromophores were measured by Hyper-Rayleigh scattering (HRS) experiments at a fundamental wavelength of 1000 nm (pulse width, 7 ns), using p-nitroaniline (p-NA, β1064 nm = 28.8 × 10−30 esu24) as an external standard.27 Since all the NLO chromophores showed very weak absorption at the second harmonic frequency (i.e. 500 nm), as shown in Fig. 2, low two-photon resonance enhancement in β can be expected. Fig. 3 shows the typical HRS spectra of P-Ti2-TCV in chloroform. Strong two-photon fluorescence was observed for all the NLO chromophores. In the study, the second harmonic signals at 500 nm were extracted from the background fluorescence by spectral deconvolution. The signal intensities were calibrated with Beer–Lambert law to correct the absorptional effect. A linear plot of the signal intensities was drawn against the concentrations of the chromophore solutions (Fig. 4). By comparing between the slopes of the chromophores and the p-NA reference and using the equation of β = (kchromophore/kp-NA)1/2βp-NA,27 β values of the NLO chromophores at 1000 nm were obtained. Table 2 summarizes the β values of the NLO chromophores at 1000 nm.
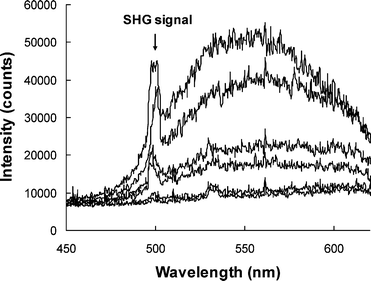 | ||
| Fig. 3 Hyper-Rayleigh scattering spectra of P-Ti2-TCV in chloroform. From bottom to top, the concentrations are 0.1 × 10−4 M, 0.2 × 10−4 M, 0.8 × 10−4 M, 1 × 10−4 M, 2 × 10−4 M, and 3 × 10−4 M. | ||
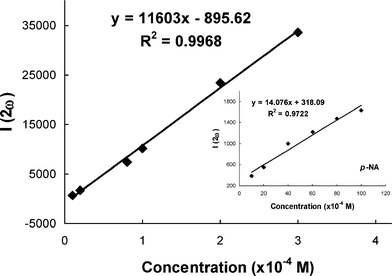 | ||
| Fig. 4 The typical linear dependence of HRS signal intensity (corrected for absorption) of P-Ti2-TCV on solution concentration. The inset shows the plot of HRS signal intensity of p-NAversus the solution concentration. | ||
For direct comparison between the optical nonlinearities of the chromophores, static β (β0) values were calculated from the β values at 1000 nm within the two-level model (Table 2).29 Correlation study of the structures and the β0 values of the NLO chromophores allows us to systematically examine the importance of the gradient electronic structures. Comparison between B-Ti2-TCV, T-Ti2-TCV and P-Ti2-TCV shows a sensitivity of the β0 value to the electron density of the auxiliary donors. P-Ti2-TCV that has pyrrole, the most electron-rich five-membered aromatic heterocycle, as the auxiliary donor exhibited a much larger β0 of 516 × 10−30 esu than its thiophene (i.e., T-Ti2-TCV, β0 = 485 × 10−30 esu) and benzene (i.e., B-Ti2-TCV, β0 = 383 × 10−30 esu) analogues. This agrees with the theoretical expectations by Albert et al.8 and Rao et al.9 In their studies, the introduction of electron-rich pyrrole or thiophene units to the donor end of the π–bridge as the auxiliary donor led to an increased optical nonlinearity of the chromophores. The amino donor also plays a role in the optical nonlinearity. Replacement of piperidin-1-yl donor with methyl 4-methoxylphenyl amine led to a drop in β value (i.e., MP-Ti2-TCVvs.P-Ti2-TCV), due probably to the decrease of donor strength. However, it is interesting to note that TPA-Ti2-TCV that have diarylamine as the donor and electron-inefficient benzene as the auxiliary donor exhibited a very large β0 value of 698 × 10−30 esu, which is even larger than the pyrrole-based P-Ti2-TCV. Marder and co-workers30 reported that bis(4-methoxyphenyl)aminophenyl group had a high donor strength comparable to that of dimethylaminophenyl group. This can be confirmed in our study by comparing the 1H NMR, CV and UV-vis spectra between B-Ti2-TCV and TPA-Ti2-TCV. However, this does not justify the observed much larger optical nonlinearity of TPA-Ti2-TCV than B-Ti2-TCV and P-Ti2-TCV. Recently, Davies et al.15 reported TPA-based NLO chromophores exhibiting very large β values comparable to those of diarylaminothiophene- and diarylaminopyrrole-based chromophores, although the latter were expected to have a stronger π-donor strength.30 In comparison of NLO chromophores with “matched” (i.e., Ti-Ti5-TCV and TPA-Ti5-TCV) and “un-matched” thiazole (i.e., Ti-Ti2-TCV and TPA-Ti2-TCV), the former exhibited much larger β0 values than the latter, demonstrating the theoretical predictions by Breitung et al.10 and by others.11
Static β0 values of the NLO chromphores were also computed with MOPAC program on geometric structures optimized by AM1 method (Table 2). Although smaller in magnitude, the calculated β0 values show essentially the same trends with the experimental results in terms of the effects of the electron density of the auxiliary donor and the regiochemistry of thiazole. Similar to the HRS measurement results, a large calculated β0 value was found for TPA-Ti2-TCV. This indicates that the very large experimental β0 values of TPA-based chromophores are not due to the resonance enhancement. However, at this stage, a rational explanation for the uniquely large β0 values of TPA-based chromophores is yet to be come up with.
Thermal and photochemical stabilities
The on-set thermal decomposition temperatures (Td) of the NLO chromophores were recorded by TGA in nitrogen (Table 2). Most of them exhibited good thermal stability over 215 °C. In comparison, however, the pyrrole-based NLO chromophores (i.e., P-Ti2-TCV and MP-Ti2-TCV) exhibited poorer stability than the thiophene and benzene analogues; and “un-matched” NLO chromophores (i.e., Ti-Ti2-TCV and TPA-Ti2-TCV) showed higher thermal stability than their “matched” counterparts. The photochemical stability of the NLO chromophores was studied by monitoring the UV-vis spectral change of the chromophore solutions in chloroform at room temperature (∼25 °C) with and without the indoor light. It is assumed that any decompositon of the conjugated structure would lead to a decrease in absorbance at λmax and appearance of new absorption peak at high energy band. In the study, no substantial spectral change was observed when the sample solutions were stored in dark for one week. However, under indoor light, NLO chromophores based on electron-rich pyrrole and thiophene showed a decrease in absorbance after 24 h and significant bleaching after one week, suggesting a relatively poor photochemical stability. In comparison, all benzene-based NLO chromophores showed good photochemical stability under the indoor light.Conclusions
A new series of heterocycle-based NLO chromophores with systematically tuned gradient electronic structures were successfully prepared. In the series, the most electron-rich pyrrole was for the first time coupled with the electron-poor thiazole in NLO chromophores, which allows for a systematic variation of the auxiliary donor from benzene to thiophene and to pyrrole. NLO chromophores with “matched” and “un-matched” thiazole at the acceptor end of the π-bridge were also for the first time prepared and compared for their nonlinear optical properties. In the study, a great sensitivity of β0 values to the electron density of auxiliary donor and to the regiochemistry of thiazole as the auxiliary acceptor was demonstrated. With the increase of electron density of the auxiliary donor from benzene to thiophene and to pyrrole, a significant increase in β value was observed; NLO chromphores with “matched” thiazole as the auxiliary acceptor exhibited much larger β0 values than their “un-matched” isomers. However, a tradeoff between the large optical nonlinearity and high thermal stability was also revealed. TGA investigations showed that pyrrole-based and “matched” thiazole-based NLO chromophores tended to have a decreased thermal stability. However, exceptions were found for TPA-based NLO chromophores (e.g., TPA-Ti5-TCV) that exhibited both very large β0 values and good thermal stability.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos.: 200504003 and 20433010) and the SRF for ROCS, SEM. The authors are very grateful for the fruitful discussions with Dr M. Sun of the Institute of Physics, Chinese Academy of Sciences.References
- L. Dalton, A. Harper, A. Ren, F. Wang, G. Todorova, J. Chen, C. Zhang and M. Lee, Ind. Eng. Chem. Res., 1999, 38, 8 CrossRef CAS; L. Dalton, Adv. Polym. Sci., 2002, 158, 1 CAS; F. Kajzar, K.-S. Lee and A. K.-Y. Jen, Adv. Polym. Sci., 2003, 161, 1 CAS; D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1984, 94, 31.
- T. Kim, J. Kang, J. Luo, S. Jang, J. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. Herman and A. K.-Y. Jen, J. Am. Chem. Soc., 2007, 129, 488 CrossRef CAS; T.-D. Kim, J. Luo, Y.-J. Cheng, Z. Shi, S. Hau, S.-H. Jang, X.-H. Zhou, Y. Tian, B. Polishak, S. Huang, H. Ma, L. R. Dalton and A. K.-Y. Jen, J. Phys. Chem. C, 2008, 112, 8091 CrossRef CAS.
- Y. V. Pereverzev, K. N. Gunnerson, O. V. Prezhdo, P. A. Sullivan, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, A. K.-Y. Jen and L. R. Dalton, J. Phys. Chem. C, 2008, 112, 4355 CrossRef CAS.
- H. Kang, A. Facchetti, P. Zhu, H. Jiang, Y. Yang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Z. Liu, S. Ho and T. J. Marks, Angew. Chem., Int. Ed., 2005, 44, 7922 CrossRef CAS.
- S. R. Marder, J. W. Perry, G. Bourhill, C. B. Gorman, B. G. Tiemann and K. Mansour, Science, 1993, 261, 186 CrossRef CAS; S. R. Marder, C. B. Gorman, F. Meyers, J. W. Perry, G. Bourhill, J.-L. Brédas and B. M. Pierce, Science, 1994, 265, 632 CAS.
- C. Zhang, L. R. Dalton, M.-C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043 CrossRef CAS; B. H. Robinson, L. R. Dalton, A. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorov, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. K.-Y. Jen, Chem. Phys., 1999, 245, 35 CrossRef CAS; S. Liu, M. A. Haller, H. Ma, L. R. Dalton, S. H. Jang and A. K.-Y. Jen, Adv. Mater., 2003, 15, 603 CrossRef CAS; K. Schmidt, S. Barlow, A. Leclercq, E. Zojer, S.-H. Jang, S. R. Marder, A. K.-Y. Jen and J.-L. Brédas, J. Mater. Chem., 2007, 17, 2944 RSC; Y.-J. Cheng, J. Luo, S. Huang, X. Zhou, Z. Shi, T.-D. Kim, D. H. Bale, S. Takahashi, A. Yick, B. M. Polishak, S.-H. Jang, L. R. Dalton, P. J. Reid, W. H. Steier and A. K.-Y. Jen, Chem. Mater., 2008, 20, 5047 CrossRef CAS.
- D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1998, 120, 11174 CrossRef CAS; H. Kang, A. Facchetti, C. L. Stern, A. L. Rheingold, W. S. Kassel and T. J. Marks, Org. Lett., 2005, 7, 3721 CrossRef CAS; H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho, E. C. Brown, M. A. Ratner and Tobin J. Marks, J. Am. Chem. Soc., 2007, 129, 3267 CrossRef CAS.
- D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1997, 119, 6575 CrossRef CAS.
- V. P. Rao, A. K.-Y. Jen, J. Chandrasekhar, I. N. N. Namboothiri and A. Rathna, J. Am. Chem. Soc., 1996, 118, 12443 CrossRef.
- E. M. Breitung, C.-F. Shu and R. J. McMahon, J. Am. Chem. Soc., 2000, 122, 1154 CrossRef CAS.
- C. S. Ra, S. C. Kim and G. Park, J. Mol. Structure (Theochem), 2004, 677, 173 CrossRef CAS; P. K. Nandi, N. Panja and T. K. Ghanty, J. Phys. Chem. A, 2008, 112, 4844 CrossRef CAS; Z. Benková, I. Cernušák and P. Zahradník, Mol. Phys., 2006, 104, 2011 CrossRef CAS.
- Y.-K. Wang, C.-F. Shu, E. M. Breitung and R. J. McMahon, J. Mater. Chem., 1999, 9, 1449 RSC; Ching-Fong Shu and Yuh-Kai Wang, J. Mater. Chem., 1998, 8, 833 RSC.
- D. M. Casmier, P. A. Sullivan, O. Clot, K. A. Firestone, B. E. Eichinger, S. Lee, S. Heller, A. Brumbaugh, B. Millard and L. R. Dalton, Organic Photonic Materials and Devices VI, Proc. SPIE, 2004, 5351, 243 CAS.
- X. Ma, R. Liang, F. Yang, Z. Zhao, A. Zhang, N. Song, Q. Zhou and J. Zhang, J. Mater. Chem., 2008, 18, 1756 RSC.
- J. A. Davies, A. Elangovan, P. A. Sullivan, B. C. Olbricht, D. H. Bale, T. R. Ewy, C. M. Isborn, B. E. Eichinger, B. H. Robinson, P. J. Reid, X. Li and L. R. Dalton, J. Am. Chem. Soc., 2008, 130, 10565 CrossRef CAS.
- Y.-J. Cheng, J. Luo, S. Hau, D. H. Bale, T.-D. Kim, Z. Shi, D. B. Lao, N. M. Tucker, Y. Tian, L. R. Dalton, P. J. Reid and A. K-Y. Jen, Chem. Mater., 2007, 19, 1154 CrossRef CAS; L. R. Dalton, Optical Materials in Defence Systems Technology V, Proc. SPIE, 2008, 7118, 711803-1; S. M. Budy, S. Suresh, B. K. Spraul and D. W. Smith, Jr., J. Phys. Chem. C, 2008, 112, 8099 CrossRef CAS.
- J. Hou, Z. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911 CrossRef CAS.
- Dondoni, G. Fantin, M. Fogagnolo, A. Medici and P. Pedrini, Tetrahedron, 1988, 44, 2021 CrossRef CAS; A. Dondoni and D. Perrone, J. Org. Chem., 1995, 60, 4749 CrossRef CAS.
- CN pat., 1483320, 2004.
- D. Alessandro, F. Giancarlo, F. Marco, M. Alessandro and P. Paola, Synthesis, 1987, 11, 998.
- J.-H. Lin, A. Elangovan and T.-I. Ho, J. Org. Chem., 2005, 70, 7397 CrossRef CAS.
- S. Suresh, H. Zengin, B. K. Spraul, T. Sassa, T. Wada and D. W. Smith Jr., Tetrahedron Lett., 2005, 46, 3913 CrossRef CAS.
- T. L. Cairns, R. A. Carboni, D. D. Coffman, V. A. Engelhardt, R. E. Heckert, E. L. Little, E. G. McGeer, B. C. McKusick, W. J. Middleton, R. M. Scribner, C. W. Theobald and H. E. Winberg, J. Am. Chem. Soc., 1958, 80, 2775 CrossRef CAS.
- J. N. Woodford, M. A. Pauley and C. H. Wang, J. Phys. Chem. A., 1997, 101, 1989 CrossRef CAS.
- K.-Y. Jen, V. P. Rao, K. Y. Wong and K. J. Drost, J. Chem. Soc., Chem. Commun., 1993, 90 RSC; V. P. Rao, A. K. -Y. Jen, K. Y. Wong and K. J. Drost, Tetrahedron Lett., 1993, 34, 1747 CrossRef CAS.
- Bosshard, G. Knöpfle, P. Prêtre and P. Günter, J. Appl. Phys., 1992, 71, 1594 CrossRef CAS.
- M. A. Pauley, H.-W. Guan, C. H. Wang and A. K.-Y. Jen, J. Chem. Phys., 1996, 104, 7821 CrossRef CAS; M. A. Pauley, C. H. Wang and A. K.-Y. Jen, J. Chem. Phys., 1995, 102, 6400 CrossRef CAS.
- K. Song, J. N. Woodford and C. H. Wang, J. Phys. Chem. A, 1997, 101, 3222 CrossRef CAS.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS.
- Kwon, S. Barlow, S. A. Odom, L. Beverina, N. J. Thompson, E. Zojer, J.-L. Brédas and S. R. Marder, J. Phys. Chem. A, 2005, 109, 9346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, solubility of the NLO chromophores, UV-vis absorption of the NLO chromophores in different solvents, UV-vis absorption spectra of the NLO chromophores and UV-vis absorption spectra of chromophore solutions. See DOI: 10.1039/b817789a |
| This journal is © The Royal Society of Chemistry 2009 |