Integration column: Microfluidic high-throughput screening
Sebastian J.
Maerkl
École Polytechnique Fédérale de Lausanne (EPFL), Institute of Bioengineering, Station 17, CH-1015, Lausanne, Switzerland. E-mail: Sebastian.maerkl@epfl.ch
First published on 9th December 2008
Abstract
Biology has always been a heavily technology limited field. Burgeoning fields such as systems biology require the development and implementation of new technologies, enabling high-throughput and high-fidelity measurements of large systems. Microfluidics promises to fulfil many of the requirements put forth. Here I will discuss the various approaches employed to date for performing high-throughput screening experiments on-chip, encompassing biochemical, biophysical, and cell-based assays.
ForewordIn this first issue of Integrative Biology we are introducing a column that provides a perspective on topics at the interface of biology and engineering. Six articles per year, written by outstanding young scientists or established leaders in their fields, will cover emerging tools for biologists, tools that facilitate new types of experiments that most biologists are currently not able to do. The focus will be on technologies and approaches (both experimental and theoretical) that are readily accessible and sufficiently robust for any biology lab to apply.In this first column entitled ‘Microfluidic high-throughput screening’, Sebastian Maerkl reviews microfluidic approaches for performing high-throughput screening experiments on-chip. This rapidly growing area promises to revolutionize the way biological screens are performed in the future. Matthias Lutolf, Editor, Integration column |
Introduction
Advances in our understanding of biological systems have invariably been linked to antecedent innovation in technology. This symbiosis dates from the early days of biology when Robert Hooke coined the term “cell” and the discovery of bacteria by Antonie van Leeuwenhoek. A preceding advance in engineering enabled both of these discoveries: the invention of the microscope by Dutch lens makers or possibly Galileo Galilei. More recent examples of how engineering and physics impact biology include the use of optics,1mass spectrometry,2,3 microarrays,4,5atomic force microscopy,6,7 optical tweezers,8–10 and micro- and nano-electromechanical systems (MEMS and NEMS).11,12 Of course biology itself gave rise to novel methodologies and approaches such as the polymerase chain reaction,13 molecular cloning, enzyme linked immunosorbent assays (ELISAs),14 chromatin immuno precipitation (ChIP),15–18 yeast two hybrid screening,19–21 and the green fluorescent protein.22In this review I will focus on microfluidics, one such recent engineering advance which has been applied to biology with ever increasing success. A microfluidic device, in its simplest form, consists of a channel with cross-sectional dimensions on the order of tens to hundreds of microns. Such devices, used for capillary electrophoresis, were first described in the early 1990’s.23,24 Since then the field has evolved ever more complex next-generation devices leading to the development of microfluidic large-scale integration (MLSI).25,26 MLSI allows for the integration of thousands of micromechanical valves on a single device with a postage-sized footprint. Microfluidics is becoming increasingly appealing to biologist due to recent trends in the biological sciences necessitating exceedingly large and comprehensive experiments.27–29 Several characteristics intrinsic to microfluidic systems make high-throughput screening of biological systems feasible and likely a standard practice in the near future:
(i) Unrivalled economy of scale. Microfluidics significantly lowers screening cost by drastically reducing reagent consumption by several orders of magnitude.
(ii) Through the reduction in length scales it is possible to perform thousands of experiments on a small footprint enhancing assay integration.
(iii) Microfluidics provides novel processes and detection mechanisms, not achievable on larger length-scales.
Because of these unique and promising characteristics microfluidic devices have been applied to essentially all major classical biological methods such as PCR,30–33 cloning,34 separations,35–38in vitro protein synthesis,39,40drug screening,41 cell based screens,42–48 small molecule synthesis,49 DNA/peptide synthesis,50–53 enzyme screens,54 protein interaction screening,55 and crystallography.56–59 Many of these examples describe the simple reduction of classical bench-top methods to microfluidic scales, which in many cases are accompanied by beneficial effects such as reduction in reaction times, increased sensitivity, and of course decreased reagent consumption. Microfluidic screens that allow for a drastically increased throughput as compared to their classic bench-top counterparts (if a classical counterpart even exists) are of particular interest, especially in light of the burgeoning field of systems biology. It is these high-throughput microfluidic screens that are discussed in this review. The goal of this review therefore is to provide an overview of recently developed microfluidic screening platforms, covering all major approaches to screening and high-throughput experimentation. This review necessarily covers a very small cross-section of the existing literature due to the large number of devices fabricated to date (Fig. 1), but nonetheless aims to achieve a comprehensive coverage of available approaches. Other reviews discuss the application of microfluidics to systems biology, particularly life cell imaging and cell culturing.60,61 Megason and Fraser specifically discuss imaging in systems biology in a separate review.28 Fluid physics, as observed on microfluidic platforms, is comprehensively summarized by Squires and Quake.62
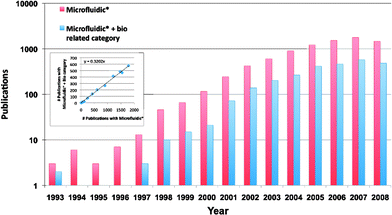 | ||
| Fig. 1 Publication history. A rough estimate of the number of journal articles published each year since 1993 as determined by using a simple keyword search (Microfluidic*) in Web of Science. The red bars show all articles recovered with microfluidic* as their topic. Blue bars show the fraction of articles falling into a biology related subject area. We can glean from this graph that: the field has grown exponentially over the last decade, and a constant 32% of microfluidic articles are biology related (inset). | ||
High-throughput microfluidic screens can be classified into two primary categories depending on whether the molecules or cells to be screened are introduced into the device randomly or systematically. Random introduction of material generally involves a heterogeneous mixture or suspension, which is directly introduced onto the device without pre-processing. The solution is aliquoted on-chip into sufficiently small volumes, such that each aliquot on average contains one quantum of the original heterogeneous mixture or suspension. Each aliquot can then be individually screened for function. Systematic approaches involve the introduction of homogeneous solutions to precise locations on a device. These solutions may then be screened directly, or can be combined to give rise to a larger screening space. Both random and systematic screening methods can be further sub-classified by whether the reactions take place in series or in parallel. Below I will give a few examples covering all four screening approaches and discuss advantages and disadvantages involved with each, providing a comprehensive overview to biologists interested in identifying and applying the most appropriate microfluidic approach to their specific experimental requirement.
Random screens
Serial devices
Probably the most familiar serial-random screening approach is flow cytometry.63–65 Here, a heterogeneous mixture of cells is introduced into a capillary channel, which permits only single cells to pass by a detector with an inter-cell distance dependent on the initial cell density. More sophisticated flow cytometers make use of laminar flow to hydro-dynamically focus the cells into the centre of the channel. One or several laser lines are used to excite specific fluorophores, which were previously attached to the cells using fluorescently labelled antibodies or fluorescent dyes,66 and the emitted photons are detected with photomultiplier tubes (PMTs). Fluorescent proteins such as GFP have also been used for detection.67 In a special type of flow cytometer, the fluorescent-activated cell sorter (FACS), cells are flown past one or more T-junctions after the detector. At these junctions cells with a specific pre-determined fluorescent fingerprint can be sorted from the bulk.The throughput of flow cytometry is one of the highest achieved to date. Commercially available flow cytometers and FACS sorters can measure and sort on the order of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells/second. No other device is more adapt at quantitative measurements of whole cell fluorescence in multiple wavelengths. Additionally, FACS is an exceedingly powerful approach for enriching rare cells, such as stem cells,68 from a heterogeneous population for downstream processing.
000 cells/second. No other device is more adapt at quantitative measurements of whole cell fluorescence in multiple wavelengths. Additionally, FACS is an exceedingly powerful approach for enriching rare cells, such as stem cells,68 from a heterogeneous population for downstream processing.
But performance comes with a hefty price tag, causing most flow cytometers and FACS instruments to be found in core facilities rather than in individual research labs. Recently, microfluidic research groups have manufactured one-time use microfluidic FACS.69–72 But even though the performance of these simple and inexpensive devices lags behind that of commercial machines, there is no fundamental reason why, with additional improvements, these microfluidic throwaway FACS can not reach acceptable levels of performance making them effective alternatives for many basic flow cytometry and sorting experiments.
A more fundamental limitation of flow cytometry is the fact that individual cells are measured only once, and can not be interrogated over a period of time (a cytometer developed by Voldman et al. based on dielectropheresis for cell trapping is one exemption to this rule69). Researchers have measured populations of cells over a period of time, but the time-dependent information from a single cell is not accessible.67 Finally, flow cytometry generally lacks the high-spatial resolution necessary for determining sub-cellular localization of proteins.28 For experiments requiring the measurement of these types of parameters it is necessary to apply either random or systematic parallel cell arrays (described below).
Parallel devices
In random-parallel devices a heterogeneous mixture is introduced and aliquoted into sufficiently small volumes so that each unit volume on average contains a single cell or molecule, thus homogenizing the mixture. We previously applied such a screening methodology to the directed evolution of an enzyme (Fig. 2).25 In directed evolution73 an open reading frame (ORF) coding for a protein (generally an enzyme) of interest is randomly mutagenized, commonly via error-prone PCR. The resulting heterogeneous mixture of PCR products is then cloned into a vector, which in turn is transformed into a host, generally Escherichia coli (E. coli). The heterogeneous mixture of cells is then “purified” by spread plating the cells. The resulting clonal colonies are picked and placed into multiwell plates, where they can be grown and the mutant proteins tested for function using an appropriate assay. This is an extremely labour intensive process, particularly because tens of thousands of clones should be picked and assayed in order to ascertain that improved versions of the original protein are found. We designed a complex microfluidic device that allowed us to commence the screening and selection step directly on the heterogeneous mixture of transformants.25 This was achieved by introducing the cell mixture into a long serpentine microfluidic channel. Once loaded, the channel could be compartmentalized into 256 chambers of 375 pL and doing so would give on average ∼1 cell/chamber (if the initial cell density was adjusted appropriately). A specific substrate was introduced into each chamber by passive diffusion which, when turned-over by the enzyme expressed in each cell, becomes fluorescent. The entire device could be quantitated using a standard DNA microarray scanner. The recovery of the clone of interest was accomplished using a microfluidic analogue of a multiplexer allowing each chamber to be individually addressed. Single E. coli cells were successfully recovered from the device followed by growth on an LB-agar plate demonstrating the feasibility of the approach. | ||
| Fig. 2 MLSI device for single cell screening. (a) An optical micrograph of the device with its channels filled with food dyes for visualization. The device contains 2056 micromechanical valves on a 2.5 cm × 2.5 cm footprint. (b) Heterogeneous populations of E. coli cells could be introduced onto the device followed by compartmentalization and imaging. (c) Once individual cells were trapped in one of the 256 chambers of the device, an enzyme assay could be performed, generating signal only in chambers containing one or more cells. (d) Quantitation of the observed signals normalized by the number of cells from which the signal originated gave rise to broadly distributed signal levels (adapted with permission from ref. 25). | ||
These measurements present one of the few single cell high-throughput enzyme measurements to-date. But, due to the large intrinsic noise of the biological system, we observed a broad distribution of signal even for a homogeneous population of cells, all expressing wild type enzyme.74 Noise on the single cell level can arise, amongst other factors, from varying plasmid copy numbers, which translate into differing enzyme concentrations, or from cells being in different parts of the cell cycle (or dead). To correct for this it is necessary to either normalize the observed signal with the exact concentration of enzyme present (rather than assuming that each cell roughly contains the same number). A second approach would be to grow clonal populations of cells on-chip starting from the compartmentalized single cells.
Throughput of less than 256 (not all chambers contain exactly a single cell due to the random seeding process) for this proof-of-principle experiment must also be improved in order to make it a feasible method for screening in directed evolution. This could be accomplished by using pooling or enrichment strategies, using a modified microfluidic design. Such an optimized screening platform would provide significant savings in assay time, labour, and cost, when compared to the currently used approach.
Another recently developed device by Lee and co-workers is capable of generating large arrays of cells, randomly immobilized in small fluidic traps (Fig. 3).43 The fluidic traps consist of small, cup-shaped polydimethylsiloxane (PDMS) posts hanging from the roof of the device. Each post is suspended roughly 2 μm above the floor of the channel. This gap is sufficient to allow fluid to flow beneath the post. If a solution of cells is added to the device the cells will be dragged next to the cup-shaped post, and trapped in that location. A better than random trap occupancy was observed for single cells on these devices. Lee and co-workers were able to trap both HeLa and Jurkat cells on the device and ran time-dependent enzyme assays on the trapped cells. This trapping device is a simple, yet efficient approach to generating large random arrays of cells. Furthermore, by turning of the flow, or reversing it, the arrays can be regenerated, allowing for multiple consecutive trapping runs. One drawback of the approach is the fact that cells being screened can not be specifically recovered from the device, as would be necessary when screening for directed evolution, or if downstream processing of a single cell is required. Also, due to the method of trapping, arraying smaller cells such as bacteria may prove difficult. But for many applications not requiring recovery or trapping of small cells, this device is a perfect solution to generating random screening diversity.
 | ||
| Fig. 3 Microfluidic cell isolation array. Eight parallel channels containing trapping arrays were used to dynamically trap both HeLa as well as Jurkat cell lines. Trapping is accomplished with cup-shaped posts hanging from the roof of the channel. Depending on the size of the cup, one or multiple cells may be immobilized in each trap (adapted with permission from ref. 43). | ||
Random screens summary
Random high-throughput screening devices work on the principle of serial analysis of small sample volumes or use homogenization by compartmentalization in combination with parallel screening to simplify a complex input mixture. Random-serial devices such as flow cytometers are capable of extremely high-throughput and have been widely applied to biology. Random-parallel devices are more complex and thus generally have drastically reduced throughput, but they allow for complex fluidic assays to be performed and permit the continuous interrogation of each compartment.It probably also has become apparent that random screening devices work well using cells as input. This is because cells nicely package their contents, and their length-scales are sufficiently matched to the length scales of the microfluidic devices that they become relatively easy to manipulate. Cells are also readily detectable using standard microscopy. There is no limiting factor that would prevent single molecules from being assayed using similar approaches as described above. Screening of single molecules does require that the genotype and phenotype remain linked throughout the assay. This can be accomplished using compartmentalization. One gedanken assay would for example include compartmentalization of single plasmids coding for an enzyme into microfluidic chambers followed by on-chip in vitro transcription/translation followed by an appropriate readout. If necessary, additional steps such as PCR for amplification before or after protein synthesis could be included as well.
Flow cytometry is not directly applicable to those types of experiments because the genotype -phenotype linkage is not preserved. But, when combined with water/oil (w/o) emulsions or microfluidic droplet generation, flow cytometry has the potential of becoming an exceedingly high-throughput approach to screening complex molecular libraries.
Systematic screens
Serial devices
Water/oil (w/o) emulsions have recently been applied with great effect to the clonal amplification of single DNA molecules for use in second-generation sequencing.76 A wide range of droplet sizes from tens of pL to nL can be formed by mixing two immiscible liquids. Monodispersed droplets or plugs can be formed precisely and on the order of thousands per second on microfluidic devices.77–81 More importantly, each droplet can be formulated systematically from a combination of several input streams .56,57,75 Once generated, droplets can be manipulated on an individual basis, such as merging adjacent droplets,82,83 dividing a single droplet,82,84 and even extracting the contents of a single droplet into a continuous fluid stream.85 These characteristics make on-chip droplet formation and handling interesting for high-throughput screening.Rustem F. Ismagilov’s group at the University of Chicago has successfully applied dynamic droplet formation to proteincrystallography (Fig. 4).56,57 One has to test a large combinatoric space of precipitating agents over a wide range of concentrations to find a condition in which a given protein will crystallize. Exacerbating that difficulty is the fact that many proteins of interest are also exceedingly difficult to purify in quantities required for standard crystallization screens. Reducing the reaction volumes from tens of microliters down to sub-nanoliter volumes entails a huge benefit by either requiring less protein for the same number of screens or allowing for a larger search space with the same amount of material.
 | ||
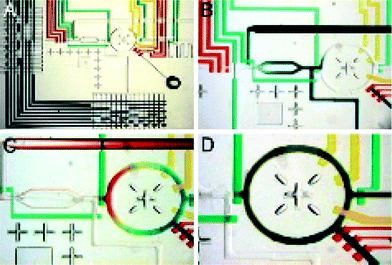
| Fig. 4 Microfluidic droplets and plugs. Two approaches for generating water/oil or water/oil/gas droplets and plugs. (a) A scheme for the rapid generation of varying ratios between three input streams, for the comprehensive screening of crystallization conditions. The photos on the left show the entire device as well as crystals contained in small water droplets. (b) Water, oil, or gas (air) plugs can also be used for high-throughput screening of crystallization conditions (adapted with permission from 57 and 75). | ||
Ismagilov and co-workers generate programmable droplets by combining 3–4 water streams containing protein, buffer and various precipitating agents. Mixing ratios are adjusted by changing the relative pressures in these feed channels. The laminar water streams are then sheared off by the oil phase and the contents of the droplets either mix diffusively or by active mixing. Droplets are routed along a holding channel where they can be stored and interrogated after a given amount of time. Glass capillaries have been used for storage channels to avoid solution evaporation,57 which was observed when channels made from PDMS were used for long term storage.56 These initial microbatch reactions spanned a low dimensional phase space of conditions consisting of two crystallization agents. Increasing the dimensionality of this system to tens of crystallization reagents should nonetheless be possible and probably necessary to make the approach more broadly applicable.
Vapour-diffusion reactions were also created by placing two droplets with differing osmotic pressures in close proximity.57Water moves by diffusion from one droplet to its neighbour causing a time-dependent change in volume and thus concentration of each droplet. With this method it is possible to dramatically increase the number of conditions tested by continuously moving through a concentration space of protein and precipitating agent, while the ratio of the two remains constant. It is thus only necessary to set up differing ratios, whose concentration can be changed post-programming via evaporation or condensation. Finally, crystals formed in droplets and stored in glass capillaries can be placed directly into a beam line and subjected to X-ray diffraction, nicely integrating the process.
Droplet generation for high-throughput screening of crystallization conditions is a powerful approach that can be extended to the screening of other biochemical reactions. One limitation of the method is the fact that to-date only a limited number of fluid streams have been used for generating low-dimensional mixtures of 3–4 substances. Higher dimensional mixtures of all commonly used crystallization reagents would be a major advance to the system, and likely achievable. A more fundamental limitation is the water/oil interface itself, which can potentially interfere with a large number of reactions, because of loss of hydrophilic molecules to the oil phase or aggregation of labile molecules at the w/o interface. This may be solved to a certain extent with appropriate chemistries.86 But, until a generic solution to this issue has been found, w/o emulsions and droplets are likely to be applicable only to a largely reduced number of reactions than would otherwise be possible.
Stephen Quake’s group at Stanford University has taken a different approach to protein crystallization by screening not directly for crystallization conditions, but rather for conditions that precipitate protein from solution (Fig. 5).59 Knowledge of the complete phase space of a protein should in principal aid crystallization efforts, as protein crystallization is likely to occur at the interface between the soluble and precipitating phases.
 | ||
| Fig. 5 Formulater device. (a) Optical micrograph of a formulator device. The central component is a mixing ring, which can be loaded with differing ratios of up to 32 solutions selected with the multiplexers seen on the left and bottom of the image. (b–d) Demonstration of how the mixing ring can be loaded with three different solutions (blue, yellow and red), followed by rapid mixing (d). (reproduced with permission from ref. 59). | ||
The microfluidic device designed by Hansen et al. is capable of rapidly combining and mixing arbitrary ratios of 32 stock reagents. Each reagent can be selected on-chip via the use of a microfluidic multiplexer. Once selected, precise amounts of the reagent can be metered out by an integrated peristaltic pump and introduced into a ring-structure. Once all reagents including the protein are added, rapid mixing (<3 s for aqueous and <6 s for viscous solutions) is achieved by pumping the solution around in a circle. Pumping causes the fluid lamina to fold over one another, effectively reducing the diffusion distance required for complete randomization of the molecules. Protein precipitation is detected by simple absorption measurements. A proof-of-principle experiment consisted of measuring the phase space of xylanase by screening ∼4000 precipitating reactions in quadruplicates, with each run requiring ∼20 h (giving rise to a throughput of roughly 3.3 Hz) and consuming about 8 μL of protein solution. Hansen et al. then went on to show that rationally chosen crystallization conditions from their phase-space diagrams resulted in an increased hit density as compared to standard sparse matrix screens. For screening protein crystallization conditions directly, the authors developed a systematic-parallel screening platform capable of performing 144 simultaneous crystallization screens by free interface diffusion.58
The formulator device is a good example of how complex fluidic manipulations can be achieved on a microfluidic platform. The fluidic complexity allowed the authors to combine roughly an order of magnitude more input streams than has been shown with droplet-based screening. On the other hand, the throughput of the formulator will most likely remain significantly below the conceptually achievable throughput of droplet-based devices. Furthermore, formulator throughput is strongly dependent on the length of time required for each reaction to take place. Slow reactions such as protein synthesis or crystallization can only be achieved if a storage loop87,88 is added to the functionality of the formulator. A fusion device consisting of a formulator upstream of a droplet generator could be an optimal solution, eliminating the major drawbacks of both approaches.
Both the droplet based devices and the formulator could in principle be applied to a plethora of screening applications. These devices are particularly appropriate if exceedingly high-throughput, on the order of tens of thousands of reactions, is required. The main limitation of these approaches is the fact that diversity is achieved, in both cases, by mixing ratios of a rather small number of input solutions. Of course this is perfectly appropriate for experiments such as protein crystallization screens. When thousands of unique samples need to be interrogated the only choice probably lies with systematic-parallel devices described below. Yet, as alluded to above, the most powerful serial screening experiments will be of the random-serial type, which could be fused with these two methods to great effect.
Parallel devices
Systematic-parallel screening devices generally consist of hundreds to thousands of unit cells. In each unit cell a unique reaction is taking place that can be either generated by pair wise combination of a small number of input reagents or programmed directly using a microarray-based method we recently developed.55Liu et al. developed a device based on pair wise combinations of input solutions to generate 400 unique PCR reactions in parallel (Fig. 6).31 In a proof-of-principle experiment Liu generated a precise pattern of 100 positive and 300 negative PCR reactions. The device allows for the separate introduction of 20 primer pairs, 20 templates, and one buffer solution. These three solutions can be mixed on-chip followed by thermal cycling. Using this matrix approach requires only (N + M) + 1 pipetting steps to generate an entire N × M matrix of combinations. N × M arrays do indeed drastically reduce the number of necessary pipetting steps, but become cumbersome when N or M become large (>20), as each input has to be individually loaded and addressed. Better integration with multiwell plate formats for this step can potentially increase this number close to 100, giving rise to a feasible matrix size of 10000. Finally, the matrix approach is only applicable to screens where complexity can be generated by combining multiple solutions (similar to the systematic-serial screens), but it cannot be used when large libraries of unique components such as drug or other molecular libraries need to be tested directly.
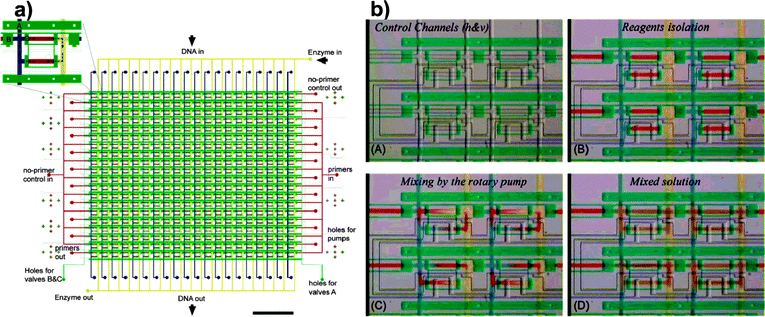 | ||
| Fig. 6 Matrix device. The device shown schematically in (a) is capable of running 400 individual PCR reactions, generated by combining 20 different primer pairs with 20 different templates. (b) The primers, DNA templates and buffer solution are combined on a chip via active mixing (adapted with permission from ref. 31). | ||
Using a similar strategy, Lee et al. devised a microfluidic device capable of generating 64 parallel nanoliter sized chambers for culturing mammalian cells (Fig. 7).42 In this approach eight columns could be individually loaded with cells followed by continuous perfusion of medium along the rows of the device. A concentration gradient89 could be generated across the rows to give rise to 64 distinct environments covering both genotypic and environmental diversity. The authors studied culturing conditions of HeLa cells covering eight different serum levels. Cell seeding density could be controlled to give rise to initial densities varying between 19 to 42 cells per chamber and was independent of chamber index. Cells were grown on the device for a week without significant drops in cell viability making this approach applicable to experiments requiring long term cell culturing. Each chamber is optically accessible allowing for the determination of cellular phenotypes such as morphology and fluorescent reporter gene expression. The approach suffers from the same limitations as the approach taken by Liu et al. in that only a limited number of unique solutions can be introduced. Additionally, the use of a gradient generator limits the second dimension to concentration gradients of a single substrate of interest, but this can be easily modified to include inputs for eight unique solutions.
 | ||
| Fig. 7 Microbioreactor array. (a) Image of the microbioreactor array device, filled with green dye for visualization of the flow channels. The device contains 64 individually addressable reactors (b). The device was used to screen cell growth at eight different concentrations of serum (c). (adapted with permission from ref. 42). | ||
Gómez-Sjöberg et al. implemented a microfluidic platform capable of 96 independent culturing conditions in 60 nL chambers (Fig. 8).44 Here the authors generate complexity by serially addressing each chamber. Cells could be loaded to precise densities by consecutive loading cycles. The precision of loading being dependent on the initial cell density, with lower densities giving rise to more precise metering, while requiring more cycles to be performed for reaching higher densities. Once inoculated, each chamber could be fed with a complex mixture of up to 16 components mixed during the loading process. The entire device, including optical readout of each chamber, was highly automated, using software based control of the feeding schedule and device readout. The authors studied the differentiation of human primary mesenchymal stem cells (hMSCs) by varying both the initial seeding densities as well as the duration of stimulation. These complex culturing conditions are difficult to achieve using classical high-throughput culturing methods based on multiwell plates. Furthermore, for rare samples, microfluidic culturing entails huge benefits due to enormous reduction in the required material.
 | ||
| Fig. 8 Micrfluidic cell culture system. A cell culturing system capable of generating 96 parallel, highly complex culturing conditions is depicted here. The device consists of 96 chambers, individually addressable by a multiplexer. Each chamber can be fed serially with a different medium recipe. Feeding recipes and schedules, as well as chip imaging are all completely automated. (Reproduced with permission from ref. 44. Copyright 2007, American Chemical Society). | ||
Both cell-culturing devices discussed above achieve medium throughput on the order of a single 96-well plate. The advantage of the microfluidic approaches lie in the reduction of sample required and the increase in assay complexity. Being able to study multiple cell reaction chambers with both high temporal as well as spatial resolution is imperative in systems biology where network responses can occur both rapidly and on a scale only observable on the single cell level.90–92 The ability to scale-up single cell measurements to include a large number of environmental conditions is paramount to understanding what the cellular responses are to these environments. Even though these examples only achieved relatively low throughput on the order of 64 and 96 parallel reactions, there is no doubt that future designs will achieve higher parallelization.
We have recently developed a high-throughput microfluidic platform for performing highly parallelized protein interaction measurements (Fig. 9) coupled to a novel detection mechanism based on the mechanically induced trapping of molecular interactions (MITOMI) capable of detecting transient interactions.55 The devices consist of thousands of reaction chambers, which are individually programmed using a microarray. Microarrays are particularly well suited for integration with microfluidics since the process of microarraying consolidates existing libraries housed in multiwell plates on a small footprint that is compatible with microfluidic devices. Aligning of these printed microarrays to microfluidic devices programs each chamber with a single spot and compartmentalizes it, eliminating any potential for downstream cross-contamination. Finally, sample consumption is minimized as spotting consumes as little as 250 nL of sample, from which hundreds of sub-nanoliter spots can be generated. Other microfluidic methods generally suffer from large dead-volumes when coupling reagent reservoirs to the fluidic devices, leading to dead-volumes in the tens of microliters. Generating microarrays using standard spotting techniques is also extremely modular, allowing for the arraying of proteins, DNA, small molecules, and even colloidal suspensions.
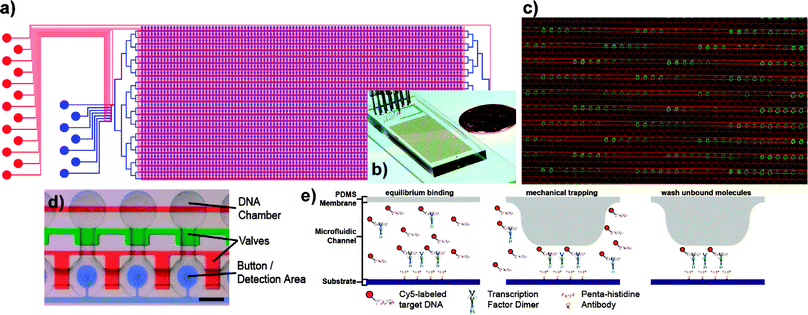 | ||
| Fig. 9 MITOMI device. (a) Schematic and (b) photo of the MITOMI device. The flow layer is shown in blue, the control layer is denoted in red. The device contains 2400 unit cells (d) and is controlled by 7233 valves. Each unit cell is programmed by aligning the device to a spotted microarray (c); DNA spots of varying sequence and concentration (green) are aligned to the microfluidic device, whose outlines are visible in red. (e) A novel detection mechanism based on the mechanically induced trapping of molecular interactions (MITOMI). MITOMI allows for thousands of parallel high-precision interaction measurements to be performed on a single device. ((a,d,e) adapted with permission from ref. 55). | ||
Using this approach of programming microfluidic devices with microarrays we measured comprehensive binding energy landscapes for several transcription factors by experimentally determining the affinity of several transcription factors to 464 different double-stranded DNA oligomers. Each oligomer was spotted in duplicate at six different concentrations. 2400 of such affinity measurements were taken in parallel on a single device, resulting in a total of over 41000 unique affinity measurements determined on 17 devices. The transcription factor to be tested is synthesized on-chip from linear expression templates by in vitro transcription/translation. Fluorescent quantitation of the trapped material is done using a DNA microarray scanner, slightly modified to accept the microfluidic device. Such comprehensive measurements of the biophysical characteristics of a transcription factor allowed us to determine basic parameters governing transcription factor–DNA interactions, as well as to reconstruct biological networks in silico using the binding energy landscapes determined in vitro.
Einav et al. applied this approach to characterizing the RNA binding of a hepatitis C membrane protein NS4B.41 NS4B was found to specifically bind the 3’UTR of the negative strand of the viral RNAgenome , implicating it in viral assembly. This in turn made it an interesting candidate for drug discovery. To this end Einav et al. spotted a commercially available small compound library encompassing 1280 members and tested each of these compounds for interference between NS4B and RNA binding. The screen yielded 18 compounds that interfered with binding in vitro, of which one was shown to inhibit viral replication in vivo without negatively impacting cell viability.
Because the spotted arrays are compartmentalized it becomes feasible to spot multiple solutions onto a single spot on the array, which is consequently isolated on-chip. This allows users to generate multiplexed assays similar to those accomplished using the matrix device described by Liu et al. at the beginning of the section.31 It is not only possible to test N × M combinations, but one can also generate higher-dimensional arrays consisting of N × M × K samples by spotting three or more unique substances on a single spot.
We took advantage of being able to generate arbitrary combinations of two samples by co-spotting a library of linear expression templates coding for 95 different transcription factor mutants with a library of 64 target DNA sequences (Maerkl and Quake, submitted). Each chamber, containing linear expression template for one mutant and one target DNA sequence, is flooded with in vitro transcription–translation reaction, which synthesizes the mutant protein. The transcription factor is consequently immobilized on the surface and binding to the target DNA sequence is measured. This approach allowed us to measure over 6000 protein–DNA interactions using in situ synthesized transcription factor mutants.
In a second example of how co-spotting can be applied to biological network characterization, Gerber et al. co-spotted all combinations of 43 linear expression templates, coding for 43 different Streptococcus pneumoniaeproteins (Gerber et al., accepted to Nature Methods). The proteins were tested for direct protein–protein interactions, uncovering a rich protein interaction network.
The use of microarrays for programming microfluidic devices is a rapid approach with many advantages, including simple integration with existing libraries, extremely low sample consumption, high-throughput, and modularity. We have shown that a number of screening assays are possible with this approach and this number is likely to broaden considerably in the future.
Meyvantsson et al. recently developed a device to allow researcher to perform basic microfluidic experiments using standard laboratory liquid handling equipment such as pipettes and robotic platforms.93 The device makes use of passive pumping to introduce liquids into the 96–192 channels located on each device. The channel interface follows the standard micro-titer plate standard to facilitate use with medium to high-throughput eight channel pipettes and robotic liquid-handling platforms, respectively. Simple fluidic manipulations such as pumping, routing, compartmentalization, and laminar flow could be achieved. Furthermore, the authors cultured mammalian cells for 5 days with media additions every 24 h. Meyvantsson et al. realized a low-tech microfluidic platform that can be used by researchers lacking specialized backgrounds in microfluidics and microfabrication, allowing these platforms to be more widely used than other more complex system (the issue of increasing accessibility to microfluidic platforms is also discussed in the conclusions).
Finally, centrifuge-based microfluidic platforms have also been broadly applied to standard methods in biology, such as ELISA assays,94 cell viability assays, PCR, and mass spectrometry, which have all been comprehensively reviewed by Madou.95 CD based systems generally consist of several dozens of parallel reaction chambers arranged radially on a CD type substrate. Assays are capable of mixing several input streams, serial addition of samples, hydrophobic and capillary based valving, and precise metering.95
Systematic devices summary
Systematic screens are useful for many applications since the exact identity of each reaction is known a priori. It is thus possible to screen a large number of reactions, which as a whole provide an answer to complex problems and systems. Systematic screens often lag behind random screens in throughput. This is acceptable though, as many libraries that exist today comprise on the order of thousands to tens of thousands of members. Systematic screens have been developed for chemical,41 biochemical,55 and cell based assays,42,44 indicating their versatility. As fields such as systems biology put ever more emphasis on global screens of cellular function and biochemistry, microfluidic based systematic screens are likely to become more broadly used in the near future.Hybrid devices
Microfluidic screens of course do not have to be exclusively serial or parallel. Hybrid devices can easily be generated by either parallelizing multiple serial screening experiments on a single platform or by running a parallel device multiple times in series. Droplet devices such as those applied to systematic crystallization screens could for example be parallelized, allowing multiple precipitating agents and combinations thereof to be screened simultaneously. Parallel devices on the other hand can often be serialized, giving rise to large gains in throughput. The cell screening devices described by Lee et al.42 and Gómez-Sjöberg et al.44 are already capable of such serialization through performing a number of varying perfusion/feeding experiments.Random and systematic screens can also be combined. One example would be an initial round of screening and selection on a heterogeneous population of cells, followed by systematic screens on the selected sub-population. Rohde et al. propose such an approach by combining a number of devices they designed for screening and handling C. elegans, including a high-throughput sorter, a parallel screening device, and a multiwell plate interface.48
Conclusions
The field of microfluidics has made significant progress over the last decade, leading to the development of highly-integrated screening platforms. A number of proof-of-principle experiments have shown that this technology can be applied to systems biology,55,92,96,97 promising to widen many bottlenecks that limit advances in biology today.Research labs will continue to develop novel and more highly-integrated devices in the future albeit dissemination of this technology has progressed at a much slower pace. It is absolutely necessary to port developments made in the research lab to commercially available platforms. Developing advanced world-to-chip interfaces, and simplifying chip usage will be paramount in achieving broad acceptance of this technology by the intended users. Arguably, these kinds of improvements are most appropriately solved by companies interested in commercializing microfluidics, while the research community continues to explore the frontiers of the field of microfluidics. Most universities have the required infrastructure for fabricating many, if not all, of the devices described herein. For those universities that lack the infrastructure or expertise, microfluidic foundries are available at Caltech (http://kni.caltech.edu/foundry/) and Stanford University (http://thebigone.stanford.edu/foundry/). These foundries will fabricate custom designed molds and PDMS based microfluidic devices at minimal cost to the user.
Another critical component in the development of fluidic technology will be the integration with detection mechanisms. Many promising detectors have recently been demonstrated, either based on solid state optics,1 MEMS/NEMS devices,11 or CMOS based chemical sensors.98 Successful integration of novel technologies with microfluidics will generate more compact and portable devices, and may result in higher detection sensitivities than those currently achievable.
The potential of microfluidics for impacting the biological sciences is apparent. A number of microfluidic based assays have already provided novel insights into the structure and function of biological systems, and the field of microfluidics will continue to impact biology, biology has long since outgrown its 8-strip PCR tubes and 96 well plates.
References
- A. M. Armani, R. P. Kulkarni, S. E. Fraser, R. C. Flagan and K. J. Vahala, Science, 2007, 317, 783–787 CrossRef CAS.
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270, 467–470 CrossRef CAS.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- A. C. Gavin, M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J. M. Rick, A. M. Michon, C. M. Cruciat, M. Remor, C. Hofert, M. Schelder, M. Brajenovic, H. Ruffner, A. Merino, K. Klein, M. Hudak, D. Dickson, T. Rudi, V. Gnau, A. Bauch, S. Bastuck, B. Huhse, C. Leutwein, M. A. Heurtier, R. R. Copley, A. Edelmann, E. Querfurth, V. Rybin, G. Drewes, M. Raida, T. Bouwmeester, P. Bork, B. Seraphin, B. Kuster, G. Neubauer and G. Superti-Furga, Nature, 2002, 415, 141–147 CrossRef CAS.
- Y. Ho, A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, L. Yang, C. Wolting, I. Donaldson, S. Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C. Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A. Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H. Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, B. D. Sorensen, J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D. Durocher, M. Mann, C. W. Hogue, D. Figeys and M. Tyers, Nature, 2002, 415, 180–183 CrossRef CAS.
- M. Rief, F. Oesterhelt, B. Heymann and H. E. Gaub, Science, 1997, 275, 1295–1297 CrossRef CAS.
- M. Radmacher, R. W. Tillmann, M. Fritz and H. E. Gaub, Science, 1992, 257, 1900–1905 CAS.
- M. D. Wang, M. J. Schnitzer, H. Yin, R. Landick, J. Gelles and S. M. Block, Science, 1998, 282, 902–907 CrossRef CAS.
- K. Svoboda, C. F. Schmidt, B. J. Schnapp and S. M. Block, Nature, 1993, 365, 721–727 CrossRef CAS.
- S. R. Quake, H. Babcock and S. Chu, Nature, 1997, 388, 151–154 CrossRef.
- R. McKendry, J. Y. Zhang, Y. Arntz, T. Strunz, M. Hegner, H. P. Lang, M. K. Baller, U. Certa, E. Meyer, H. J. Guntherodt and C. Gerber, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9783–9788 CrossRef CAS.
- T. P. Burg, M. Godin, S. M. Knudsen, W. Shen, G. Carlson, J. S. Foster, K. Babcock and S. R. Manalis, Nature, 2007, 446, 1066–1069 CrossRef CAS.
- J. M. Bartlett and D. Stirling, Methods Mol. Biol., 2003, 226, 3–6 Search PubMed.
- R. S. Yalow and S. A. Berson, J. Clin. Invest., 1960, 39, 1157–1175 Search PubMed.
- M. H. Kuo and C. D. Allis, Methods, 1999, 19, 425–433 CrossRef CAS.
- D. K. Pokholok, C. T. Harbison, S. Levine, M. Cole, N. M. Hannett, T. I. Lee, G. W. Bell, K. Walker, P. A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Lewitter, D. K. Gifford and R. A. Young, Cell, 2005, 122, 517–527 CrossRef CAS.
- S. Impey, S. R. McCorkle, H. Cha-Molstad, J. M. Dwyer, G. S. Yochum, J. M. Boss, S. McWeeney, J. J. Dunn, G. Mandel and R. H. Goodman, Cell, 2004, 119, 1041–1054 CAS.
- G. Euskirchen, T. E. Royce, P. Bertone, R. Martone, J. L. Rinn, F. K. Nelson, F. Sayward, N. M. Luscombe, P. Miller, M. Gerstein, S. Weissman and M. Snyder, Mol. Cell. Biol., 2004, 24, 3804–3814 CrossRef CAS.
- P. James, J. Halladay and E. A. Craig, Genetics, 1996, 144, 1425–1436 CAS.
- P. Uetz, L. Giot, G. Cagney, T. A. Mansfield, R. S. Judson, J. R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, A. Qureshi-Emili, Y. Li, B. Godwin, D. Conover, T. Kalbfleisch, G. Vijayadamodar, M. Yang, M. Johnston, S. Fields and J. M. Rothberg, Nature, 2000, 403, 623–627 CrossRef CAS.
- T. Ito, T. Chiba, R. Ozawa, M. Yoshida, M. Hattori and Y. Sakaki, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4569–4574 CrossRef CAS.
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802–805 CrossRef CAS.
- D. J. Harrison, K. Fluri, K. Seiler, Z. H. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895–897 CrossRef CAS.
- A. T. Woolley and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 11348–11352 CAS.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS.
- H. Kitano, Science, 2002, 295, 1662–1664 CrossRef CAS.
- S. G. Megason and S. E. Fraser, Cell, 2007, 130, 784–795 CrossRef CAS.
- T. Ideker, T. Galitski and L. Hood, Annu. Rev. Genom. Hum. Genet., 2001, 2, 343–372 Search PubMed.
- J. Liu, M. Enzelberger and S. Quake, Electrophoresis, 2002, 23, 1531–1536 CrossRef CAS.
- J. Liu, C. Hansen and S. R. Quake, Anal. Chem., 2003, 75, 4718–4723 CrossRef CAS.
- E. A. Ottesen, J. W. Hong, S. R. Quake and J. R. Leadbetter, Science, 2006, 314, 1464–1467 CrossRef CAS.
- E. T. Lagally, C. A. Emrich and R. A. Mathies, Lab Chip, 2001, 1, 102–107 RSC.
- J. W. Hong, V. Studer, G. Hang, W. F. Anderson and S. R. Quake, Nat. Biotechnol., 2004, 22, 435–439 CrossRef CAS.
- R. Ashton, C. Padala and R. S. Kane, Curr. Opin. Biotechnol., 2003, 14, 497–504 CrossRef CAS.
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS.
- L. R. Huang, J. O. Tegenfeldt, J. J. Kraeft, J. C. Sturm, R. H. Austin and E. C. Cox, Nat. Biotechnol., 2002, 20, 1048–1051 CrossRef CAS.
- B. H. Weigl and P. Yager, Science, 1999, 283, 346–347 CrossRef.
- P. S. Dittrich, M. Jahnz and P. Schwille, ChemBioChem, 2005, 6, 811-+ CAS.
- S. J. Maerkl and S. R. Quake, Science, 2007, 315, 233–237 CrossRef CAS.
- S. Einav, D. Gerber, P. D. Bryson, E. H. Sklan, M. Elazar, S. J. Maerkl, J. S. Glenn and S. R. Quake, Nat. Biotechnol., 2008, 26, 1019–1027 CrossRef CAS.
- P. J. Lee, P. J. Hung, V. M. Rao and L. P. Lee, Biotechnol. Bioeng., 2006, 94, 5–14 CrossRef CAS.
- D. Di Carlo, N. Aghdam and L. P. Lee, Anal. Chem., 2006, 78, 4925–4930 CrossRef.
- R. Gomez-Sjoberg, A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–8563 CrossRef.
- R. F. Ismagilov, J. M. Ng, P. J. Kenis and G. M. Whitesides, Anal. Chem., 2001, 73, 5207–5213 CrossRef CAS.
- W. Gu, X. Y. Zhu, N. Futai, B. S. Cho and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15861–15866 CrossRef CAS.
- A. Huebner, M. Srisa-Art, D. Holt, C. Abell, F. Hollfelder, A. J. Demello and J. B. Edel, Chem. Commun., 2007, 1218–1220 RSC.
- C. B. Rohde, F. Zeng, R. Gonzalez-Rubio, M. Angel and M. F. Yanik, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13891–13895 CrossRef CAS.
- C. C. Lee, G. Sui, A. Elizarov, C. J. Shu, Y. S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake and H. R. Tseng, Science, 2005, 310, 1793–1796 CrossRef CAS.
- Y. Y. Huang, P. Castrataro, C. C. Lee and S. R. Quake, Lab Chip, 2007, 7, 24–26 RSC.
- X. C. Zhou, S. Y. Cai, A. L. Hong, Q. M. You, P. L. Yu, N. J. Sheng, O. Srivannavit, S. Muranjan, J. M. Rouillard, Y. M. Xia, X. L. Zhang, Q. Xiang, R. Ganesh, Q. Zhu, A. Matejko, E. Gulari and X. L. Gao, Nucleic Acids Res., 2004, 32, 5409–5417 CrossRef CAS.
- M. J. Moorcroft, W. R. A. Meuleman, S. G. Latham, T. J. Nicholls, R. D. Egeland and E. M. Southern, Nucleic Acids Res., 2005, 33, e75 CrossRef.
- X. L. Gao, X. C. Zhou and E. Gulari, Proteomics, 2003, 3, 2135–2141 CrossRef CAS.
- Y. Rondelez, G. Tresset, K. V. Tabata, H. Arata, H. Fujita, S. Takeuchi and H. Noji, Nat. Biotechnol., 2005, 23, 361–365 CrossRef CAS.
- S. J. Maerkl and S. R. Quake, Science, 2007, 315, 233–237 CrossRef CAS.
- B. Zheng, L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 11170–11171 CrossRef CAS.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 2508–2511 CrossRef CAS.
- C. L. Hansen, E. Skordalakes, J. M. Berger and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16531–16536 CrossRef CAS.
- C. L. Hansen, M. O. Sommer and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14431–14436 CrossRef CAS.
- H. Andersson and A. van den Berg, Sens. Actuators, B, 2003, 92, 315–325 CrossRef.
- D. Di Carlo and L. P. Lee, Anal. Chem., 2006, 78, 7918–7925 CrossRef.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977–1026 CrossRef CAS.
- P. K. Horan and L. L. Wheeless, Science, 1977, 198, 149–157 CAS.
- J. A. Steinkamp, Rev. Sci. Instrum., 1984, 55, 1375–1400 CrossRef.
- L. A. Herzenberg, D. Parks, B. Sahaf, O. Perez, M. Roederer and L. A. Herzenberg, Clin. Chem., 2002, 48, 1819–1827 CAS.
- S. Bruno, P. Lassota, W. Giaretti and Z. Darzynkiewicz, Oncol. Res., 1992, 4, 29–35 CAS.
- C. S. Chin, V. Chubukov, E. R. Jolly, J. DeRisi and H. Li, PLoS Biol., 2008, 6, 1343–1356 Search PubMed.
- K. W. Johnson, A. Dooner and P. J. Quesenberry, Curr. Pharmaceut. Biotechnol., 2007, 8, 133–139 Search PubMed.
- J. Voldman, M. L. Gray, M. Toner and M. A. Schmidt, Anal. Chem., 2002, 74, 3984–3990 CrossRef CAS.
- A. Y. Fu, C. Spence, A. Scherer, F. H. Arnold and S. R. Quake, Nat. Biotechnol., 1999, 17, 1109–1111 CrossRef CAS.
- A. Y. Fu, H. P. Chou, C. Spence, F. H. Arnold and S. R. Quake, Anal. Chem., 2002, 74, 2451–2457 CrossRef CAS.
- J. Kruger, K. Singh, A. O'Neill, C. Jackson, A. Morrison and P. O'Brien, J. Micromech. Microeng., 2002, 12, 486–494 CrossRef.
- F. H. Arnold, Nature, 2001, 409, 253–257 CrossRef CAS.
- M. B. Elowitz, A. J. Levine, E. D. Siggia and P. S. Swain, Science, 2002, 297, 1183–1186 CrossRef CAS.
- B. Zheng and R. F. Ismagilov, Angew. Chem., Int. Ed., 2005, 44, 2520–2523 CrossRef CAS.
- J. Shendure, G. J. Porreca, N. B. Reppas, X. X. Lin, J. P. McCutcheon, A. M. Rosenbaum, M. D. Wang, K. Zhang, R. D. Mitra and G. M. Church, Science, 2005, 309, 1728–1732 CrossRef CAS.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- M. Prakash and N. Gershenfeld, Science, 2007, 315, 832–835 CrossRef CAS.
- T. Nisisako, T. Torii and T. Higuchi, Lab Chip, 2002, 2, 24–26 RSC.
- T. Thorsen, R. W. Roberts, F. H. Arnold and S. R. Quake, Phys. Rev. Lett., 2001, 86, 4163–4166 CrossRef CAS.
- V. Cristini and Y. C. Tan, Lab Chip, 2004, 4, 257–264 RSC.
- D. R. Link, E. Grasland-Mongrain, A. Duri, F. Sarrazin, Z. D. Cheng, G. Cristobal, M. Marquez and D. A. Weitz, Angew. Chem., Int. Ed., 2006, 45, 2556–2560 CrossRef CAS.
- D. R. Link, S. L. Anna, D. A. Weitz and H. A. Stone, Phys. Rev. Lett., 2004, 92, 054503-1–054503-4.
- L. M. Fidalgo, C. Abell and W. T. S. Huck, Lab Chip, 2007, 7, 984–986 RSC.
- L. M. Fidalgo, G. Whyte, D. Bratton, C. F. Kaminski, C. Abell and W. T. S. Huck, Angew. Chem., Int. Ed., 2008, 47, 2042–2045 CrossRef CAS.
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785–796 CrossRef CAS.
- J. U. Shim, G. Cristobal, D. R. Link, T. Thorsen and S. Fraden, Cryst. Growth Des., 2007, 7, 2192–2194 CrossRef CAS.
- J. U. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. W. Jia, K. Piattelli and S. Fraden, J. Am. Chem. Soc., 2007, 129, 8825–8835 CrossRef CAS.
- N. L. Jeon, S. K. W. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. M. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef CAS.
- L. Cai, C. K. Dalal and M. B. Elowitz, Nature, 2008, 455, 485–U416 CrossRef CAS.
- J. M. Skotheim, S. Di Talia, E. D. Siggia and F. R. Cross, Nature, 2008, 454, 291–U212 CrossRef CAS.
- M. R. Bennett, W. L. Pang, N. A. Ostroff, B. L. Baumgartner, S. Nayak, L. S. Tsimring and J. Hasty, Nature, 2008, 454, 1119–1122 CrossRef CAS.
- I. Meyvantsson, J. W. Warrick, S. Hayes, A. Skoien and D. J. Beebe, Lab Chip, 2008, 8, 717–724 RSC.
- S. Lai, S. N. Wang, J. Luo, L. J. Lee, S. T. Yang and M. J. Madou, Anal. Chem., 2004, 76, 1832–1837 CrossRef CAS.
- M. Madou, J. Zoval, G. Y. Jia, H. Kido, J. Kim and N. Kim, Annu. Rev. Biomed. Eng., 2006, 8, 601–628 CrossRef CAS.
- E. M. Lucchetta, J. H. Lee, L. A. Fu, N. H. Patel and R. F. Ismagilov, Nature, 2005, 434, 1134–1138 CrossRef CAS.
- S. Paliwal, P. A. Iglesias, K. Campbell, Z. Hilioti, A. Groisman and A. Levchenko, Nature, 2007, 446, 46–51 CrossRef CAS.
- A. Hierlemann, O. Brand, C. Hagleitner and H. Baltes, Proc. IEEE, 2003, 91, 839–863 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |