An in-situ X-ray absorption spectroelectrochemistry study of the response of artificial chloride corrosion layers on copper to remedial treatment
Annemie
Adriaens
*a,
Mark
Dowsett
b,
Gareth
Jones
b,
Karen
Leyssens
a and
Sergey
Nikitenko
c
aDepartment of Analytical Chemistry, Ghent University, B-9000, Ghent, Belgium. E-mail: annemie.adriaens@ugent.be; Fax: +32 9 264 4960; Tel: +32 9 264 4826
bDepartment of Physics, Warwick University, Coventry, UK CV4 7AL
cESRF, DUBBLE, 6 rue Jules Horowitz, F-38043, Grenoble, Cedex, France
First published on 24th October 2008
Abstract
In the experiments described in this study, we make use of X-ray absorption spectroscopy (XAS) at the K-edge of copper for monitoring the surface–solution interface of corroded metals in-situ in a sodium sesquicarbonate solution. We show conclusively that, unlike previously published XRD measurements, the XAS spectra are almost wholly characteristic of the contaminated solution and not the corroded surface, in cases where the sesquicarbonate is effective at removing chlorides. This is true even when there is only ∼125 µm of fluid over the sample, which demonstrates that there is very effective absorption of the Cu Kα radiation from the surface by copper in solution. As a function of time we observe a rising intensity in the XANES and EXAFS regions which is accompanied by a proportional increase in the degree of EXAFS modulation. The latter is due to the fact that a significant proportion of the corrosion product goes into solution, rather than being converted to a stable compound on the surface. Moreover, the spectra show that the local atomic environment of the copper in solution is extremely similar to that in the surface. Overall from both XAS and previously performed XRD data we can conclude that the chlorine containing corrosion products become detached from the surface and go partly into solution, while a thin cuprite layer forms (or is already present at) at the original metal surface.
Introduction
Copper has played an important role in human development. The earliest use of native copper was at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years ago, and the first copper-based alloys were produced in the 4th millennium BC. The intrinsic advantages of copper, such as easy extraction and handling, as well as excellent heat conduction, make it an ideal utilitarian metal. Additionally, copper and its alloys were (and still are) appreciated as decorative metals which have been used throughout history for the realization of various art objects, from jewellery to statuary.1
000 years ago, and the first copper-based alloys were produced in the 4th millennium BC. The intrinsic advantages of copper, such as easy extraction and handling, as well as excellent heat conduction, make it an ideal utilitarian metal. Additionally, copper and its alloys were (and still are) appreciated as decorative metals which have been used throughout history for the realization of various art objects, from jewellery to statuary.1
As with many other metals, copper corrodes once it comes into contact with an aggressive environment, for example the sea or the atmosphere.2 In the field of art, copper-based objects are often preferred in the corroded state, not only because of the aesthetically pleasing colours, but also because the presence of corrosion products provides evidence of time past and time passing, thereby adding extra value to the object.1 However, corrosion may also become a problem, especially when specific corrosion products (such as cuprous chlorides) are in contact with the metal core. Under certain conditions, the deterioration of the underlying metal will continue and will lead to the destruction of the object.
Corroded archaeological cupreous artefacts recovered from wet saline environments are, in particular, very susceptible to further corrosion when exposed to air. As a result they are stored in solution, such as tap water or dilute sodium sesquicarbonate, for a period of up to several years.2 In spite of these precautions, experience shows that the conserved patina may still be unstable: chemical transformation of the natural patina and the development of active corrosion can take place.3,4 As a result, continuous monitoring of the system and the conserved artefact remains essential.
The conservation process is usually monitored by measuring the chloride level at regular time intervals in the solution. Whilst this is a simple and inexpensive process suitable for use in a conservation laboratory, it gives no information on the surface state or surface/solution interface during immersion. In previous experiments we monitored the surface in solution as a function of time using in-situsynchrotron radiation X-ray diffraction5,6 in a novel electrochemical cell (eCell) designed for use with rough heterogeneous surfaces.7 There, we were able to observe the replacement of cuprous chlorides with the more benign cuprite (Cu2O) in real time when immersed in a 1% (wt/v) sodium sesquicarbonate solution. In this study we report of the use of X-ray absorption spectroscopy (XAS) in the vicinity of the copper K-edge in order to study the dynamic processes in more detail. This provides an independent means of surface characterization as the technique is not only sensitive to the presence and evolution of amorphous surface compounds, but will also give information on the possible presence of complex ions in the solution. XAS was first used in-situ in an electrochemical cell8 in around 1990, some 10 years after the first similar use of X-ray diffraction (XRD).9 Subsequently, there have been numerous reports of the use of both XAS and X-ray diffraction (XRD) on single crystal and atomically flat metal surfaces and reaction products measured whilst immersed in electrolyte, for example: passivating films,10–12 battery electrodes,13fuel cells,14 fundamental corrosion studies,15 and biological applications.16 Recently,17 Kvashnina et al. have used XAS at the Cu 2p edge to study the effect of ground water on copper in a special fluid cell. We use the K-edge as this X-ray energy will penetrate ∼1 mm paths in the electrolyte and therefore allows us to study realistically rough surfaces.
Here, we describe the time-dependent behaviour of artificial corrosion layers on copper in a 1% (wt/v) sesquicarbonate solution, thereby imitating the storage conditions for copper-based artefacts recovered from marine environments. The immersion of freshly polished and cleaned copper metal, cuprite, nantokite and atacamite were all studied with a 20 min time-resolution. The first two because they are possible end points for a conservation process, and the others to observe the chloride removal in real time. The behaviour of atacamite is discussed in detail. Simultaneously with the atacamite measurements we measured the corrosion potential, Ecorr, as the open circuit potential (OCP) of the cell to see if this parameter correlated with the chloride removal. In principle it could form the basis of a simple sensor for end point detection in the conservation process.18
Experimental
Prior to immersing the corroded copper samples in a 1% (wt/v) sesquicarbonate solution and performing in-situ time-resolved XAS measurements, XAS data were acquired from pure corrosion products in the form of powders, and for copper(II) chloride in solution. The aim was to determine how distinctive the spectra of different corrosion products are, and to obtain a set of reference spectra to aid future identification.Sample preparation
Powder samples of Cu2O (for comparison with cuprite, Fluka, > 99%) and Cu(I)Cl (for comparison with nantokite, Fluka, > 97%) were purchased. Atacamite powder is not commercially available and was prepared19 by adding a 50 mL 0.1 M Na2CO3 (Fluka, > 99.5%) solution dropwise to a stirred solution of 100 mL 0.1 M CuCl2·2H2O (Aldrich, > 99%) boiling at reflux. After 5 h the slurry was filtered, washed and dried at 333 K. The samples were crushed to a very fine powder, mixed with a light silicone-free grease (Apiezon M, M & I Materials Ltd.) and fixed between two thin Kapton® sheets. They were measured in transmission mode.Copper(II) chloride (3.5 M CuCl2) solution was made from 60 g CuCl2·2H20 (Aldrich, > 99%) per 100 mL of water. Both the source material and the solution were measured in fluorescence mode, the latter in a 4 mm thick fluid cell with new 60 µm thick Kapton® windows. Only the results for the solution are reported here.
Electrodes: Circular copper coupons, 12 mm in diameter (Advent, purity 99.9%), were made into electrodes to fit the electrochemical cell described below. The coupons were connected to a brass rod by means of conducting glue, and encapsulated in acrylic resin so that only one surface was exposed to the electrolyte. A brass stud in a tapped hole in the rod attaches the electrode to the cell, and an O ring seals against the acrylic to exclude electrolyte from the electrical contact thus formed.
The electrodes were ground on 1200 grit SiC paper to obtain a fresh surface. Further smoothing of the surface was carried out using a polishing cloth covered with alumina powder of 1 µm particle size. Any adherent Al2O3 particles on the surface were removed by immersing the samples in an ultrasonic bath for 15 min and rinsing them thoroughly with deionized water.
The samples were corroded artificially using various corrosion protocols19 to obtain different chloride-containing corrosion products, including cuprite (Cu2O), nantokite (CuCl) and atacamite (Cu2(OH)3Cl). To obtain a cuprite layer (Cu2O), the copper based samples were polarized anodically at −360 mV (vs. MSE) for 16 h in a 0.1 M Na2SO4 solution (Fluka). Copper covered with nantokite (CuCl) was obtained by immersing pure copper coupons for 1 h in a saturated CuCl2·2H2O solution (Aldrich). After rinsing with deionised water they were exposed to the air overnight. For the atacamite, a solution of 15.07 g ammonium carbonate (NH4)2CO3 (Sigma-Aldrich, ammonia > 30%) and 10.02 g ammonium chloride NH4Cl (Sigma-Aldrich, 99.998%) in 100 mL deionised water was prepared. One drop of the solution was used to wet the surface twice a day for 5 days. Between each wetting, the samples were left to dry in air. After the period of 5 days, the samples were left in the air for a further 5 days.
Immersion experiments
The 1% (wt/v) sodium sesquicarbonate solution was prepared by dissolving 11.89 g L−1 of Na2CO3·NaHCO3·2H2O (Sigma-Aldrich) in deionised water (pH = 10).The immersion experiments were performed in a specialized cell compatible with synchrotron radiation X-ray based methods. The cell is designed for combined electrochemical and surface analyses (spectroelectrochemistry). In this work we made open circuit potential (OCP) measurements to examine the time dependence of the corrosion potential Ecorr. These were simultaneous with the XAS measurements. The cell is made from PCTFE for good chemical resistance and reasonable mechanical stability. It is a cylinder 60 mm in diameter and 100 mm high containing a piston which is driven along the bore by a stepper motor through a dynamic seal. The working electrode, whose surface forms the sample for X-ray analysis, is mounted on the piston. The cell contains 35 mL of electrolyte. In this work the electrode surface was kept in a fixed position 125 µm from a flexible 12 µm thick LDPE window. A rigid polyethylene teraphthalate inner window with a rectangular or elliptical hole in the middle was used as a spacer. The two windows are tightly fastened onto the cell body by an O-ring. This way, the path length for X-rays in the electrolyte is a fraction of a millimetre to reduce scattering and X-ray absorption. The electrode was not moved between XAS spectra because of the time taken for each acquisition (20 min) and the need to get the highest density of spectra possible because of the rapid changes which were observed. More details on the cell and the possibility to perform electrochemical measurements are described in detail elsewhere.7
The system is complimented by a webcam which allows video or stills of the metal surface to be observed and captured.
In the case of copper, cuprite, and nantokite, the results of a single immersion lasting between 3 and 16 h are reported in outline. The atacamite immersion was done in several stages: A 14 h immersion with XAS and OCP recorded in parallel, was followed by immersion for a similar period in fresh electrolyte with a new window. This was followed by a short immersion to see if the trend continued. The electrolyte produced in the first stage was filtered to remove loose insoluble debris, and also analysed using XAS in fluorescence mode in the 4 mm thick fluid cell.
XAS analyses
Experiments were performed at DUBBLE (Station BM26A, European Synchrotron Radiation Facility, Grenoble). Cu K-edge XAS spectra were recorded as a function of energy over the range 8.9 to 9.6 keV defined by the stepping of an Si (1 1 1) double-crystal monochromator. Measurements were performed either in transmission (powders) or in fluorescence mode for electrodes measured dry or immersed in the eCell, and for fluids. The scan time was 60 min for the powders, and 20 min for the other samples. The latter is a compromise between obtaining adequate statistics in the EXAFS region, and sufficient time-resolution to follow the solution chemistry. Two Oxford Instruments ion chambers were used for transmission experiments. Fluorescence measurements were made using the X-ray beam at 80° to the sample surface with a nine channel monolithic Ge fluorescence detector (E&G Ortec Inc.)20 at 90° to the beam in order to minimize the collected flux of backscattered X-rays. The result is a minimum path length in the fluid of 0.8 mm, but sample roughness can increase this. The electrochemical cell was mounted so that the sample surface was in a vertical plane.For the immersion experiments, 20 min XAS scans were continuously repeated over a period of several hours. For the copper, cuprite, nantokite and atacamite, we studied immersions starting from a fresh surface, and continuing until there were no further changes in the spectra–either because the process was complete (e.g. in the case of nantokite as indicated by earlier XRD experiments5–7) or because the electrolyte was saturated. For the atacamite, which showed the most dramatic effects, this procedure was also followed, with the addition that, in some experiments the electrolyte in the cell was changed after the spectra had stabilized, and the experiment was repeated. Simultaneous OCP measurements were also made. All measurements were repeated several times during different beam time allocations with similar results.
Results and discussion
Fig. 1–3 show, respectively, the near-edge (XANES) region of the powder references, the dry corrosion products on copper, and the corrosion products on copper immediately after immersion in the sesquicarbonate solution. The data were background corrected using a linear fit to the region between 150 eV and 30 eV below the edge, and normalized to a spline fitted between 10 eV and 500 eV above the edge. Athena v. 8.05 was used to process the data.21 | ||
| Fig. 1 XAS spectra for copper, cuprite, CuCl (the analogue for nantokite) and synthesized atacamite. The inset shows the edge region in detail. | ||
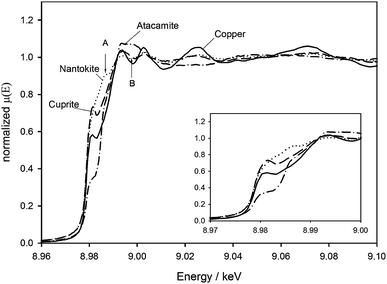 | ||
| Fig. 2 XAS spectra for a clean copper electrode, and copper electrodes covered with cuprite, nantokite and atacamite corrosion layers. | ||
 | ||
| Fig. 3 The copper electrode, and the cuprite, nantokite and atacamite corroded electrodes upon first immersion in a 1% (wt/v) sodium sesquicarbonate solution. | ||
Fig. 1 shows spectra for the three powder references and a bare copper sample, with an inset to show the details of the edge region. The copper and Cu2O spectra, and the edge shift of ∼0.4 eV agree well with others in the literature.22,23 A small peak is clearly visible on the edge itself for the copper and cuprite sample and to a lesser extent also for the atacamite sample. This has been attributed to a core electron transition into an unoccupied bound state above the Fermi level,24 an effect which is slightly more pronounced in the Cu2O which is a wide bandgap semiconductor. For the chlorides, the 1.2 eV and 6.2 eV shifts between the copper and CuCl (CuI) and copper and atacamite (CuII) edges agree with the expected behaviour, which is a shift to higher energy with increasing oxidation state for the metal ion. Above the edge, the spectra show clear characteristic differences. Together, all of these features provide a distinctive fingerprint for each solid sample.
Fig. 2 shows the data obtained in fluorescence mode from the corrosion products on the copper surface. The cuprite and nantokite spectra are strongly influenced by the presence of the underlying copper, simply because the layers are thin. Nevertheless, the shoulder in the nantokite at 8985 eV (A) is clearly a relic of the post edge peak in the CuCl, and the spectrum from the cuprite coated sample is modified from its dominant copper shape at (B). The thicker atacamite spectrum remains distinctive, although it too is clearly influenced somewhat by the underlying copper.
XANES spectra recorded immediately for the samples immersed in a 1% (wt/v) sodium sesquicarbonate solution are shown in Fig. 3. The spectra start approximately 2 min after immersion, and the region displayed took 3 min to acquire (although the repeat time was 20 min since EXAFS data were collected as well). Although the noise levels are higher in comparison to the dry samples, the spectra for bare copper and atacamite on copper remain distinctive. The samples covered with cuprite and nantokite, are more dominated by the underlying copper than in the dry case. From XRD data5–7 we know that the initial removal of nantokite is quite rapid and, in any case, it is obvious from visual inspection that these corrosion layers (particularly the cuprite) are much thinner than the atacamite.
Fig. 4 shows a comparison between XAS from synthetic atacamite powder, atacamite on copper and the material after immersion for 14 h in the sesquicarbonate solution and an analysis of the same material after drying in air. The layer post-immersion shows a marked similarity to the atacamite powder, whereas the layer as produced shows differences both in the XANES and EXAFS regions. Together with the behaviour observed during immersion (described later) this suggests that the synthesized atacamite incorporates another component at the surface.
 | ||
| Fig. 4 Comparison of the post-edge region from atacamite powder, atacamite on copper, and the same layer after 14 h immersion in 1% (wt/v) sodium sesquicarbonate. | ||
Time dependence
In complete contrast to the case for our XRD measurements, the XAS spectra of all four samples collected during their immersion in the sesquicarbonate solution show increasing signals as a function of time, whereas in XRD the chloride related peaks decrease monotonically, whilst the cuprite and copper peaks increase.5,7 In the XAS experiments, the increase is observed both at the Cu K edge, and in a proportionate increase in the amplitudes of the XANES and EXAFS modulation, whilst the shapes of the spectra either remain constant within the noise, or evolve slightly. In other words, XRD shows evolution in the surface chemistry, whilst XAS remains approximately characteristic of the original corrosion product, or even pure copper. This observation, and direct measurements on the spent electrolyte itself show that (i) the XAS is characteristic of the 0.8 mm of fluid on the exit path to the detector, and (ii) on a timescale from minutes to days, copper ions in the 1% sesquicarbonate solution retain a very similar nearest neighbour structure to that in the solid surfaces used here, surprisingly even where the source is copper metal. The former is due to the fact that there is a rapidly increasing concentration of complex octahedrally coordinated copper ions immediately behind the window. Absorption due to the solute between the solid sample surface and the window quickly masks the contribution of the surface.Table 1 gives an overview of the changes seen in the signal for immersed and dry samples measured at scan number 7 (i.e. after 140 min). While only a random variation is seen in successive scans of the dry atacamite sample, results show a small increase in signal for copper, a larger increase for cuprite and nantokite and the largest increase (>60 times) for the atacamite sample. In the latter case there was considerable variation in the increase at this point between repeated experiments, the smallest recorded being 4 times. The behaviour of the nantokite and cuprite may simply be due to the fact that the layers are powdery and particles become suspended in the fluid. The copper behaviour is hard to account for. Further measurements are in progress for all three.
| Sample | Condition | Increase (times) |
|---|---|---|
| Copper | Immersed | 1.3 |
| Cuprite | Immersed | 1.8 |
| Nantokite | Immersed | 1.9 |
| Atacamite | Immersed | 63 |
| Atacamite | Dry | none |
Fig. 5(a) and (b) show the successive XAS spectra recorded for the atacamite sample. From the ninth spectrum on, the intensity tends to saturation, however, repeat measurements with different X-ray fluxes show that this is not detector saturation. One underlying effect, which is a continuous increase in the height of the copper edge, results in continuously increasing background slopes in the EXAFS region. Samples measured in air over comparable time periods show only random intensity variations.
 | ||
| Fig. 5 Sequential XAS spectra of a copper/atacamite sample in solution. Each scan takes 20 min. (a) First immersion. (b) immersion of same sample in fresh electrolyte with new window. | ||
The cell was charged with fresh electrolyte, and the window was changed prior to the recording of the data in Fig. 5(b). A significant, but less dramatic increase in the signal intensity occurs—at scan 7 the change is 5 times compared to scan 1. A similar saturation is behaviour is observed. If the experiment is repeated again, the results are similar, albeit with a smaller rate of signal increase still, consistent with a diminishing source. However, the atacamite layer remains almost identical visually throughout a process lasting many days when the work is performed ex-situ, and its XAS spectrum is also changed very little. Clearly, the corrosion product has at least two components—one which is relatively soluble (or which goes easily into colloidal suspension) and one which remains attached to the copper.
Whilst spectral acquisition is in progress, eCell makes a continuous visual record of the process using a webcam. Fig. 6 shows two stills recorded after 20 min (left-hand plate) and at the end (13 h 40 min) during the sequence in Fig. 5(a). The images show the detachment of part of the atacamite layer from the copper substrate, in the form of blue “smokey” material streaming downwards before going into solution as a complex ion (or possibly forming a colloidal suspension). Large crystalline fragments which become detached are also seen. After a while (right-hand plate) the former becomes distributed throughout the fluid. The XAS spectrum remains similar to that of solid throughout this process. When the same sample is re-immersed in fresh solution, and despite the fact that the bulk of the atacamite still remains as a thick crust, no more visible material is removed.
 | ||
| Fig. 6 Webcam images of the eCell during the sequence in Fig. 5(a). Left hand plate shows the blue haze flowing off the sample after 20 min immersion. The right hand plate shows the situation at the end of the first immersion. | ||
As a check on the origins of the spectrum, we carried out analyses on the LDPE window and the used electrolyte after a 4 h immersion. The window measurement is shown in Fig. 7 in comparison with the last spectrum taken during the immersion, and an unused window. It is clear that the LDPE absorbs some of the copper complex, but the signal is 30 times less intense than that of the final scan. A similar result was obtained for a 6 µm thick Kapton window. The contamination could not be removed by rinsing in deionised water. The spent electrolyte from this experiment was filtered to remove particles and analysed in fluorescence. The result is shown in Fig. 8, where it is compared with the atacamite on copper and XAS from the 3.5 M CuCl2 solution. Again, the spectrum from the electrolyte is similar to, but not identical with, its source, and the CuCl2.
 | ||
| Fig. 7 XAS of an LDPE window before and after a 4 h immersion of a copper/atacamite electrode, showing the absorption of copper chloride into the window. For comparison, the last in-situXAS is also shown. | ||
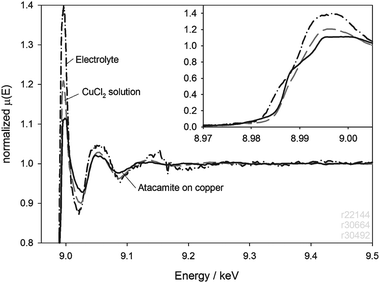 | ||
| Fig. 8 XAS spectrum of the spent electrolyte after the 4 h immersion of Fig. 7. The electrolyte was filtered before analysis to remove solid debris. | ||
The crystallographic structure of Atacamite was established in 1949 by Wells25 using XRD. Copper atoms lie at the centre of one of two distorted octahedron with 6 nearest neighbours: 2 chlorine atoms, one at either vertex, and 4 OH groups in the equitorial plane, or with the vertices containing one Cl and one OH. It is this structure which gives rise to the characteristic XAS spectrum. Copper ions in solution also tend to form octahedrally coordinated and other complexes. For example, D'Angelo et al. have investigated aqueous solutions of CuCl2 with increasing Cl−/Cu2+ ratios using EXAFS and shown that the spectra are consistent with Cu2+ octahedrally coordinated with oxygen and chlorine in the equitorial and axial positions in various combinations.26 Our data suggest something similar occurs in the atacamite case with the difference that the source is far less soluble than CuCl2 which can be made up to a 5 M solution at room temperature. One possibility is that the CuCl2(OH)4 or CuClOH(OH)4 octahedra in the atacamite persist in the sesquicarbonate solution as [CuCl (Cl or OH) (OH)4]4−. Another possible hypothesis to cover this behaviour would be the efflorescence of colloidally sized crystalline fragments from the surface into the fluid. In either case, it is clear that the simulated corrosion layer consists of an external thin crust which is removed rapidly by dissolution or efflorescence, and which is removed by the sesquicarbonate solution. Rutherford backscattering data which will be reported more fully elsewhere show that this crust is chlorine rich compared to the bulk atacamite. However, a persistent atacamite layer remains on the surface. Lamy19 has also noted the relative stability of synthetic atacamite under this treatment.
Fig. 9 shows the Ecorr data taken from each part of the immersion shown in Fig. 5. This shows a rapid rise in first 3 h or so, but it stabilizes after 7 h and is rather unresponsive to the electrolyte changes. Indeed, although chloride removal continues throughout the experiment, Ecorr actually becomes more negative after 20 h. This suggests that the chemistry at the copper atacamite interface stabilizes rather independently of removal of chlorides from the atacamite layer itself. A fresh atacamite layer is hard, and adheres well to the copper. However, if this is carefully chipped away, the surface spectrum reveals the presence of cuprite formed during the synthesis. This layer appears to thicken by direct interaction between the copper surface and the sesquicqarbonate, lowering the adhesion of the atacamite to the copper substrate; whilst the original layer can only be chipped off, treated layers can be peeled smoothly away. The Ecorr is probably characteristic of the cuprite layer but gives no information on the chloride removal.
 | ||
| Fig. 9 E corr data taken simultaneously with each immersion stage of the experiment in Fig. 5. The reference electrode was 3 M Ag/AgCl. | ||
Conclusions
Taken together, XRD and XAS of a surface with a thin layer of electrolyte above give the possibility of probing both the surface structure and the complex ions or colloidal material in the liquid. The addition of Ecorr measurements shows that changes in chemistry of the bulk corrosion product are not necessarily linked to this parameter which appears to be dominated by the chemistry of a (semi) conducting interfacial layer of cuprite. Therefore, Ecorr is not a good indicator of chloride removal.The XAS of synthetic atacamite and its response to a standard conservation treatment indicates that something in addition to simple atacamite is present in the layer. One component is rapidly removed into solution as a suspension or a complex ion with a very similar copper coordination to that in the solid. The other is resistant to treatment, and essentially insoluble, but may become detached as an interfacial layer of cuprite between the atacamite and the copper, originally due to the synthesis, becomes thicker.
Other corrosion products such as nantokite and cuprite also appear to go into the fluid, whilst retaining the local coordination of the solid material. Nantokite and cuprite are insoluble, although the former is believed to react with the sesquicarbonate to form cuprite if it remains on the copper surface. Here, we are probably seeing loss of particles of the corrosion product into suspension.
The precise chemistry of the synthetic atacamite on copper is under further investigation using XRD, XAS and RBS. The original techniques used to check the synthetic product19 would not have detected thin interfacial layers of cuprite or thin coatings on the bulk atacamite. The usefulness of testing a conservation technique on a material whose response is determined by a macroscopic structure which may be entirely different from that of natural atacamite is open to question.
Acknowledgements
The authors gratefully acknowledge the following for their help: Bart Schotte, Derrick Richards, Pieter van Hoe, Adrian Lovejoy (cell construction); Dr C. Degrigny, J. Robinson and R.F. Pettifer and Prof. E. Temmerman. The eCell was developed using private funds from EVA Surface Analysis (UK) and lately from the Paul Instrument fund. The work was supported by Ghent University (BOF grants) and would not have been possible without COST Action G8.References
- D. A. Scott, Copper and Bronze in Art. Corrosion, Colorants, Conservation, Getty Publications, Los Angeles, 1990 Search PubMed.
- D. L. Hamilton, Methods of Conserving Underwater Archaeological Material Culture. Conservation Files: ANTH 605, Conservation of Cultural Resources I. Nautical Archaeology Program, Texas A&M University, 1998. http://nautarch.tamu.edu/class/ANTH605 (last visited July 2006) Search PubMed.
- C. V. Horie and J. A. Vint, Stud. Conserv., 1982, 27, 185–186 CrossRef CAS.
- A. M. Pollard, R. G. Thomas and P. A. Williams, Stud. Conserv., 1990, 35, 148–152 CrossRef CAS.
- K. Leyssens, A. Adriaens, M. Dowsett, B. Schotte, I. Oloff, E. Pantos, A. Bell and S. Thompson, Electrochem. Commun., 2005, 7, 1265–1270 CrossRef CAS.
- A. Adriaens, M. Dowsett, K. Leyssens and B. Van Gasse, Anal. Bioanal. Chem., 2007, 387(3), 861–868 CrossRef CAS.
- M. Dowsett and A. Adriaens, Anal. Chem., 2006, 78, 3360–3365 CrossRef CAS.
- M. Kerkar, J. Robinson and A. Forty, J. Faraday Discuss. Chem. Soc., 1990, 89, 31–40 Search PubMed.
- M. Fleischmann, P. J. Hendra and J. Robinson, Nature, 1980, 288, 152–154 CrossRef CAS.
- A. J. Davenport, H. S. Isaacs, J. A. Bardwell, B. MacDougall, G. S. Frankel and A. G. Schrott, Corros. Sci., 1993, 35(1–4), 19–25 CrossRef CAS.
- S. Shah, A. Ward and A. J. Davenport, Corrosion, 1995, 51(1), 67–70 Search PubMed.
- S. Virtanen, P. Schmuki, A. J. Davenport and C. M. Vitus, J. Electrochem. Soc., 1997, 144, 198–204 CAS.
- A. Deb and E. J. Cairns, Fluid Phase Equilib., 2006, 241, 4–19 CrossRef CAS.
- R. J. K. Wiltshire, C. R. King, A. Rose, P. P. Wells, M. P. Hogarth, D. Thompsett and A. E. Russell, Electrochim. Acta, 2006, 50, 5208–5217.
- F. U. Renner, A. Stierle, H. Dosch, D. M. Kolb, T.-L. Lee and J. Zegenhagen, Nature, 2006, 439, 707–710 CrossRef CAS.
- I. Ascone, S. Zamponi, A. Cognigni, F. Marmocchi and R. Marassi, Electrochim. Acta, 2005, 50, 2437–2443 CrossRef CAS.
- K. O. Kvashnima, S. M. Butorin, A. Modin, I. Soroka, M. Marcellini, J. Nordgren, J.-H. Guo and L. Werne, Chem. Phys. Lett., 2007, 447, 54–57 CrossRef.
- K. Leyssens, A. Adriaens, C. Degrigny and E. Pantos, Anal. Chem., 2006, 78(8), 2794–2801 CrossRef CAS.
- C. Lamy, Stabilisation d'Objets Archéologiques Chlorurés en Alliage Cuivreux, Report ARC'Antique, Nantes, France, 1997 Search PubMed.
- G. Derbyshire, K. C. Cheung, P. Sangsingkeow and S. S. Hasnain, J. Synchrotron Radiat., 1999, 6, 62–63 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- A. Singh, K. Baur, S. Brennan, T. Homma, N. Kubo and P. Pianetta, Mater. Res. Soc. Symp. Proc., 2002, 716, B1.4.1–1.4.6 Search PubMed.
- A. Moen, D. G. Nicholson and M. Rønning, J. Chem. Soc., Faraday Trans., 1995, 91(18), 3189–3194 RSC.
- A. Bianconi, in X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES, ed. D. C. Koningsberger and R. Prins, Wiley and Sons, New York, 1998, pp. 573–662 Search PubMed.
- A. F. Wells, Acta Crystallogr., 1949, 2, 175–180 CrossRef CAS.
- P. D'Angelo, E. Bottari, M. R. Festa, H.-F. Noltung and N. V. Pavel, J. Chem. Phys., 1997, 107, 2807–2812 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |