Speciation, formation, stability and analytical challenges of human arsenic metabolites
Lucy
Yehiayan
a,
Mahesh
Pattabiraman
a,
Konstantinos
Kavallieratos
a,
Xiaotang
Wang
a,
Lawrence H.
Boise
c and
Yong
Cai
*ab
aDepartment of Chemistry & Biochemistry, Florida International University, 11200 SW 8th St, Miami, Florida 33199, USA. E-mail: cai@fiu.edu; Fax: +1 305-348-3772; Tel: +1 305-348-6210
bSoutheast Environmental Research Center, Florida International University, Miami, Florida 33199, USA
cDepartment of Microbiology and Immunology and The Sylvester Comprehensive Cancer Center, University of Miami, Miami, Florida 33136, USA
First published on 21st July 2009
Abstract
Human arsenic metabolism produces a number of species with varying toxicities; the presence of some has been identified while the existence of others has been postulated through indirect evidence. Speciation methods for the analysis of arsenite (AsIII), monomethylarsonous acid (MMAIII), dimethylarsinous acid (DMAIII), arsenate (AsV), monomethylarsonic acid (MMAV), dimethylarsinic acid (DMAV), arsino-glutathione (As(GS)3), monomethylarsino-glutathione (MMA(GS)2) and dimethylarsino-glutathione (DMA(GS)) were developed in this study through the use of cation exchange and reverse phase chromatography in a complementary manner. Electrospray ionization mass spectrometry (ESI-MS) was used for molecular identification of the arsenicals while inductively coupled plasma mass spectrometry (ICP-MS) was employed for quantitation purposes. Validation of the developed methods against each other for the quantitation of trivalent and pentavalent arsenicals was performed. The effect of reduced glutathione (GSH) concentration on the formation of arsenic-glutathione (As-GSH) complexes was studied. In the presence of glutathione, the occurrence of chromatographic artifacts on the cation exchange column was observed. The stability of trivalent arsenicals and As-GSH complexes was studied at various pH conditions. The results shed light on the importance of sample preparation, storage and proper choice of analytical column for the accurate identification of the As species. Reinvestigation of some of the previously reported As speciation studies of glutathione-rich biological samples needs to be performed for the verification of occurrence of As-GSH complexes and DMAIII.
Introduction
Arsenic (As) is a global environmental contaminant with potent health effects. It is a well-established human carcinogen.1–3 Ingestion of As-contaminated water in the West Bengal area and Bangladesh has led to serious arsenicosis in humans. Diseases like hypertension, keratosis, skin pigmentation, diabetes, cardiovascular disorders, and cancer of the bladder, lung and skin have been reported after As exposure.2–5 Paradoxically, As derivatives have been used as therapeutic agents since the 1900's for the treatment of syphilis, psoriasis and rheumatosis.6 The first As drug (Trisenox) in the US for the treatment of acute promyelocytic leukemia, was approved by the US FDA in 2000 and NIH in 2001.7 Currently arsenic trioxide (ATO) is being used for the treatment of various cancers including but not limited to ovarian and colon cancer, Hodgkin's lymphoma, myeloid leukemia and multiple myeloma.8,6,9,10 Patents have been applied for new As derivatives that are believed to be more potent on cancer cells and less toxic on normal cells.11,12 As uptake often occurs through inhalation and/or ingestion. Following its uptake, As is absorbed through the gastrointestinal tract and lungs, is distributed in the organs, tissues and blood stream, and then metabolized and excreted through urine, keratin, and feces.13,14 Arsenic metabolism in humans involves the reduction of arsenate followed by methylation in the presence of s-adenosyl methionine, cytochrome-19 and reducing agents. Thiol-containing compounds such as glutathione, glutaredoxin and thioredoxin have been proposed as the reducing agents.15,16 Arsenic-thiol affinity has been proposed to be the cause for As accumulation, toxicity17–20 and ironically, effectiveness in cell detoxification and cancer treatment.6,21,22 In addition to its cell regulatory functions (reduction of hydroperoxides and detoxification of electrophiles),23 glutathione, present in large concentrations in cells (0.1–10 mM),23,24 has been proposed to be the reducing agent involved in the As metabolism of humans by two separately proposed mechanisms.25,26 The As-containing species produced through these mechanisms include: i) the inorganic arsenicals; arsenite (AsIII) and arsenate (AsV), ii) the methylated arsenicals; monomethylarsonous acid (MMAIII), monomethylarsonic acid (MMAV), dimethylarsinous acid (DMAIII) and dimethylarsinic acid (DMAV)25,26 and iii) glutathione-bound arsenicals; arsino-glutathione (As(GS)3), monomethylarsino-glutathione (MMA(GS)2) and dimethylarsino-glutathione (DMA(GS)).26 The distributions of these metabolites in biological matrices depend on factors such as availability of reduced glutathione (GSH) and/or other thiol-containing compounds,25 the presence of As-methylating enzymes,1,27,28 the matrix pH29,30 and the species hydrophobicity and penetrability.18 Different As species have different toxicities. AsIII is more toxic than AsV and methylated trivalent arsenicals are more toxic than their pentavalent counterparts. MMAIII and DMAIII are genotoxic.31 The reported order of As toxicity in Chang human hepatocytes is MMAIII > DMAIII = AsIII > AsV > DMAV and MMAV.32,33 Recent studies have also demonstrated that MMA(GS)2 and DMA(GS) are toxic to cells through their dissociation into MMAIII and DMAIII in the absence of GSH.12,34,35 In addition to the common As metabolites (AsIII, AsV, MMAV and DMAV), the presence of MMAIII and DMAIII in urine14,36–39 and cultured human hepatocellular carcinoma cells have been declared.39 Reports of the presence of As(GS)3, MMA(GS)2 in rat bile and urine also exist,40,41 and indirect evidence has been provided for the presence of As(GS)3 in human blood cells and plasma.9 These studies have investigated on either the occurrence of trivalent and pentavalent arsenicals or As-GSH complexes. No comprehensive study for the occurrence of all As species (trivalent, pentavalent and GSH complexed) in biological samples, specifically GSH-rich matrices has been reported. Therefore, a thorough understanding of As speciation as a function of GSH is warranted since As species exist in equilibrium with their corresponding GSH complexes.26Since As toxicity and bioavailability is species dependant, the development of efficient and selective speciation technique is crucial for the correct identification and quantification of all As metabolites in biological and clinical matrices. Due to the instability of As-GSH complexes and trivalent methylated arsenicals, very few speciation methods have been reported for the identification of AsIII, AsV, MMAIII, MMAV, DMAIII, DMAV, As(GS)3, MMA(GS)2 and DMA(GS).36,38,42–44 The reported methods have used ion-exchange or ion-pairing chromatography for the separation of trivalent and pentavalent arsenicals and reverse phase chromatography for the separation of As-GSH complexes. The detection methods employed with these separation techniques were inductively coupled mass spectrometry (ICP-MS), atomic fluorescence spectroscopy (AFS) or electrospray ionization mass spectrometry (ESI-MS). The limitations of the reported methods for the speciation of trivalent and pentavalent arsenicals include poor resolution of chromatographic peaks,36,44 employment of MMAIII synthetic procedures that lead to the production of thiolated MMAIII in addition to the target product MMAIII,44 and the need for the use of two analytical columns.38 These methods did not test for elution of As-GSH complexes with other As species in the presence of GSH-rich matrices. Also, in these methods, identification of As species is based solely on retention times. Since As can exist in many forms, the use of a molecule-specific detector is indispensable for correct species identification. With respect to separation and identification of As-GSH complexes, a method has been reported based on reverse phase chromatography with the use of a C18 column.42,43 Unfortunately, this method is incapable of separating each trivalent As species from its corresponding pentavalent and GSH complexed form. Moreover, the use of a C18 column, results in coelution of AsIII, AsV and MMAIII with MMAV even though separation of DMAV, DMAIII and the complexes could be achieved (data not shown). Ionization of inorganic arsenicals at electrospray conditions is very inefficient compared to the organic arsenicals. In addition, trivalent organic arsenicals in general are easily oxidized in the electrospray stage. Therefore, identification of these species through ESI-MS is not possible with this method. Other limitations of the method used for the separation of As-GSH complexes include the use of high percentage organic mobile phase (up to 40% acetonitrile) and addition of oxygen into the carrier gas for reducing the effect of organic solvent. Since organic solvents are not ICP-MS friendly, a decrease in sensitivity is typically observed due to interferences from formation of polyatomic adducts and continuous instrumental maintenance of cones, lenses and vacuum gauge is required. Random decomposition of the As-GSH complexes into species of unknown identity has also been reported with the use of this chromatographic method.42
To date, no single chromatographic technique can separate all previously-reported As species probably because of the diverse characteristics among different As species. The pKa values for AsIII, AsV, MMAV and DMAV are known, while those for MMAIII, DMAIII, As(GS)3, MMA(GS)2 and DMA(GS) are not. The pKa values of GSH are 2.1 and 3.5 for the carboxylic groups and 9.6 for the thiol group.42 Since As bonding to GSH is through sulfur, the possibility of existence of these complexes in neutral and anionic forms at pH values of 2 or higher theoretically enables the use of ion-exchange chromatography for the separation of As-GSH complexes.
To accurately detect and quantify arsenicals in biological and clinical matrices, we have developed two speciation methods to be used in a complementary manner. A cation exchange chromatographic method is developed for the separation and identification of AsIII, AsV, MMAV and DMAV from MMAIII and DMAIII with ICP-MS and ESI-MS (for organic arsenicals only) as detectors. A reversed phase chromatographic method coupled with ICP-MS and ESI-MS is developed for the separation and identification of each trivalent As species from its corresponding pentavalent and GSH complexed form. Specifically, i) AsIII from AsV and As(GS)3, ii) MMAIII from MMAV and MMA(GS)2, and iii) DMAIII from DMAV and DMA(GS)). The reversed phase method can also be used to quantify glutathione in both its reduced and oxidized forms. Validation of the two methods for the quantitation of trivalent and pentavalent As species in the presence or absence of GSH was performed with the use of a Deming plot. The analytical figures of merit were determined. The effect of GSH concentration on the formation of As-GSH complexes was studied and the occurrence of analytical artifacts was observed during speciation of As species with cation exchange chromatography. To further postulate the occurrence of As-GSH complexes in biological matrices and to identify the optimum pH conditions for stabilizing methylated trivalent arsenicals and As-GSH complexes in extraction media, the percent abundances of these species at different pH values were determined and the half-lives for As-GSH complexes were estimated.
Materials and methods
Reagents and standards
All reagents were of analytical grade. Deionized (DI) water (18 MΩ Barnstead Nanopure Diamond) was used throughout the experiments. Sodium metaarsenite, 98% (AsIII), sodium arsenate dibasic, 99% (AsV), cacodylic acid sodium salt, 98% (DMAV), L-glutathione reduced, 98–100% (GSH), phosphorous pentoxide (P2O5), sulfur dioxide lecture bottle, diethylether anhydrous and deuterated water (D2O) were purchased from Sigma-Aldrich, USA. Monosodium acid methane arsonate, 99.5% (MMAV) was purchased from Chem Service, USA. Oxidized glutathione (GSSG) was purchased from MP Biomedicals, USA. Hydrochloric acid (HCl), trace metal grade, potassium iodide (KI), acetonitrile, methanol and optima LC/MS-grade water were purchased from Fisher, USA. Calcium chloride anhydrous (CaCl2) and formic acid, 99% were purchased from Acros Organics, USA. Phosphate buffer saline (PBS) (pH = 7.4), was prepared by dissolving 4.00 g of sodium chloride (NaCl) with 1.36 g of sodium phosphate, dibasic (Na2HPO4·7H2O), 0.10 g of potassium chloride (KCl) and 0.12 g of potassium dihydrogen phosphate (KH2PO4) (Fisher, USA) in 500 mL of DI water.45 For the study of pH effect on As species stability and half-life, pH adjustments were made with either ammonium hydroxide (NH4OH) or nitric acid (HNO3), trace metal grade both purchased from Fisher, USA.Instrumentation
A Hamilton PRPX-200 cation exchange column (250 × 4.1 mm in dimension and 10 micron particle size) was employed for the separation of trivalent and pentavalent arsenicals. The mobile phase flow rate was set at 1 mL min−1 and the sample injection volume was 50 µL.
A Waters Spherisorb C8 column (150 × 4.6 mm in dimension and 5 micron particle size) was employed for the separation of As-GSH complexes from each other and from their corresponding trivalent and pentavalent species. The mobile phase flow rate was set at 1 mL min−1 and the sample injection volume was 100 µL. Column effluent was split either into a ratio of 1:1 (one part going into ESI-MS and one part going to waste) or 1:2 (one part going into ICP-MS and two parts going to waste). For a constant flow split, the waste tubing was either controlled by the ICP-MS peristaltic pump set at a speed of 30 rpm or an IPC pump (ISMATEK, Switzerland) with a speed set-up of 3.5% of the flow. All HPLC connections between pump, column and detectors were made of inert PEEK material. Mobile phase compositions are listed in Table 1.
| HPLC columns | ||
| Cation exchange column | Hamilton PRPX-200 | |
| Isocratic | 0.05% Formic acid | |
| Separation time | 10 min. | |
| Reverse Phase Column | Waters Spherisorb C8 | |
| Gradient program | ||
| Time | 0.1% Formic acid (%) | Acetonitrile (%) |
| 0 min. | 95 | 5 |
| 10 min. | 85 | 15 |
| Equilibrate 5 min. | 95 | 5 |
| ICP-MS | ||
| Rf power (W) | 1350 | |
| Plasma gas flow rate (L/min.) | 15.00 | |
| Auxiliary gas flow rate (L/min.) | 1.20 | |
| Nebulizer gas flow rate (L/min.) | 0.96–1.04 | |
| Dwell time (ms) | 600 | |
| Lens voltage (V) | 8 | |
| Acquisition time (min.) | 15 | |
| Post column injection time (min.) | 13 | |
| Post column injection duration (sec.) | 25 | |
| ESI-MS | ||
| ISpray Voltage (kV) | 5 | |
| Sheath gas (arb) | 80 | |
| Auxiliary gas (arb) | 20 | |
| Capillary Voltage (V) | 46 | |
| Temperature (°C) | 350 | |
| Tube lens offset (V) | 55 | |
| Acquisition time (min.) | 15 | |
Procedures
Results and discussion
Synthesis of methylated trivalent arsenicals
To verify the success of the reduction process of DMAV and MMAV, 1H-NMR spectra of the starting material and the corresponding product were measured in degassed D2O. Upfield shifts were observed for the synthesized trivalent species compared to their pentavalent starting precursors. After reduction of MMAV, the chemical shift changed from 1.80 ppm to 1.37 ppm, while after reduction of DMAV, the chemical shift changed from 1.61 ppm to 1.35 ppm. These values are in good agreement with those reported in the literature and confirm the formation of the trivalent species.30,49 The purities obtained for the synthesized compounds were determined to be around 100% for MMAIII and 82% for DMAIII with 1H-NMR.Synthesis of arsenic-glutathione complexes
To verify the synthesis of arsenic-glutathione complexes, direct infusion on ESI-MS was performed for the starting material and the products obtained. Oxidation of trivalent methylated arsenicals in the electrospray was observed; a molecular ion peak at m/z = 140.97 was observed for MMAIII analysis, which is the same m/z as MMAV. Similarly, for DMAIII analysis, m/z = 138.98 was observed which is the same m/z as DMAV. To confirm the oxidation process of the trivalent organic arsenicals at the electrospray stage, chromatographic separation of MMAIII from MMAV and DMAIII from DMAV was performed. Two separate peaks on the chromatogram were observed for MMAIII and MMAV with same m/z = 140.97. Similar observation was obtained for DMAIII and DMAV. With respect to the complexes, each solution was composed of the molecular ion peak for the product formed (m/z = 993.99 for As(GS)3, 702.94 for MMA(GS)2 and 411.95 for DMA(GS)), GSH, GSSG and the starting As species. The main mass spectral peaks obtained for each solution are listed in Table 2.| Species | m/z | Molecular formula |
|---|---|---|
| a GS stands for the monodeprotonated form of glutathione. | ||
| As(GS)3 | 993.99 | [As(GS)3+H]+ |
| 687.03 | [As(OH)(GS)2–H2O+H]+ | |
| 497.80 | [As(GS)3+2H]2+ | |
| 613.21 | [GSSG+H]+ | |
| 307.44 | [GSSG+2H]2+ | |
| 308.7 | [GSH+H]+ | |
| MMA(GS)2 | 702.94 | [CH3(As)(GS)2+H]+ |
| 395.98 | [CH3(As)(OH)(GS)–H2O+H]+ | |
| 352.15 | [CH3(As)(GS)2+2H]2+ | |
| 613.21 | [GSSG+H]+ | |
| 307.44 | [GSSG+2H]2+ | |
| 308.7 | [GSH+H]+ | |
| 140.97 | [CH3(As)(OH)2(O)+H]+ | |
| DMA(GS) | 411.95 | [(CH3)2(As)(GS)+H]+ |
| 308.7 | [GSH+H]+ | |
| 613.21 | [GSSG+H]+ | |
| 307.44 | [GSSG+2H]2+ | |
| 138.96 | [(CH3)2(As)(OH)(O)+H]+ | |
| DMAV | 138.98 | [CH3)2(As)(OH)(O)+H]+ |
| 276.71 | [2((CH3)2(As)(OH)(O))+H]+ | |
| MMAV | 140.97 | [CH3(As)(OH)2(O)+H]+ |
| DMAIII | 139.00 | [(CH3)2(As)(OH)(O)+H]+ |
| 276.77 | [2((CH3)2(As)(OH)(O))+H]+ | |
| 298.77 | [2((CH3)2(As)(OH)(O))+Na]+ | |
| 436.62 | [3((CH3)2(As)(OH)(O))+Na]+ | |
| MMAIII | 140.97 | [CH3(As)(OH)2(O)+H]+ |
Speciation of trivalent and pentavalent arsenicals using cation exchange chromatography
The separation of AsIII, AsV, MMAV and DMAV from MMAIII and DMAIII was successfully achieved by using PRPX-200 column (Fig. 1). Formic acid was the mobile phase of choice because of its compatibility with ESI-MS and ICP-MS. Variations in concentration and ionic strength of formic acid were performed to obtain optimum separation of the As species at the shortest time interval. The change in formic acid concentration had a large effect on AsV retention time. With 0.1% formic acid, DMAV and AsV chromatographic peaks were not well resolved, while at 0.01%, the retention time of AsV was tripled. The separation of MMAIII and DMAIII was not possible at all tested conditions. The identification of MMAIII and DMAIII on this column was performed by ESI-MS as separate molecular ion peaks (data not shown). The optimum separation of AsIII, AsV, MMAV and DMAV from MMAIII and DMAIII was achieved with 0.05% formic acid (pH = 2.60) within 10 minutes.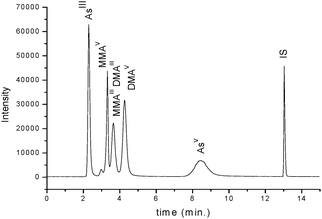 | ||
| Fig. 1 Separation chromatogram of AsIII, AsV, MMAV and DMAV from MMAIII and DMAIII on a Hamilton PRPX-200 column with 0.05% formic acid as mobile phase and ICP-MS as a detector. The injection volume was 50 µL of a mixture of 40 µg L−1 of each species. | ||
Tests were also performed to check the elution of As-GSH complexes with the cation exchange column. On the ICP-MS, for each As-GSH complex solution injected, only two peaks were observed corresponding to the trivalent and pentavalent counterparts of the complexes. To determine whether the complexes were coeluting with their corresponding trivalent or pentavalent counterparts, ESI-MS detection was performed. Molecular ion peaks for As(GS)3, MMA(GS)2 and DMA(GS) were not detectable, while a constant elution of GSH was observed, as demonstrated by the presence of a high baseline peak with a m/z = 308.7 (data not shown). Therefore, it is plausible that the complexes were retained by the column and slowly decomposed to free GSH.
Speciation of arsenic species and arsenic glutathione complexes using reverse phase chromatography
With the use of a C8 column, separation of As(GS)3, MMA(GS)2 and DMA(GS) was possible within 8 minutes (Fig. 2B and 2C). Although it was impossible to separate all trivalent and pentavalent arsenicals on this column, the separation of each individual trivalent arsenic species from its corresponding pentavalent and glutathione complexed form was plausible (Fig. 2A). Since As-GSH complexes exist in equilibrium with their corresponding pentavalent and trivalent species, this method could be used for toxicological and clinical trials of arsenic derivatives on cells after proper validation with cell matrices. The equilibrium between each trivalent, pentavalent and glutathione complexed form of As is highly dependant on the oxidation state of the starting material and GSH concentration. At low GSH concentrations, equilibrium exists between the pentavalent and the GSH-complexed forms. As concentration of GSH increases, the equilibrium is shifted towards coexistence of trivalent and GSH-complexed forms. Due to this matter, superimposition of the chromatograms of i) MMAV, MMAIII and MMA(GS)2 and ii) DMAV, DMAIII and DMA(GS) was performed to show the separation. | ||
| Fig. 2 Typical separation chromatograms of A) each trivalent As species from its corresponding pentavalent and GSH-complexed form obtained by HPLC-ICP-MS. B) GSH, GSSG and As-GSH complexes obtained by HPLC-ESI-MS. Single injection for a mixture of the standards was performed with SIM chromatograms superimposed on top of each other, and C) GSSG, As(GS)3, MMA(GS)2 and DMA(GS) obtained by HPLC-UV. All separations were obtained on a Spherisorb C8 column with formic acid and acetonitrile in gradient mode as mobile phase. The injection volume was 100 µL and the concentrations of the species correspond to 10–75 µg L−1 on ICP-MS, 0.25–0.50 mg L−1 on ESI-MS and UV detectors. The column temperature was maintained constant at 10 °C. | ||
Formic, trifluoroacetic or acetic acid are solvents commonly used for reverse phase chromatography. Since trifluoroacetic acid causes signal suppression in the electrospray and acetic acid has a strong pungent odor, formic acid was selected as the mobile phase. With the use of 0.1% formic acid (pH = 2.67), a long separation time window for the As-GSH complexes was observed and the DMA(GS) complex eluted as a very broad peak (data not shown). Organic modifiers were then added to formic acid to test their effect on the separation. Addition of 5–15% methanol caused decomposition of DMA(GS) on the column and the chromatographic separation did not reach the baseline (data not shown). Therefore, further optimization attempts with methanol were not performed. Acetonitrile was the second solvent of choice. Addition of low percentages of acetonitrile (1–5%) to formic acid, in both isocratic and gradient modes, promoted the fast elution of the As-GSH complexes (10–14 minutes). However, DMA(GS) peak broadening did not completely disappear. Optimum separation was achieved with the addition of acetonitrile as a gradient from 5–15%. Within 10 minutes, the complexes were separated and DMA(GS) eluted with the least broadening. The peak broadening of DMA(GS) could be attributed to its decomposition or interactions with the column during separation. Since increase in GSH concentration could reduce the decomposition of the complex, variations in GSH concentration in solution were performed. Addition of 5 mM GSH to the standard mixture of DMA(GS) did decrease the peak fronting, indicating an improvement in the stability of the DMA(GS) complex. The changes in acetonitrile percentage did not affect the retention times of AsIII, AsV, MMAIII, MMAV, DMAIII and DMAV. Although AsIII and MMAV coelute and MMAIII and DMAV coelute on the C8 column, the identification and quatitation of these species is still possible by ESI-MS. With this method the separation and quantitation of glutathione species in both their reduced and oxidized forms is also plausible with SIM mode (Fig. 2B).
Analytical figures of merit
The analytical performance using HPLC-ESI-MS and HPLC-ICP-MS was characterized by the linearity of the calibration curves, detection limits and repeatability. Five point-calibration plots for each As species were obtained in triplicate on both columns. The different As species concentrations were in the range 0.25–11.00 mg L−1 of elemental As on the ESI-MS and in the range between 2–200 µg L−1 of elemental As on the ICP-MS. The calibration curves were linear with correlation coefficients in the range 0.96–1.00 for all the As species. Detection limits were calculated as 3 times the standard deviation of seven replicate measurements of a standard solution. The concentrations used for detection limit calculations were 0.25 mg L−1 of elemental As for DMAV and MMAV and 0.60 mg L−1 of elemental As for the complexes on ESI-MS and 4 µg L−1 of elemental As on ICP-MS with the C8 column and 2 µg L−1 with the cation exchange column. Quantitation on ICP-MS was performed against an internal standard of AsV solution. Method repeatability was calculated as relative standard deviation of triplicate consecutive analysis of the lowest calibration point. Retention times, detection limits and method repeatability are summarized in Table 3. The detection limits on the ESI-MS obtained in the SIM mode fell in the low mg L−1 range while those on the ICP-MS, were in the very low µg L−1 range with lower values obtained using the cation exchange column compared to the C8 column. This can be attributed to the fact that the column effluent of the C8 column was split to sustain the plasma. Taking consideration of the high instability of the As-GSH complexes, the method repeatability obtained is good. Chromatographic recoveries for the standards eluting from the columns were reasonable, lying in the range between 89–97%. From Fig. 2C it can be observed that GSSG, As(GS)3, MMA(GS)2 and DMA(GS) show absorption peaks on UV. Linear calibration plots were also obtained with a UV detector but detection limits were not calculated for this method due to the low sensitivity of UV compared to MS for these species.| AsIII | AsV | MMAIII | MMAV | DMAIII | DMAV | GSH | GSSG | As(GS)3 | MMA(GS)2 | DMA(GS) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Spherisorb C8 | |||||||||||
| Ret. time (min.) UV | 2.83 | 4.17 | 5.26 | 6.73 | |||||||
| Ret. time (min.) ESI-MS | 2.48 | 2.2 | 4.8 | 2.78 | 2.71 | 2.97 | 4.37 | 5.43 | 6.90 | ||
| DL by ESIMS (mg L−1 As) | 0.094 | 0.073 | 0.175 | 0.072 | 0.039 | ||||||
| Repeatability ESI-MS (%) | 0.17 | 1.67 | 5.41 | 6.57 | 2.43 | ||||||
| Ret. time (min.) ICP-MS | 2.39 | 1.93 | 2.75 | 2.34 | 5.91 | 2.86 | 5.15 | 6.36 | 7.73 | ||
| DL by ICPMS (µg L−1As) | 0.67 | 1.34 | 0.28 | 0.81 | 1.51 | 1.11 | 1.02 | 5.37 | 6.14 | ||
| Repeatability ICP-MS (%) | 2.48 | 0.38 | 0.77 | 0.30 | 3.95 | 1.29 | 1.61 | 1.58 | 1.41 | ||
| Hamilton PRPX-200 | |||||||||||
| Ret. time (min.) ICP-MS | 2.33 | 8.17 | 3.75 | 3.33 | 3.75 | 4.05 | |||||
| DL by ICPMS (µg L−1As) | 0.24 | 0.69 | 0.64 | 0.49 | 0.48 | 0.47 | |||||
| Repeatability ICP-MS (%) | 1.16 | 4.94 | 2.56 | 1.20 | 3.05 | 0.78 | |||||
Method validation and GSH concentration effect on the formation of As-GSH complexes and analytical artifacts with cation exchange chromatography
Experiments were conducted to ensure the possibility of using the two developed methods in a complementary manner for the quantitation of trivalent and pentavlent arsenicals. A Deming plot was performed which takes into consideration the uncertainties in both methods. A 34 different sample analysis by the two methods for various As species (AsIII, AsV, MMAIII, MMAV, DMAIII and DMAV) in the presence or absence of GSH in the mixture (Fig. 3A) yielded a linear plot with a slope of 0.99 and an intercept of 0.013. The 95% confidence limit range for the slope is from 0.92 to 1.05, which provides evidence that the slope is not significantly different from 1. While the 95% confidence limit range for the intercept is from −0.086 to 0.11, which provides evidence that the intercept is not significantly different than 0. Hence, it was confirmed that the two methods could be used in a complementary manner. With respect to GSH effect on the formation of As-GSH complexes, the results obtained with the C8 column (Fig. 3B) indicate that higher concentrations of As-GSH complexes were produced with trivalent arsenicals as starting materials due to the fact that pentavalent As species are first reduced by GSH before complexation. Maximum formation was observed with solutions of 5.6 mM GSH, the highest tested. Complexes were formed in the range 10–95% depending on the identity of the As species incubated. At lower GSH concentration, reduction and complexation was still possible but with lower yields. With respect to GSH effect on analytical artifact with the PRPX-200 column, it was observed that As solutions with higher GSH concentration, yielded lower quantitation recoveries for the sum of trivalent and pentavalent species compared to the injected concentration (as low as 5%) (data not shown). However, GSH-free As solutions injected yielded ∼ 100% recoveries. These results further support our previous hypothesis that As-GSH complexes are retained by the cation exchange column, hence generating analytical artifacts. Similar results were also reported by Slejkovec et al.9 who used anion exchange chromatography. These results indicate the importance of selecting appropriate columns for As speciation when dealing with samples containing high concentration of GSH (e.g. biological matrices). | ||
| Fig. 3 A) Deming plot obtained for trivalent and pentavalent As species quantitation using C8 and PRPX-200 columns (line represents linear regression line while dotted line represents Deming plot). A plot with a slope of 0.99 and an intercept of 0.013 was obtained based on analysis of 34 different samples of various As species. B) Effect of GSH concentration on As-GSH species formation and elution on C8. The species incubated were AsIII, AsV, MMAIII, MMAV, DMAIII and DMAV respectively. | ||
Stability of AsIII, MMAIII, DMAIII and As-GSH complexes
As shown in Fig. 4A, the percent abundance of DMAIII was around 55%, analyzed immediately after the preparation of the standard, indicating DMAIII is highly unstable even in argon purged media. Variations in solution pH did not affect its abundance. AsIII and MMAIII were the most stable of the species with a relative abundance of ∼100%. AsIII is also stable in non-degassed solutions. High instabiliy of DMAIII was also observed by Gong et al., and a plausible explanation was related to the increase in the electron density of the species due to the presence of methyl groups and hence higher tendency for oxidation.50 With respect to the complexes, As(GS)3 was more stable at pH 2.0 while MMA(GS)2 and DMA(GS) had higher stabilities at pH 4.3 (Fig. 4A). The complexes were higly unstable at neutral and basic conditions. The half-lives for the As-GSH complexes were estimated. DMA(GS) had the longest half-life among the three complexes at all times (Fig. 4B). At pH 2.0, DMA(GS) had a half-life of 55 minutes, while at neutral or slightly basic pH values, its half-life ranged between 23 and 37 minutes. This could be one of the reasons why DMA(GS) has been suggested as a potentially more favorable anti-leukemic drug candidate compared to others and has been developed for clinical trials.51 As(GS)3 was the least stable, with a half-life of 35 minutes at pH 2.0 and less than 6 minutes at pH 6.0 and above. The half life of MMA(GS)2 was 37 minutes at pH 2.0 and less than 6 minutes at pH 8.4. Comparing the overall stability of the complexes, DMA(GS) has the highest stability followed by MMA(GS)2, and As(GS)3 being the least stable of the three. This is the opposite trend than the one observed for trivalent arsenicals, for which AsIII is highly stable and MMAIII shows high stability when prepared in oxygen free water compared to DMAIII. The half-life results for the complexes obtained at pH 8.4 lie in the middle range between those of Raab et al.42 and Kala et al.43 The half-lives reported by Raab et al. for all the complexes were between 5 and 10 minutes while those reported by Kala et al. were 40 minutes. In our study, the half-lives of As(GS)3 and MMA(GS)2 were less than 6 minutes, while that for DMA(GS) was 23 minutes. The variations in the half-life could be attributed to the variations in the matrices in which As-GSH complexes were present. Kala et al. studied the complex stability in bile (pH = 8.0) and Raab et al. prepared the solutions in ammonium carbonate (pH = 8.3), while we prepared the solutions in a PBS buffer to mimic biological conditions and salinity (pH = 8.4). Further experiments using specific matrices of interest need to be performed to determine the stability of these species in biological samples. Overall, the results show that the complexes and DMAIII are highly unstable and biological matrices containing these molecules should be analyzed immediately after proper sample collection since during storage complex decomposition and further oxidation is highly susceptible. | ||
| Fig. 4 pH effect on A) the percentage abundance of AsIII, MMAIII, DMAIII, As(GS)3, MMA(GS)2 and DMA(GS) in solution, B) the half-life of As-GSH complexes. | ||
Conclusions
A preliminary separation and identification of nine standards of possible human arsenic metabolites was achieved through the use of reverse phase and cation exchange chromatography. The use of ESI-MS as a molecule specific detector and ICP-MS as an element specific detector are necessary for correct identification and quantitation of the species. The two developed methods can be used in a complementary manner for the identification and quantitation of the As species of interest. Further validation experiments in matrices of interest need to be performed based on requirements of a specific application. Glutathione concentration has significant effect on the formation of the As-GSH complexes and formation of analytical artifacts with cation exchange column. Since GSH is ubiquitous in most biological and clinical samples, the use of both cation and reverse phase chromatographic techniques is indispensible for the prevention of analytical artifacts. Stability experiments show that MMAIII is more stable than DMAIII in argon purged solutions with pH having no effect on the stability of both standards. As-GSH complexes show higher stability at acidic conditions (pH = 2.0–4.3). Because the complexes have very short half-lives, samples suspected of containing these species should be analyzed immediately after sampling. Although trivalent methylated arsenicals are more stable compared to As-GSH complexes, proper sample storage measures still need to be taken to prevent species oxidation. Based on our results, reinvestigation of some of the previously reported As-speciation studies of glutathione rich biological samples needs to be performed for the verification of occurrence of As-GSH complexes and DMAIII.Acknowledgements
This study was partially supported by NIEHS ARCH (S11 ES11181) and NIH-MBRS (3 S06 GM008205-20S1) programs. This is contribution # 444 of Southeast Environmental Research Center at FIU. We would like to thank Mr. Yali Hsu for his assistance in NMR analysis. Ms. Yehiayan thanks Florida International University Graduate School for her dissertation year fellowship.References
- Y. Kobayashi, T. Hayakawa and S. Hirano, Environ. Toxicol. Pharmacol., 2007, 23, 115–120 CrossRef CAS.
- N.R.C. (NRC) Arsenic in Drinking Water; The National Academies Press: Washington, DC, 1999 Search PubMed.
- N.R.C. (NRC) Arsenic in Drinking Water: 2001 Update; The National Academies Press: Washington, DC, 2001 Search PubMed.
- J. C. Ng, J. Wang and A. Shraim, Chemosphere, 2003, 52, 1353–1359 CrossRef CAS.
- C.-J. Chen, S.-L. Wang, J.-M. Chiou, C.-H. Tseng, H.-Y. Chiou, Y.-M. Hsueh, S.-Y. Chen, M.-M. Wu and M.-S. Lai, Toxicol. Appl. Pharmacol., 2007, 222, 298–304 CrossRef CAS.
- C. Anderson Kenneth, H. Boise Lawrence, R. Louie and S. Waxman, Cancer journal (Sudbury, Mass.), 2002, 8, 12–25 Search PubMed.
- Y.-L. Lai; Y.-J. Chen; Y.-F. Hu; C.-L. Kan; K.-C. Chiu; (TTY Biopharm Ltd., Taiwan). Application: US, 2005, p 8 pp.
- Y.-L. Kwong; (The University of Hong Kong, Hong Kong). Application: US, 2008, p 19pp, Cont -in-part of U S Ser No 549,347.
- Z. Slejkovec, I. Falnoga, W. Goessler, J. T. van Elteren, R. Raml, H. Podgornik and P. Cernelc, Anal. Chim. Acta, 2008, 607, 83–91 CrossRef CAS.
- J. Hu, J. Fang, Y. Dong, S. J. Chen and Z. Chen, Anti-Cancer Drugs, 2005, 16, 119–127 CrossRef CAS.
- R. A. Zingaro; E. L. Freireich; H. Dukale; H. Kantarjian; S. Verstovsek; M. Sotelo-Lerma; (Board of Regents, the University of Texas System, USA; Texas A & M University). Application: WO, 2003, p 107 pp.
- T. Sakurai, C. Kojima, Y. Kobayashi, S. Hirano, M. H. Sakurai, M. P. Waalkes and S. Himeno, Br. J. Pharmacol., 2006, 149, 888–897 CrossRef CAS.
- S. Mann, P.-O. Droz and M. Vahter, Toxicol. Appl. Pharmacol., 1996, 140, 471–486 CrossRef CAS.
- B. K. Mandal, Y. Ogra, K. Anzai and K. T. Suzuki, Toxicol. Appl. Pharmacol., 2004, 198, 307–318 CrossRef CAS.
- R. A. Zakharyan and H. V. Aposhian, Chem. Res. Toxicol., 1999, 12, 1278–1283 CrossRef CAS.
- J. Messens and S. Silver, J. of Mol. Biol., 2006, 362, 1–17 Search PubMed.
- M. Styblo and D. J. Thomas, Toxicol. Appl. Pharmacol., 1997, 147, 1–8 CrossRef CAS.
- M. Lu, H. Wang, X.-F. Li, X. Lu, W. R. Cullen, L. L. Arnold, S. M. Cohen and X. C. Le, Chem. Res. Toxicol., 2004, 17, 1733–1742 CrossRef CAS.
- G. Jiang, Z. Gong, X.-F. Li, W. R. Cullen and X. C. Le, Chem. Res. Toxicol., 2003, 16, 873–880 CrossRef CAS.
- T. T. Ngu and M. J. Stillman, J. Am. Chem. Soc., 2006, 128, 12473–12483 CrossRef CAS.
- T. W. Kensler, N. Wakabayashi and S. Biswal, Annual Rev. Pharmacol. Toxicol., 2007, 47, 89–116 Search PubMed.
- X. He, M. G. Chen, G. X. Lin and Q. Ma, J. of Biol. Chem., 2006, 281, 23620–23631 Search PubMed.
- G. Bellomo, M. Vairetti, L. Stivala, F. Mirabelli, P. Richelmi and S. Orrenius, Proc. Natl. Acad. Sci. USA, 1992, 89, 4412–16 CrossRef CAS.
- A. Meister, J. Biol. Chem., 1988, 263, 17205–8.
- W. R. Cullen, B. C. McBride and J. Reglinski, J. Inorg. Biochem., 1984, 21, 179–94 CrossRef CAS.
- T. Hayakawa, Y. Kobayashi, X. Cui and S. Hirano, Arch. Toxicol., 2005, 79, 183–191 CrossRef CAS.
- R. Zakharyan, Y. Wu, G. M. Bogdan and H. V. Aposhian, Chem. Res. Toxicol., 1995, 8, 1029–38 CrossRef CAS.
- R. A. Zakharyan, F. Ayala-Fierro, W. R. Cullen, D. M. Carter and H. V. Aposhian, Toxicol. Appl. Pharmacol., 1999, 158, 9–15 CrossRef CAS.
- N. A. Rey, O. W. Howarth and E. C. Pereira-Maia, J. Inorg. Biochem., 2004, 98, 1151–1159 CrossRef CAS.
- T. Nakayama, J. S. Edmonds and et al., J. Chem. Res., 2006, 3, 185–187 Search PubMed.
- M. J. Mass, A. Tennant, B. C. Roop, W. R. Cullen, M. Styblo, D. J. Thomas and A. D. Kligerman, Chem. Res. Toxicol., 2001, 14, 355–361 CrossRef CAS.
- J. S. Petrick, F. Ayala-Fierro, W. R. Cullen, D. E. Carter and H. Vasken Aposhian, Toxicol. Appl. Pharmacol., 2000, 163, 203–207 CrossRef CAS.
- M. Styblo, L. M. Del Razo, L. Vega, D. R. Germolec, E. L. LeCluyse, G. A. Hamilton, W. Reed, C. Wang, W. R. Cullen and D. J. Thomas, Arch. Toxicol., 2000, 74, 289–299 CrossRef CAS.
- C. Kojima, T. Sakurai, M. P. Waalkes and S. Himeno, Biol. Pharmac. Bull., 2005, 28, 1827–1832 Search PubMed.
- S. Hirano and Y. Kobayashi, Toxicology, 2006, 227, 45–52 CrossRef CAS.
- X. C. Le, X. Lu, M. Ma, W. R. Cullen, H. V. Aposhian and B. Zheng, Anal. Chem., 2000, 72, 5172–5177 CrossRef CAS.
- Z. Wang, J. Zhou, X. Lu, Z. Gong and X. C. Le, Chem. Res. Toxicol., 2004, 17, 95–103 CrossRef CAS.
- R. Xie, W. Johnson, S. Spayd, G. S. Hall and B. Buckley, Anal. Chim. Acta, 2006, 578, 186–194 CrossRef CAS.
- L. M. Del Razo, M. Styblo, W. R. Cullen and D. J. Thomas, Toxicol. Appl. Pharmacol., 2001, 174, 282–293 CrossRef.
- S. V. Kala, G. Kala, C. I. Prater, A. C. Sartorelli and M. W. Lieberman, Chem. Res. Toxicol., 2004, 17, 243–249 CrossRef CAS.
- X. Cui, Y. Kobayashi, T. Hayakawa and S. Hirano, Toxicol. Sci., 2004, 82, 478–487 CrossRef CAS.
- A. Raab, A. A. Meharg, M. Jaspars, D. R. Genney and J. Feldmann, J. Anal. At. Spectrom., 2004, 19, 183–190 RSC.
- S. V. Kala, M. W. Neely, G. Kala, C. I. Prater, D. W. Atwood, J. S. Rice and M. W. Lieberman, J. Biol. Chem., 2000, 275, 33404–33408 CrossRef CAS.
- K. T. Suzuki, B. K. Mandal and Y. Ogra, Talanta, 2002, 58, 111–119 CrossRef CAS.
- J. Gailer and W. Lindner, J. Chromatograp., B, 1998, 716, 83–93 Search PubMed.
- G. J. Burrows and E. E. Turner, J. Chem. Soc., Transactions, 1920, 117, 1373–80 Search PubMed.
- R. A. Zingaro, Synthetic Methods of Organometallic and Inorganic Chemistry, 1996, 3, 198–200 Search PubMed.
- N. Scott, K. M. Hatlelid, N. E. MacKenzie and D. E. Carter, Chem. Res. Toxicol., 1993, 6, 102–6 CrossRef CAS.
- A. M. Spuches, H. G. Kruszyna, A. M. Rich and D. E. Wilcox, Inorg. Chem., 2005, 44, 2964–2972 CrossRef CAS.
- Z. Gong, X. Lu, W. R. Cullen and X. C. Le, J. Anal. At. Spectrom., 2001, 16, 1409–1413 RSC.
- S. Verstovsek and Z. Estrov, Leukemia Res., 2004, 28, 901–903 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |