Perturbation of single hematopoietic stem cell fates in artificial niches†
Matthias P.
Lutolf‡
*ab,
Regis
Doyonnas‡
*ac,
Karen
Havenstrite
a,
Kassie
Koleckar
a and
Helen M.
Blau
*a
aBaxter Laboratory in Genetic Pharmacology, Department of Microbiology and Immunology, BioX and Stem Cell Institute, Stanford University School of Medicine, Stanford, CA 94305, USA. E-mail: hblau@stanford.edu; Fax: +1 650-736-0080; Tel: +1 650-723-6209
bLaboratory of Stem Cell Bioengineering and Institute of Bioengineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), Switzerland. E-mail: matthias.lutolf@epfl.ch; Fax: +41 (0)21 693 96 65; Tel: +41 (0)21 693 18 76
cGenetically Modified Models Center of Emphasis, Pfizer Global Research & Development, Groton, CT 06340, USA. E-mail: regis.doyonnas@pfizer.com; Fax: +1 860-686-1164; Tel: +1 860-686-4186
First published on 21st November 2008
Abstract
Hematopoietic stem cells (HSCs) are capable of extensive self-renewal in vivo and are successfully employed clinically to treat hematopoietic malignancies, yet are in limited supply as in culture this self-renewal capacity is lost. Using an approach at the interface of stem cell biology and bioengineering, here we describe a novel platform of hydrogel microwell arrays for assessing the effects of either secreted or tethered proteins characteristic of the in vivo microenvironment, or niche, on HSC fate in vitro. Time-lapse microscopic analyses of single cells were crucial to overcoming inevitable heterogeneity of FACS-enriched HSCs. A reduction in proliferation kinetics or an increase in asynchronous division of single HSCs in microwells in response to specific proteins (Wnt3a and N-Cadherin) correlated well with subsequent serial long-term blood reconstitution in mice in vivo. Single cells that divided once in the presence of a given protein were capable of in vivo reconstitution, providing evidence of self-renewal divisions of HSCsin vitro. These results validate the hydrogel microwell platform as a broadly applicable paradigm for dissecting the regulatory role of specific signals within a complex stem cell niche.
Insight, innovation, integrationAdult stem cell microenvironments, termed niches, are crucial for stem cell function. They are complex, multifactorial regulatory entities that are difficult to study using conventional experimental in vivo or vitro paradigms. We have combined biomaterials technology with microengineering to produce protein-functionalized hydrogel-microwell arrays. Analyses of single hematopoietic stem cells (HSCs) by time-lapse microscopy resolved kinetic differences obscured in bulk cultures and showed that in vitro behavior could predict in vivostem cell function. By demonstrating that single cells undergo self-renewal divisions in microwells in response to selected soluble proteins or immobilized proteins that mimic cell–cell interactions typical of HSC niches, we could deconstruct a complex niche and parse out the role of certain individual components using novel ‘artificial niches’. |
Introduction
Hematopoiesis relies on the life-long self-renewal and differentiation capacity of sparse populations of hematopoietic stem cells (HSCs). Although HSC function is exemplary in the intact organism, in that single HSCs can reconstitute all of the cell types of the blood,1HSCs tend to rapidly specialize and lose their stem cell properties when grown in conventional culture conditions. In vivo, a specific stem cell microenvironment, or niche, has been shown to play a critical role in the maintenance of stem cell function, particularly in Drosophila germ line,2 mammalian skin3 and blood.4,5 Yet, the specific function of HSC niche components, in particular their role in orchestrating the delicate balance between self-renewal and differentiation, remains poorly understood or is a matter of debate.6In vivo studies using knockout and transgenic mouse models have provided important insights,7–13 but have also led to results that are contradictory, due in part to the complexity of cell–cell and protein interactions in the intact niche. On the other hand, in vitro analyses of FACS-enriched HSCs in bulk cultures have been hindered by unavoidable cell population heterogeneity. Thus, the behavior of the enriched stem cells may be masked by highly proliferative progenitor populations.14–16
We reasoned that in vitro assays that enable single-cell resolution in a high-throughput format such as microwell arrays17–24 could shed light on the effects of specific niche proteins on HSC function. Since, in the niche, HSCs are in contact not only with soluble proteins but also membrane components provided by support cells, we developed a novel micropatterning technique to mimic such interactions by presenting appropriately oriented tethered proteins to cells in arrays of hydrogel microwells. We show that self-renewal and long-term blood reconstitution can be induced by exposure of single HSCs to either soluble or tethered proteins. Here we show that cells that divided once in the presence of a given protein led to serial long-term reconstitution of the blood in HSC-depleted lethally irradiated mice, providing evidence of self-renewal divisions in vitro. Together, these data provide insights into the roles of selected proteins implicated in the HSC niche and they validate the hydrogel microwell array platform for studies of extrinsic regulators of single stem cell fate in general.
Experimental
Isolation and purification of hematopoietic stem/progenitor cells by flow cytometry
All animal experiments were performed in compliance with the laws and institutional guidelines of Stanford University. The protocols and experiments were approved by the Administrative Panel on Laboratory Animal Care (Assurance #A3213-01) and verified by Stanford's Institutional Animal Care and Use Committee. Hematopoietic stem/progenitor cells were isolated from bone marrow donors of 8- to 12-week-old GFP+C57BL/6-Ly5.1 mice, stained and purified as described in detail in the ESI.† The lineage depleted cell population was separated by Flow cytometry on a Vantage SE FACS instrument (BD Bioscience, USA) at the Stanford FACS core facility. Single viable (propidium iodide negative) Lin−ckit+Sca1+CD150+ cells were triple sorted using the gates shown in the FACS plots in Fig. 2 and directly deposited in microwell arrays. FACS data were plotted using FlowJo (TreeStar Inc., USA).Long-term reconstitution assays
In order to assess self-renewal potential of HSCs, long-term blood reconstitution assays were conducted in mice lethally irradiated to deplete their endogenous HSCs. We transplanted 10, 20, 40, 100 or 500 GFP+ cells (C57BL/6, Ly5.1) of the LKS or LKS-CD150+ population per animal together with 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GFP−Sca1−CD150− bone marrow ‘helper’ cells incapable of long-term reconstitution (C57BL/6, Ly5.1) into lethally irradiated wild-type host mice (C57BL/6, Ly5.2), as previously described.25 Reconstitution was measured by assessment of GFP+Ly5.1+ cells in the CD45 gated peripheral blood derived from retro-orbital bleeding 4, 8, 12, and 24 weeks after transplantation as described in the ESI.†
000 GFP−Sca1−CD150− bone marrow ‘helper’ cells incapable of long-term reconstitution (C57BL/6, Ly5.1) into lethally irradiated wild-type host mice (C57BL/6, Ly5.2), as previously described.25 Reconstitution was measured by assessment of GFP+Ly5.1+ cells in the CD45 gated peripheral blood derived from retro-orbital bleeding 4, 8, 12, and 24 weeks after transplantation as described in the ESI.†
Fabrication of hydrogel microwell arrays for high-throughput analysis of single HSC behavior
Poly(ethylene glycol) (PEG). Linear PEG-(SH)2 (mol. wt. 3400 g mol−1, 100% substitution as indicated by the manufacturer) was purchased from Nektar (Huntsville, AL, USA). 8arm-PEG-vinylsulfones were produced from 8arm-PEG-OH (mol. wt. 40000 g mol−1) as described elsewhere26 and characterized as described in the ESI.†
Gelation of PEG precursors (Fig. 1A) to form hydrogels was performed by adapting a previously developed mild and versatile chemistry.26 Details of the procedure are available in the ESI.†
 | ||
| Fig. 1 Microwell hydrogel array platform to systematically explore the function of single HSCs in high-throughput. (A) Reactive thiol- and vinylsulfone end-groups on poly(ethylene glycol) (PEG) precursors react under mild conditions to form hydrogel matrices that can be used to form microwell arrays. (B) Overview of multistep process to fabricate hydrogel microwell arrays. A PDMS stamp containing an array of micropillars is cast on a silicon master (step 1). This stamp is used as a template to crosslink a PEGgel containing the complementary microwell array topography (step 2 and 3). Upon swelling and washing, the hydrogel surface is used to trap large numbers of individual HSCs (step 4). Typical dimensions of microwells are indicated on the right. (C) Hydrogel microwell arrays can be placed on the bottom of any standard well plate (here: 96-well) to culture single HSCs and track their behavior by time-lapse videomicroscopy over many days (see Supplementary Movie 1† ). | ||
Hydrogel microwell array formation (Fig. 1B). Hydrogel microwell arrays were fabricated by a multistep soft lithography process. PDMS microwell array masters of the size of an entire Si wafer were a generous gift from Dr Marc Dusseiller (ETH Zurich, Switzerland). Immediately after mixing, the PEG precursor solution was pipetted onto the microstructured PDMS surfaces and polymerized (30 min, 37 °C in a humidified incubator) between two hydrophobic glass slides separated by a 0.7 mm spacer at both ends. The PEG hydrogel microwell arrays were peeled off, washed thoroughly and left to swell overnight in PBS. Prior to cell culture, the swollen PEG hydrogel microwell arrays were fixed on the bottom of plastic wells of a desired well plate using the above solution of gel precursors as efficient ‘glue’, and the arrays were equilibrated at 37 °C in cell culture medium. Further details are available in the ESI.†
Hematopoietic stem cell culture
LKS-CD150+ were cultured under sterile conditions in a serum-free basal media containing Stemline II hematopoietic expansion medium (Sigma, USA) supplemented with 100 ng ml−1 SCF and 2 ng ml−1 Flt-3 ligand in 10% CO2 at 37 °C in a humidified incubator. In a typical experiment, 300 individual LKS-CD150+ cells were seeded per well of a 96-well plate containing a total of 400 microwells in 200 μl of medium (or else to facilitate micromanipulation, 1000 to 2000 cells per well of a 24-well plate containing 4000 microwells in 1 ml of medium). After 1 h during which individual cells randomly sedimented to the bottoms of microwells, the plate was transferred to the incubator of the microscope and further cultured under the same sterile conditions for at least four days. The selected putative soluble HSC regulatory proteins that were tested for their effects on HSC fate are described in Supplementary Table 1.†Time-lapse microscopy and image analysis
LKS-CD150+ cells were directly sorted into wells each of which contained hundreds of microwells, as described above. The plate was then placed in the environmental chamber of an inverted microscope (Zeiss Axiovert 200) equipped with a motorized stage. After cells were randomly distributed and trapped in microwells, the XYZ stage was programmed to repeatedly raster across the microwell array surface, acquiring phase contrast images at 5× (in some cases 10×) magnification of multiple locations in defined time intervals for a period of up to 7 days. The entire surface of the microwell arrays was scanned and the resulting images of the time-lapse experiment were then automatically compiled into a stack (library) using the Volocity software (Improvision). Cells were scored dead when they ceased to move on the microwell surface. PI staining was utilized to confirm the death read-out. Scoring of time-lapse movies was facilitated by a Matlab program as described (see the Results section and the ESI† ).Analysis of HSC maintenance and self-renewal in vitro by transplantation of cultured cells into mice
In a first experimental paradigm, the progeny of 100 stem cells generated over 4 days in culture were pooled and then transplanted into lethally irradiated mice as described above. In another set of experiments, micromanipulation was employed to pick progeny of single stem cells that underwent a defined number of divisions in culture. Single cells, doublets or larger colonies (>3 divisions) were collected with a glass micropipette using an inverted microscope equipped with a micromanipulator and micro-injector. The micromanipulated cells were transferred to Eppendorf tubes and then transplanted as described above.In situ patterning of biomolecules on microstructured gels by ‘reactive microcontact printing’
In order to control both topography and localized presentation of extracellular matrix and transmembraneHSC regulatory proteins on gel microwell arrays, protein immobilization was restricted to selected areas on the gel surface via a novel hydrogel microfabrication process termed ‘reactive microcontact printing’ (Fig. 4) described in detail in the ESI.† Briefly, the above PDMS masters were first ‘inked’ using PEG-modified ProteinA (PIERCE, USA) to adsorb the ProteinA just on the tip of the PDMS pillars, followed by hydrogel microwell casting as described above, transferring to and locally covalently immobilizing ProteinA from the PDMS surface onto the microstructured gel matrix. Subsequently, ProteinA-modified PEG hydrogel microwells were incubated (1 h, 37 °C, humidified incubator) with a solution of a desired Fc-chimeric protein (at 10 μg ml−1) in PBS and extensively washed to remove non-immobilized proteins. Confocal laser scanning microscopy was utilized to test the extent, uniformity, and stability of the microcontact printing process. The selected putative Fc-chimeric HSC regulatory proteins that were tested for their effects on HSC fate are listed in Supplementary Table 2.†Statistical analysis
Student’s T test was used to determine significance, set at p < 0.05.Results
Fabrication of hydrogel microwell arrays for cultivation of single stem cells
We used a combination of biomaterials engineering27,28 and microfabrication29 to generate arrays of hydrogel microwells to track the proliferative behavior of single stem cells. Single cell analysis was critical, given the inevitable heterogeneity of even FACS-enriched HSC populations.30 The platform was designed to be amenable to live-cell microscopic analysis of hundreds of single HSCs exposed to the same protein, and compared simultaneously with HSCs exposed to other proteins. Such ‘microwell arrays’ can spatially segregate individual cells and recently have become useful tools for studies of the biological properties of single cells.17–24We selected poly(ethylene glycol) (PEG)-based hydrogels as microwell substrates due to their biomimetic physicochemical properties.31,32 In contrast to tissue culture plastic or many previously developed microwell arrays, crosslinked PEG networks are soft (elastic modulus in the range of hundreds of Pa28) and have a high water content (>95%). We therefore reasoned that PEG hydrogels could simulate the soft and hydrated microenvironment of HSCs in the bone marrow.
Arrays of hydrogel microwells were fabricated using standard photolithography to etch the microwell pattern into a silicon substrate19,29 onto which liquid thermocurable poly(dimethylsiloxane) (PDMS) was polymerized (Fig. 1B1). The elastomeric PDMS template was then used to cast hydrophilic polymer precursors of PEG that were covalently crosslinked, as previously described,33 to form inert hydrogels (Fig. 1B2). PEG hydrogel microwell arrays were removed from the PDMS, equilibrated in PBS, and attached to the bottom of tissue culture wells (Fig. 1B3–4). Since each well was supplemented with an individual protein, the several hundred single HSCs trapped within microwells (100–130 μm diameter) of a given well were all exposed to the same protein. Since the unmodified PEG hydrogel substrates are non-adhesive for cells,28 even highly migratory HSCs were not able to escape the microwells, facilitating long-term live-cell microscopic analyses (Fig. 1C and Supplementary Movie 1† ).
Isolation and characterization of murine hematopoietic stem cells
To select for a highly enriched HSC population for use in our studies, we adapted two published FACS protocols. Specifically, we compared the in vivo reconstitution potential of double-sorted Lin−ckit+Sca1+ (LKS) cells,34 and a subpopulation of LKS cells expressing the SLAM receptor CD150 (LKS-CD150+)35 (Fig. 2A). Reconstitution was measured by assessment of GFP+Ly5.1+ cells at 1, 2, 3, and 6 month after transplantation (Fig. 2B–D). When 10–40 cells were transplanted, LKS-CD150+ demonstrated multilineage reconstitution potential in 100% of the recipients analyzed; by contrast, as the transplanted LKS cells decreased in number, a progressive loss of reconstitution potential was observed, in agreement with previously reported findings.35 These data provide functional evidence that the majority of cells in the LKS-CD150+ population are long-term reconstituting HSCs (LT-HSC), while the LKS fraction is more heterogeneous presumably due to an increased proportion of multipotent progenitor cells (MPP) with reduced self-renewal function. | ||
| Fig. 2 Purification and long-term reconstitution efficiency of LKS versus LKS-CD150+ (A) Freshly isolated murine bone marrow cells were magnetically depleted for Lineage markers, FACS-sorted for Lin−ckit+Sca1+ (LKS), then resorted twice for CD150+. LKS comprise 1% ± 0.5% of the magnetically depleted Lin− fraction and the CD150+ fraction comprises 20% ± 7% of the LKS population. (B) To assess long-term reconstitution and thus self-renewal potential of the LKS and LKS-CD150+ populations, 10, 20, 40, 100 and 500 GFP+ cells (C57BL/6, Ly5.1) of each population were transplanted into lethally irradiated wild-type host mice (C57BL/6, Ly5.2) together with 500000 CD150−Sca1− helper cells (C57BL/6, Ly5.1). Percentages of mice that sustained >0.5% PB chimerism up to 24 weeks post-transplant were determined for each population (Black bars: LKS, grey bars: LKS-CD150+) (C) Peripheral blood (PB) from each transplanted mouse was analyzed for reconstitution for at least 6 months by assessing the proportion of GFP+Ly5.1+ circulating white blood cells. (D) Both lymphoid (B220/CD3) and myeloid (Mac1/Gr1) lineages were reconstituted in these mice. | ||
Stem cells exhibit slower in vitro division kinetics than multipotent progenitors
We postulated that the extent of division of the stem cell enriched LKS-CD150+ cell population would be reduced, as the cells would divide more slowly in culture than LKS cells. To test this hypothesis we assessed the division kinetics of each population by seeding 300 single cells per well of a 96-well plate containing a total of 400 microwells (Fig. 1C). Single cells randomly sedimented within minutes to the bottoms of microwells. Microwells with multiple cells at the onset of the experiment were eliminated retrospectively from the analysis. At least 100 single cells of both populations were tracked by automated time-lapse microscopy and the kinetic proliferation profiles quantified as the distribution of numbers of HSC progeny generated per microwell as a function of time. When cultured in a basal serum-free culture medium supplemented with only stem cell factor (SCF, or c-kit ligand; 100 ng ml−1) and Flt-3 ligand (2 ng ml−1),36 cells of the two phenotypes exhibited marked differences in division kinetics (Fig. 3). LKS proliferated rapidly compared to LKS-CD150+, as shown in the still images of representative time-lapse analyses and a quantification of the distribution of cells per microwell at various time points (Fig. 3A and Supplementary Movies 2&3† ). | ||
| Fig. 3 Single LKS-CD150+ are marked by slow division kinetics compared to LKS. (A) In hydrogel microwells, growth of single cells of both populations was monitored via time-lapse microscopy. Still images selected at the indicated time points taken from representative movies of LKS (top panels) and LKS-CD150+ (bottom panels) cultures are shown (see corresponding Supplementary Movies 2&3† ). LKS were highly proliferative, while LKS-CD150+ displayed slow proliferation kinetics. Coloured circles indicate microwells hosting clones that underwent variable numbers of divisions. Quantification of the distribution of cells per microwell at the indicated time point confirmed these visual differences: for example, 50% of all microwells comprised 8 or more cells in the LKS population, while 70% of microwells of LKS-CD150+ contained only 2 cells (220 microwells per condition analyzed; n = 102 cells per histogram for LKS and n = 103 for LKS-CD150+, respectively). (B) Quantification of distributions of the time to the 1st and times between 1st and 2nd divisions of single LKS or LKS-CD150+ (220 microwells per condition analyzed; n = 100 and 153 cells per histogram for LKS and n = 90 and 40 for LKS-CD150+, respectively). The differences in cell cycle distributions confirm that stem cells can be distinguished from progenitors by their slow division kinetics. | ||
Single cell analysis enables not only studies of the extent of division as shown above, but also allows the measurement of time between divisions for each individual HSC to be assessed, assays not possible with bulk cultures. LKS cells relatively homogeneously entered their first division after 35 h on average, whereas LKS-CD150+ displayed a higher degree of heterogeneity (Fig. 3B), with a large number of cells entering their first division at very late time points and an average time to first division of ca. 50 h. For LKS cells the distribution in the time between the first and second division (average 22 h) and between the second and third division (average 13 h, not shown) became narrower as the cells cycled faster during the two consecutive rounds of division. By contrast, LKS-CD150+ cells displayed a time between the first and second division of 42 h and rarely underwent a third division within the period analyzed. Taken together, this analysis of single cell growth kinetics revealed prolonged cell cycle times for stem cells compared to progenitors when cultured on microwell arrays in a serum-free basal culture medium supplemented with only two growth factors essential to cell survival.
We tested whether the LKS-CD150+ cells grown in PEG hydrogel microwells under basal conditions described above, with no additional factors, would support HSC function using an in vivo assay. The progeny of 100 single GFP+ cells grown for 7 days in microwells were tested for stem cell function by transplantation of the cultured cells into lethally irradiated mice. In contrast to freshly isolated LKS-CD150+ cells which led to high peripheral blood reconstitution upon transplantion in mice (5/5), none of the mice injected with cells from microwells exhibited reconstitution (0/5), indicating that essential niche factors were lacking in the culture system. These data provided the impetus for the subsequent systematic studies of individual soluble and tethered factors implicated in the HSC niche on HSC function.
Presentation of tethered proteins in hydrogel microwell arrays
The hydrogel microwell platform described above can be readily used to probe effects of soluble factors on the behavior of populations of single stem cells. However, it does not fully recapitulate the presentation mode of protein cues within the niche, as stem cells are anchored to the niche via insoluble cell–cell adhesion interactions with support cells, as well as receptor-ligand interactions from a protein- and sugar-rich extracellular matrix.37 These adhesive interactions likely not only localize the stem cell, but also bring it in close proximity to soluble self-renewal signals secreted by the support cells.38,39In order to recapitulate HSC-niche interactions in vitro in a near-physiologic fashion, we developed a novel technique to spatially pattern and immobilize proteins onto our hydrogel matrices (Fig. 4). Protein tethering was achieved by attaching a heterofunctional PEG linker to a protein of interest and then crosslinking this complex into the gel network. To ensure site-selectivity in protein immobilization, we focused on engineered Fc-chimeric proteins that could be linked via binding to an intermediate auxiliary protein, ProteinA, that contains four high-affinity binding sites (Ka = 108 per mole) for the Fc-region of human, mouse and rabbit immunoglobulins . To specifically functionalize gels and immobilize proteins only at the bottom of microwells, rather than homogeneously distributing proteins across the entire array (bulk modification), the microwell fabrication process (Fig. 1B) was augmented by adding a reactive microcontact printing step (Fig. 4B): PEG-functionalized ProteinA was adsorbed onto the posts of the PDMS stamp (step 1,2) and the hydrogel polymerized onto the ProteinA/PDMS (step 3,4), transferring both the topographic pattern and protein pattern onto the gel surface.
 | ||
| Fig. 4 Microcontact printing to generate ‘artificial niches’ via site-selective attachment of niche proteins. (A) A heterofunctional PEG linker is used to covalently attach ProteinA to the gel networks of Fig. 1. Subsequently, ProteinA site-selectively anchors Fc-chimeric proteins on hydrogel microwell arrays. (B) Overview of multistep process to locally immobilize Fc-chimeric proteins to the bottom of hydrogel microwells. A PDMS stamp containing an array of micropillars is inked at the pillar tips with PEG-modified ProteinA (step 1 and 2). Similar to the protein-free process, this stamp is used as a template to crosslink a PEGgel to generate the complementary microwell array topography. Simultaneously, PEG-ProteinA is transferred to the surface of the gel as is formed, covalently linked to the polymer network (step 3 and 4). Upon swelling and washing, an Fc-chimeric protein is incubated and selectively binds to ProteinA (step 5). Hydrogel microwell surfaces selectively modified with regulatory proteins of choice are used to trap and study HSCs as single cells (step 6). (C) Proof-of-principle experiments demonstrating the spatial control of protein immobilization afforded by the novel hydrogel microcontact printing process. Immobilized FITC-labelled BSA serves as a model protein to demonstrate anchoring on the bottom of individual microwells (right panel) rather than on the entire surface of the microwell array (left panel). 3D confocal micrographs of projection of 84 stacks acquired at a constant slice thickness of 1.8 μm. The small panels at the bottom of the 3D projections represent (x,z)-cross-sections through the gel revealing the resulting dispersed (bulk) or localized (microcontact printed) topography. (D) Immobilization of Fc-chimeric proteinsvia selective binding to ProteinA. Alexa-conjugated Fc-fragment and Fc–N-Cadherin were tethered and detected viafluorescence microscopy according to the schemes (left & middle panels). As negative controls (right panels), microwell arrays are shown that are not tethered with Protein A or treated with isotype control primary antibody . | ||
We found that selective modification of microwells with Fc-chimeric adhesion proteins such as V-CAM ensured efficient confinement and tracking of HSCs over long culture periods, whereas the cells escaped from the microwells within a few hours when the bulk of the surface was modified (not shown). Immunofluorescence microscopy revealed that microcontact printed proteins, such as a BSA-FITC model protein were localized at the bottom of the microwells (Fig. 4C). When ProteinA was used, Fc-chimeric proteins such as N-Cadherin (N-Cad) were shown via immunostaining to be effectively tethered (Fig. 4D). The platform which we generated is advantageous as it is versatile: microwell arrays containing microcontact-printed ProteinA can be incubated with any Fc-chimeric protein of interest, yielding microwells with the properly oriented, immobilized protein localized to the bottom of each well.
HSC division kinetics change in response to selected soluble and immobilized protein cues
We next systematically tested the effects of selected soluble and immobilized proteins on the proliferation kinetics of LKS-CD150+ (designated hereon as HSCs). Although the candidate proteins selected for study here had been implicated by others to have a role in HSC regulation, the specific function of these proteins in the niche remains largely unknown. In order to maximize the sensitivity in detecting responses to individual factors, we again used basal growth factor conditions, supplemented with a series of seven different soluble protein morphogens or cytokines and six Fc-chimeric transmembraneproteins. Soluble protein doses were selected based on published concentrations (see ESI† ), and all factors were tested separately, but simultaneously in multiple experiments in 96-well plates (Fig. 1A).Strikingly, the addition of single proteins to the basal medium markedly altered proliferation kinetics, as is evident from the distribution of total cells per microwell over a period of 4 days in culture (Fig. 5A). These data are derived from a quantification of the extent of proliferation, or total cell number, at time intervals for each microwell. Since the microwell platform in conjunction with time-lapse microscopy is designed to perform high-throughput experiments, a means of facilitating cell counting is critical. To obtain proliferation data at a clonal level and count cells in individual microwells we used Matlab (Mathworks Company) to design a customized, semi-automated cell counting program. Starting with a master image containing all 400 microwells within an array, edge detection was used to locate all microwells and then segment each microwell into its own image, yielding 400 separate images per array. For each microwell containing a single cell, a series of images corresponding to that microwell was generated automatically at each time point (every 24 h). This program allowed us to rapidly select the microwells for analysis and automatically visualize successive frames of time-lapse movies of the same microwell on the computer screen, enabling rapid and precise visual evaluation and recording of cell division in an annotated Excel format.
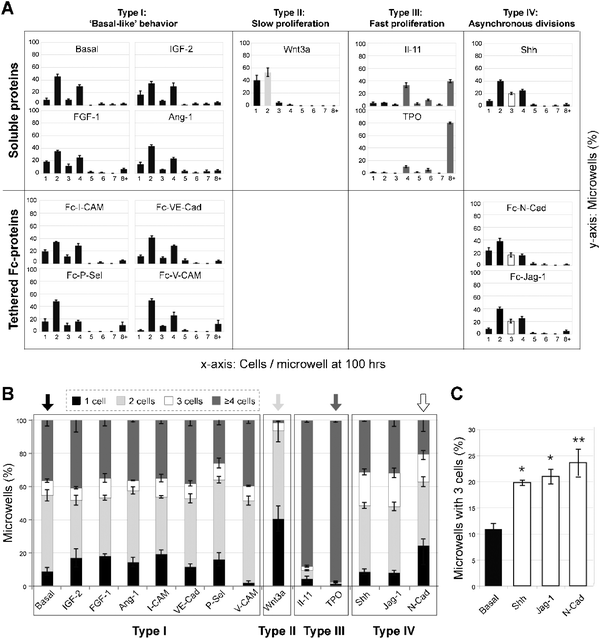 | ||
| Fig. 5 Identification of proteins that significantly influence HSC proliferation kinetics at the clonal level. Quantification of the distribution of cells per microwell after 100 h in culture reveals HSC responsiveness to soluble and immobilized Fc-chimeric protein cues characteristic of the niche (n = 70–100 microwells per condition and per experiment analyzed; averages of three independent experiments with standard deviations are shown). Candidate factors were separated into four groups according to their growth behavior compared to the basal conditions: “basal-like” (Type I), slow proliferation (Type II; Wnt3a), fast proliferation (group III; IL-11 and TPO), and asynchronous divisions (group IV; Shh, N-Cad and Jag-1). (B) Identification of soluble and tethered putative niche cues that alter proliferation kinetics compared to the basal medium control via binning of the number of progeny of single HSCs into four groups at 100 h. (C) An additional analysis was performed for Type IV proteins to determine the percentage of microwells that contained 3 cells at 24 h time intervals over a period of one week compared to basal conditions. Care was taken to only score novel appearances of 3 cells per microwell at each time point in order to avoid counting the same data twice. T test for unequal sample size was used (n = 57, 175, 65, and 80 microwells for basal, N-Cad, Jag-1, and Shh, respectively, ±SEM with the significance level *p <.05 and **p < .01). | ||
The kinetic proliferation profiles, quantified by the distribution of HSC progeny generated per microwell per day in response to specific proteins, revealed four distinct patterns (Fig. 5A). Most proteins (IGF-2, FGF-1, Ang1, I-CAM, VE-Cad, P-Sel, V-CAM) exhibited a proliferation profile similar to that of the basal media, i.e., they had no noticeable effect (Type I). By contrast, one protein, Wnt3a resulted in relatively small clone sizes of primarily one or two cells per microwell (Type II). Two proteins (TPO, IL-11) resulted in relatively large clone sizes of >8 cells per microwell (Type III). In general, Types I–III exhibited a prevalence of clones with even numbers of 2, 4 and 8 cells. However, for three proteins (Shh, Jag-1, and N-Cad), designated as Type IV, the number of clones with an odd number of 3 cells microwell was increased above basal Type I conditions, indicative of a higher frequency of asynchronous division of daughter cells. For Type IV proteins, we performed an additional analysis to determine the percentage of microwells that contained 3 cells at 24 h time intervals and an increase in the proportion of microwells with three cells was consistently observed over a period of one week compared to basal conditions (Fig. 5C). Care was taken to only score novel appearances of 3 cells per microwell at each time point in order to avoid counting the same data twice. To facilitate comparisons, a histogram for proteins of Types I–IV is shown depicting the relative proportions of microwells with non-dividing cells (1 cell), slow dividing clones (2 cells), fast dividing clones (≥4 cells) or asynchronously dividing clones (3 cells) (Fig. 5B). From these data, the four distinct proliferative patterns are particularly clear. Representative proteins of the three types that differed from basal were selected for further analysis, Wnt3a (Type II), TPO (Type III), and N-Cad (Type IV), of which the first two are soluble and the last is tethered.
An analysis of the detailed time course, in particular the time between divisions in culture, revealed profound differences among the three types of proteins (Supplementary Fig. S1 and corresponding Supplementary Movies 4–6† ). For these studies, we performed time-lapse experiments with 1 h time intervals for a period of up to 7 days. Representative images taken from the resulting movies of HSCs exposed to the four types of protein conditions (basal, TPO, Wnt3a, and N-Cad) are shown in microwells at 20 h intervals (see Supplemental Fig. S1A† ) exemplifying the differences in proliferation kinetics depicted quantitatively in Fig. 5. Compared to basal conditions, TPO-exposed HSCs exhibited a relatively homogeneous distribution, entering their first division on average at 37 h (Supplemental Fig. S1B† ). Most cells that underwent a first division in the presence of TPO, divided a second time with an average time to division of 21 h. Notably, TPO proliferation kinetics resembled those observed with multipotent progenitors (MPP/LKS in Fig. 3) with similar peaks, and times to first division and between first and second divisions. In contrast, cells exposed to Wnt3a and N-Cad revealed a higher degree of heterogeneity, with some cells dividing almost immediately and others entering their first division after as much as 80 h.
Notably, HSCs exposed to Wnt3a and N-Cad displayed average times between first and second divisions and time to first division (50 h vs. 47 h; and 47 h vs. 34 h, respectively) that were not reduced, but instead somewhat prolonged. These results show that exposure of single HSCs to single extrinsic cues has a marked effect on stem cell proliferation kinetics in vitro.
Slow cell proliferation kinetics induced by Wnt3a correlates with long-term reconstitution in vivo
The disparate proliferation behaviors observed with TPO, Wnt3a and N-Cad suggested that HSCs cultured in the presence of these three factors might have different biological properties. Accordingly, we tested whether the cells exposed to these factors differed with respect to self-renewal, multipotency, and engraftment assessed by long-term blood reconstitution. 100 HSCs were seeded in microwell arrays, exposed to TPO, Wnt3a, N-Cad or basal medium in culture for ≥4 days and all progeny harvested, pooled, and transplanted into lethally irradiated hosts (Fig. 6A).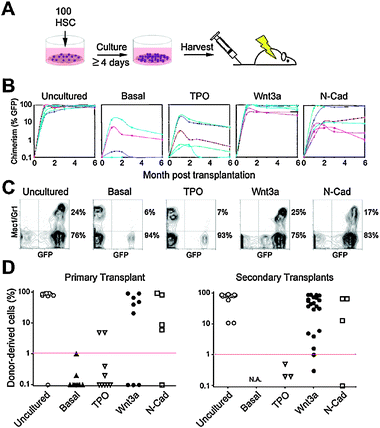 | ||
| Fig. 6 Transplantation experiments reveal in vitro self-renewal divisions of single Wnt3a- and N-Cad-exposed HSCs. (A) Bulk transplantation assay to assess stem cell function after culture. 100 freshly isolated GFP+HSCs were cultured for 4–7 days in TPO, Wnt3a, N-Cad or basal medium only (Basal) and all progeny transplanted into lethally irradiated CD45-congenic C57Bl6 recipient mice (1 well per recipient mouse, n = 10 animals per condition). (B) Peripheral blood chimerism in individual recipients as a function of time. Data points represent individual mice repopulated from 3 separate experiments. (C) Representative examples of peripheral blood FACS analyses for all conditions. A quantification of the normalized percentages of myeloid and lymphoid compartments is shown in Supplementary Fig. S2.† (D) Peripheral blood (PB) chimerism in primary and secondary transplants 24 weeks post-transplant for uncultured HSCs or the progeny of cultured cells in presence of TPO, Wnt3a, N-Cad or basal medium. | ||
After 6 months, a high efficiency of reconstitution with robust peripheral blood (PB) chimerism was obtained in mice transplanted with GFP+HSC that were cultured in the presence of Wnt3a (6/9, up to 93% PB chimerism) or N-Cad (4/5, up to 95% PB chimerism), whereas a low efficiency of reconstitution with low PB chimerism was obtained for the basal medium control (1/9, up to 5% PB chimerism) and TPO (2/9, up to 21% PB chimerism) (Fig. 6B). Moreover, donor-derived peripheral blood chimerism persisted for six months in all mice reconstituted with cells exposed to Wnt3a or N-Cad, but declined progressively in mice reconstituted with cells exposed to basal medium or TPO (Fig. 6B). Wnt3a- or N-Cad-treated cells yielded normal lymphoid and myeloid ratios, whereas cells exposed to basal medium and TPO-treated cells, gave rise primarily to lymphoid lineages (Supplemental Fig. S2† ). These differences were even more pronounced upon secondary transplantation of HSCs from reconstituted mice into lethally irradiated recipients (Fig. 6D). Whereas uncultured, Wnt3a- and N-Cad-treated cells led to reconstitution in most secondary recipients (17/17, up to 95% PB chimerism; 20/21, up to 91% PB chimerism; 3/4 up to 67%) none (0/3) of the TPO-treated cells from the poorly reconstituted primary transplants led to successful reconstitution upon secondary transplantation, and cells exposed to the basal medium yielded such low reconstitution that secondary transplants were not possible. These results show that exposure to Wnt3a or N-Cad in vitro in hydrogel microwells leads to retention of stem cell function. These data also provide evidence that both the rate and synchrony of stem cell division induced by single extrinsic factors in vitro correlated with in vivoHSC reconstitution potential in mice, suggesting that these characteristics could serve as predictors of maintenance of stem cell function.
Evidence for self-renewal: Wnt3a and N-Cad maintain stem cell multipotency after division in culture
We postulated that the effect of Wnt3a or N-Cad shown in Fig. 6 could result either from a retention of stem cell function in non-dividing cells or from self-renewal and the production of another stem cell in the course of cell division in culture. To distinguish between these two possibilities, we analyzed the in vivo function of HSCs that never divided (singlets), divided once (doublets) or divided more than three times (clones). For this purpose, a series of transplantation experiments was carried out using micromanipulation to harvest HSC progeny from individual microwells after exposure to TPO, Wnt3a or N-Cad (Fig. 7A). Notably, TPO-treated singlets could not be tested, as cells that did not undergo division within 4 days were exceedingly rare, except when they formed giant megakaryocytes (Supplementary Fig. S3 and corresponding Supplementary Movies 7&8† ). Strikingly, despite substantial technical challenges, upon transplantation of 40 singlets exposed to N-Cad (10 per lethally irradiated mouse), long-term blood reconstitution was detected in 1 of 4 mice. These results demonstrate that stem cell multipotency can be maintained for up to one week in the absence of cell division in culture in the presence of N-Cad, but not Wnt3a. | ||
| Fig. 7 Micromanipulation reveals HSC self-renewal divisions induced by Wnt3a- or N-Cad-exposure. (A) Transplantation of micromanipulated clones that underwent variable division numbers to discriminate between HSC maintenance in the absence or presence of division, and expansion. Individual clones tracked in microwell arrays by time-lapse microscopy were selected based on the number of divisions they underwent, picked and transplanted into lethally irradiated recipients as indicated. (B) Peripheral blood chimerism in primary transplants 24 weeks post-transplant for singlets, doublets or clones. Wnt3a and N-Cad, but not TPO, maintains HSCs in a stem cell state in the absence of division (singlets) and induces self-renewal divisions (doublets). | ||
To address whether exposure to Wnt3a or N-Cadherin was correlated with self-renewal, we transplanted cells that underwent one division (doublets; 5–10 per lethally irradiated mouse) (Fig. 7A). This experiment required monitoring the cultures by continuous time-lapse microscopy to ascertain when division occurred. Notably, none of the animals (0/16) transplanted with a total of 100 TPO-stimulated doublets exhibited blood reconstitution. In contrast, out of the 115 Wnt3a-stimulated HSC doublets transplanted into 19 recipients, 3 mice exhibited high reconstitution potential with up to 92% PB chimerism (115 doublets transplanted, 3/19 mice), comparable to N-Cad stimulated cells (90 doublets transplanted, 2/15 mice). These experiments demonstrate that soluble Wnt3a or immobilized N-Cad can maintain stem cells by self-renewal, whereas TPO cannot. By contrast, transplantation of larger clone sizes never resulted in bone marrow reconstitution, irrespective of the factors to which HSCs were exposed, suggesting that even in the presence of Wnt3a or N-Cad, cells characterized by a faster proliferation rate in culture had lost their stem cell capacity. These results show that stem cell function is maintained after one division in the presence of Wnt3a or N-Cad, and provide evidence of self-renewal in response to single proteinsin vitro.
Discussion
A dissection of the regulatory role of specific signals within a complex somatic stem cell niche is crucial for developing therapeutic strategies using adult stem cells. In the case of HSCs, identification of cues that induce HSC self-renewal and expansion can be envisaged, overcoming the shortage of donor cells available for transplantation from adults and facilitating the use of autologous cord blood HSCs. Here we present an approach at the interface of stem cell biology, biomolecular materials engineering and microfabrication technologies. The novel cell culture platform we designed is fabricated from a soft and inert substrate that imbibes large amounts of water, thus mimicking critical physicochemical aspects of the HSC niche. The inertness precludes non-specific adsorption of proteins. Yet, proteins of interest can be specifically presented by incorporating in the polymer network a heterofunctional PEG linker to which ProteinA is covalently crosslinked, allowing Fc-chimeric proteins to be site-selectively anchored on the hydrogel surface. In this manner, specific Fc-chimeric proteins typical of cell–cell interactions in the niche can be tested without the complexity of co-culture. While cell trapping and high-throughput single cell experimentation is afforded by several other microwell array systems,17–22 these arrays were generally made with more rigid and hydrophobic substrates than PEG hydrogels, such as PDMS or glass.Although some other groups have previously used photopolymerized PEG to fabricate hydrogel microwell arrays for the study of embryonic stem cell cultures,23,24 the selective tethering of proteins to this substrate has not been achieved. To explore the crosstalk of adult stem cells with their niche, and elucidate the role of factors that direct their self-renewal or differentiation, both soluble and immobilized extracellular matrix and transmembraneproteins must be studied. With the platform presented in this report, this will now be possible. Our minimalist approach, a ‘deconstruction of the niche’, enables the predominant role of specific proteins to be evaluated in the absence of the complexity of the in vivo microenvironment. Further studies using this platform will build upon the results presented here, revealing not only proteins involved in self-renewal, but also expansion, presumably via presentation of protein combinations.
The single cell analyses presented here using our hydrogel microwell platform support previous studies on the role of Wnt3a on HSC fate in tissue culture. Our experiments reveal, in accordance with an earlier study by Willert et al.40 that soluble Wnt3a protein plays a role in the self-renewal of HSCs. When clones from 100 microwells were pooled and transplanted into lethally irradiated mice, reconstitution of the blood was observed (Fig. 6), a finding that could have been due to either persistence of the stem cell state or to self-renewal in these culture conditions. To distinguish between these two possibilities, we transplanted doublets, cells that divided in culture once. The finding that doublets reconstituted the blood upon transplantation is evidence that self-renewal occurred. However, since we did not separate daughter cells to probe their reconstitution potential separately, we could not determine whether Wnt3a induced asymmetric self-renewal divisions leading to stem cell maintenance or induced symmetric self-renewal divisions leading to stem cell expansion. That Wnt3a leads to HSC expansion has been suggested based on studies that either employed retroviral delivery and overexpression of active β-catenin41,42 or HSCs from BCL-2 transgenic mice.40 However, the absence of self-renewal upon transplantation of clones (≥two divisions) shown here for unmodified HSCs from wild type recipients, supports a role for Wnt3a in stem cell maintenance by asymmetric divisions rather than stem cell expansion.
Studies of the role of the Wnt pathway in HSC self-renewal in vivo using genetic gain or loss of function of catenins within HSCs themselves have yielded contradictory results. Conditional Cre-mediated deletion of β-catenin in bone marrow progenitors43 or double knockout of β- and γ-catenin44 had no impact on HSC maintenance and multilineage reconstitution, yet Wnt signaling assayed by reporter genes was functional, suggesting that catenins are not essential to canonical Wnt signal transduction.44 Moreover, in two recent studies, conditional over-expression of β-catenin led not only to forced entry of HSCs into the cell cycle and multilineage reconstitution of the blood, but also to loss of a functional HSC pool capable of long-term reconstitution and ultimately death.45,46 Notably, these studies explored the impact of intrinsic effects of the lack or excess of one component of the Wnt signaling pathway, catenins, by genetic alteration of the stem cells.
However, Wnt signaling is known to be highly context-dependent47 and perturbations of Wnt levels produced by cells other than HSCs in the niche may yield more physiologic results. Indeed, a recent study by Scadden and colleagues,48 underscored the importance of the microenvironment in Wnt signalling. When transgenic mice were made in which Dickkopf1 (Dkk1), an antagonist of Wnt/β-catenin signalling, was expressed by osteoblasts, support cells neighboring HSCs in the bone marrow, a progressive decline in long-term reconstitution potential was observed and this effect was not reversed upon transplantation of the HSCs into wild-type recipients. These data indicate that when wild-type HSCs are not exposed to Wnt in the niche, self-renewal is impaired and cannot be rescued upon transplantation into normal recipients. The failure to reconstitute in the absence of Wnt signalling appears to be due to a depletion of HSCs in the G0 phase of the cell cycle, leading ultimately to a loss of the stem cell pool. These results provide strong evidence that Wnt3a in the microenvironment is crucial for sustaining HSC quiescence and long-term self-renewal capacity, in support of our in vitro findings with cells exposed to Wnt3a in hydrogel microwells.
Our single cell analyses showed that faster proliferation as seen with TPO, can correlate with a loss of HSC self-renewal function and shifts differentiation preferentially toward a lymphoid fate. A similar inhibition of self-renewal has been reported when HSCs were exposed to TPO in commercially available cytokine cocktails.49 However, recent reports using transgenic and knockout mice have suggested that TPO plays a role in stem cell quiescence in the HSC niche as well as in expansion.12,13 The difference between our in vitro findings and these mouse genetic models likely reflects the complexity of the in vivo microenvironment and the interplay of numerous secreted and cellular factors. Exposure of HSCs to TPO alone in microwells led to one dominant behavior: excessive proliferation and loss of stem cell potential. In a multi-factorial in vivo environment TPO may have dual effects including a critical role in quiescence. Future studies using microwell arrays can now investigate the interactions of two or more proteins, for example probing the effects of TPO together with Wnt3a or N-Cadherin, simultaneously or sequentially, on HSC function, an interplay that may be necessary to induce not only self-renewal leading to HSC maintenance, as shown here, but also to HSC expansion and increased stem cell numbers.
Our results suggest that N-Cadherin, when presented in vitro immobilized on hydrogel surfaces that mimic physicochemical properties of the niche, can maintain single stem cells in a multipotent, self-renewing state. The asynchrony in divisions of stem cell daughters (Fig. 5), would suggest the involvement of asymmetric divisions; a single stem cell fate that needs to be investigated further on our microwell array platform.
N-Cadherin has been shown to be present on a subset of osteoblasts in the endosteal niche.8,9,50 Indeed, with low level reactive oxygen, osteoblastic N-Cadherin is increased in parallel with HSC quiescence and self-renewal. However, a functional role in HSC maintenance has not previously been documented. A recent report suggested that HSCs do not express N-Cadherin and that its absence, or blocking via antibodies, does not alter hematopoiesis.6 However, heterophilic interactions between N-Cadherin and other classical Cadherins, protocadherins, and atypical cadherins51 expressed by HSCs cannot be ruled out and could play an important role in the HSC niche. Moreover, recently, N-Cadherin was shown to be expressed by HSCs.52 Remarkably, N-Cadherin expression levels appear to reflect specific functional ‘states’ of HSCs; HSCs that express low amounts of N-Cadherin exhibit significantly reduced cell-cycle entry rates, whereas HSCs that express intermediate amounts of N-Cadherin appear to function as ‘reserve’ cells in the niche responsible for long-term maintenance of hematopoiesis. These studies suggest that the effects on self-renewal of HSCs observed in response to extrinsic N-Cadherin, as provided in our hydrogel microwell platform, is likely to reflect its biological role in HSC function in the niche.
Conclusions
Taken together, these experiments suggest that (1) the kinetic behavior of rare populations of adult stem cells can be systematically studied and manipulated at the single cell level using arrays of hydrogel microwells in conjunction with time-lapse microscopy, (2) cell–cell interactions can be mimicked without the complexity of co-culture by tethering properly oriented transmembrane niche proteins, (3) exposure of single HSCs for ≥4 days to single extrinsic cues typical of the niche have profound effects on stem cell function in vitro enabling reconstitution of the blood in vivo, (4) division kinetics (slow or asynchronous proliferation) of single HSCs in response single proteins (Wnt3a and N-Cadherin, respectively) can correlate with in vivostem cell function. The identification of specific molecules that influence HSC maintenance by self-renewal and ultimately HSC expansion without genetic manipulation should have clinical utility in overcoming the limitation of cell numbers currently available for transplantation.Acknowledgements
We thank our collaborators Drs M. Textor and M. Dusseiller, ETH Zurich, for the generous gift of PDMS replicas, D. Klang, ETH Zurich for help with the development of microcontact printing. In the Blau laboratory, we thank P. Kraft for help with micromanipulation, Dr P. Gilbert for help with microwell array fabrication. For critical reading of the manuscript, we especially thank Drs Seung Kim and A. Sacco. For thoughtful comments on the work, we thank Irv Weissmen. M. P. L. was supported by fellowships from the Swiss National Science Foundation and the Leukemia and Lymphoma Society. This work was supported by the Baxter Foundation and NIH grants AG009521, AG020961, AG024987 to H. M. B.References
- S. H. Orkin and L. I. Zon, Cell, 2008, 132, 631–644 CrossRef CAS.
- A. Spradling, D. Drummond-Barbosa and T. Kai, Nature, 2001, 414, 98–104 CrossRef CAS.
- E. Fuchs, T. Tumbar and G. Guasch, Cell, 2004, 116, 769–778 CrossRef CAS.
- D. T. Scadden, Nature, 2006, 441, 1075–1079 CrossRef CAS.
- K. A. Moore and I. R. Lemischka, Science, 2006, 311, 1880–1885 CrossRef CAS.
- M. J. Kiel, G. L. Radice and S. J. Morrison, Cell. Stem Cell, 2007, 1, 204–217 CrossRef CAS.
- L. M. Calvi, G. B. Adams, K. W. Weibrecht, J. M. Weber, D. P. Olson, M. C. Knight, R. P. Martin, E. Schipani, P. Divieti, F. R. Bringhurst, L. A. Milner, H. M. Kronenberg and D. T. Scadden, Nature, 2003, 425, 841–846 CrossRef CAS.
- J. Zhang, C. Niu, L. Ye, H. Huang, X. He, W. G. Tong, J. Ross, J. Haug, T. Johnson, J. Q. Feng, S. Harris, L. M. Wiedemann, Y. Mishina and L. Li, Nature, 2003, 425, 836–841 CrossRef CAS.
- F. Arai, A. Hirao, M. Ohmura, H. Sato, S. Matsuoka, K. Takubo, K. Ito, G. Y. Koh and T. Suda, Cell, 2004, 118, 149–161 CrossRef CAS.
- S. K. Nilsson, H. M. Johnston, G. A. Whitty, B. Williams, R. J. Webb, D. T. Denhardt, I. Bertoncello, L. J. Bendall, P. J. Simmons and D. N. Haylock, Blood, 2005, 106, 1232–1239 CrossRef CAS.
- T. Sugiyama, H. Kohara, M. Noda and T. Nagasawa, Immunity, 2006, 25, 977–988 CrossRef CAS.
- H. Qian, N. Buza-Vidas, C. D. Hyland, C. T. Jensen, J. Antonchuk, R. Mansson, L. A. Thoren, M. Ekblom, W. S. Alexander and S. E. W. Jabobsen, Cell. Stem Cell, 2007, 1, 671–684 CrossRef CAS.
- H. Yoshihara, R. Arai, K. Hosokawa, T. Hagiwara, K. Takubo, Y. Nakamura, Y. Gomei, H. Iwasaki, S. Matsuoka, K. Miyamoto, H. Miyazaki, T. Takahashi and T. Suda, Cell. Stem Cell, 2007, 1, 685–697 CrossRef CAS.
- J. C. Young, A. Varma, D. DiGiusto and M. P. Backer, Blood, 1996, 87, 545–556 CAS.
- T. Cheng, N. Rodrigues, H. M. Shen, Y. G. Yang, D. Dombkowski, M. Sykes and D. T. Scadden, Science, 2000, 287, 1804–1808 CrossRef CAS.
- B. Dykstra, J. Ramunas, D. Kent, L. McCaffrey, E. Szumsky, L. Kelly, K. Farn, A. Blaylock, C. Eaves and E. Jervis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8185–8190 CrossRef CAS.
- A. Revzin, R. G. Tompkins and M. Toner, Langmuir, 2003, 19, 9855–9862 CrossRef CAS.
- W. G. Koh, A. Revzin, A. Simonian, T. Reeves and M. Pishko, Biomed. Microdev., 2003, 5, 11–19 CrossRef CAS.
- M. R. Dusseiller, D. Schlaepfer, M. Koch, R. Kroschewski and M. Textor, Biomaterials, 2005, 26, 5917–5925 CrossRef CAS.
- V. I. Chin, P. Taupin, S. Sanga, J. Scheel, F. H. Gage and S. N. Bhatia, Biotechnol. Bioeng., 2004, 88, 399–415 CrossRef CAS.
- J. C. Mohr, J. J. de Pablo and S. P. Palecek, Biomaterials, 2006, 27, 6032–6042 CrossRef CAS.
- A. Khademhosseini, L. Ferreira, J. Blumling, J. Yeh, J. M. Karp, J. Fukuda and R. Langer, Biomaterials, 2006, 27, 5968–5977 CrossRef CAS.
- J. M. Karp, J. Yeh, G. Eng, J. Fukuda, J. Blumling, K. Y. Suh, J. Cheng, A. Mahdavi, J. Borenstein, R. Langer and A. Khademhosseini, Lab Chip, 2007, 7, 786–794 RSC.
- H. C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung and A. Khademhosseini, Biomaterials, 2008, 29, 752–763 CrossRef CAS.
- S. Y. Corbel, A. Lee, L. Yi, J. Duenas, T. R. Brazelton, H. M. Blau and F. M. Rossi, Nat. Med., 2003, 9, 1528–1532 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
- D. B. Weibel, W. R. DiLuzio and G. M. Whitesides, Nat. Rev. Microbiol., 2007, 5, 209–218 Search PubMed.
- D. Bryder, D. J. Rossi and I. L. Weissman, Am. J. Pathol., 2006, 169, 338–346 Search PubMed.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Adv. Mater., 2003, 15, 888–892 CrossRef CAS.
- G. J. Spangrude, S. Heimfeld and I. L. Weissman, Science, 1988, 241, 58–62 CrossRef CAS.
- M. J. Kiel, O. H. Yilmaz, T. Iwashita, O. H. Yilmaz, C. Terhorst and S. J. Morrison, Cell, 2005, 121, 1109–1121 CrossRef CAS.
- K. Levac, F. Karanu and M. Bhatia, Haematologica, 2005, 90, 166–172 CAS.
- D. L. Jones and A. J. Wagers, Nat. Rev. Mol. Cell Biol., 2008, 9, 11–21 CrossRef CAS.
- X. Song, C. H. Zhu, C. Doan and T. Xie, Science, 2002, 296, 1855–1857 CrossRef CAS.
- H. Wang, S. R. Singh, Z. Zheng, S. W. Oh, X. Chen, K. Edwards and S. X. Hou, Dev. Cell, 2006, 10, 117–126 CrossRef CAS.
- K. Willert, J. D. Brown, E. Danenberg, A. W. Duncan, I. L. Weissman, T. Reya, J. R. Yates and R. Nusse, Nature, 2003, 423, 448–452 CrossRef CAS.
- T. Reya, A. W. Duncan, L. Ailles, J. Domen, D. C. Scherer, K. Willert, L. Hintz, R. Nusse and I. L. Weissman, Nature, 2003, 423, 409–414 CrossRef CAS.
- Y. Baba, T. Yokota, H. Spits, K. P. Garrett, S. I. Hayashi and P. W. Kincade, J. Immunol., 2006, 177, 2294–2303 CAS.
- M. Cobas, A. Wilson, B. Ernst, J. C. Mancini, H. R. MacDonald, R. Kemler and F. Radtke, J. Exp. Med., 2004, 199, 221–229 CrossRef CAS.
- G. Jeannet, M. Scheller, L. Scarpellino, S. Duboux, N. Gardiol, B. J., F. Kuttler, I. Malanchi, W. Birchmeier, A. Leutz, J. Huelsken and W. Held, Blood, 2008, 111, 142–149 CAS.
- M. Scheller, J. Huelsken, F. Rosenbauer, M. M. Taketo, W. Birchmeier, D. G. Tenen and A. Leutz, Nat. Immunol., 2006, 7, 1037–1047 CrossRef CAS.
- P. Kirstetter, K. Anderson, B. T. Porse, S. E. W. Jacobsen and C. Nerlov, Nat. Immunol., 2006, 7, 1048–1056 CrossRef CAS.
- M. J. Nemeth and D. M. Bodine, Cell. Research, 2007, 17, 746–758 Search PubMed.
- H. E. Fleming, V. Janzen, C. L. Celso, J. Guo, K. M. Leahy, H. M. Kronenberg and D. T. Scadden, Cell. Stem Cell, 2008, 2, 274–283 CrossRef CAS.
- J. Audet, C. L. Miller, C. J. Eaves and J. M. Piret, Biotechnol. Bioeng., 2002, 80, 393–404 CrossRef CAS.
- Y.-Y. Jang and S. J. Sharkis, Blood, 2007 Search PubMed.
- J. M. Halbleib and W. J. Nelson, Genes Dev., 2006, 20, 3199–3214 CrossRef CAS.
- S. J. Haug, X. C. He, J. C. Grindley, J. P. Wunderlich, K. Gaudenz, J. T. Ross, A. Paulson, K. P. Wagner, Y. Xie, R. Zhu, T. Yin, J. M. Perry, M. J. Hembree, E. P. Redenbaugh, G. L. Radice, C. Seidel and L. L., Cell. Stem Cell, 2008, 2, 367–379 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and supplementary figures and movies. See DOI: 10.1039/b815718a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2009 |