Raman tweezers and their application to the study of singly trapped eukaryotic cells
Richard D.
Snook
*,
Timothy J.
Harvey
,
Elsa
Correia Faria
and
Peter
Gardner
School of Chemical Engineering and Analytical Science, Manchester Interdisciplinary Biocentre, 131 Princess Street, Manchester, UK M1 7DN
First published on 19th November 2008
Abstract
In this review the recent emergence of Raman tweezers as an analytical technique for single eukaryotic cell analysis is described. The Raman tweezer technique combines Raman spectroscopy as a diagnostic tool with optical tweezers by which means single cells can be trapped and manipulated in a laser beam using a high numerical aperture imaging microscope. Necessary instrumental requirements to facilitate Raman tweezer experiments are discussed together with practical considerations such as the potential for photodamage of cells subjected to trapping and Raman excitation. Specific applications of Raman tweezers to the analysis of cancer cells, erythrocytes and lymphocytes, micro-organisms and sub-cellular componentse.g.chromosomes and mitochondria are then discussed followed by a summary of the future potential of the technique for single cell analysis.
Insight, innovation, integrationRaman tweezers can be used to trap and interrogate single eukaryotic cells to provide a Raman spectrum of their chemical components. The technique promises to be a powerful aid to cell biologists and integrative biology because it allows species such as proteins, nucleic acids, amides and lipids to be identified in an isolated cell that is free from the influence of other cells and extrinsic factors such as inter-cell signalling events. Furthermore dynamic changes in cell composition can be observed with higher spatial resolution than obtainable with infrared spectra. An exciting application of the technique can be found in the characterisation of eukaryotic cells in different states of development such as cancer cells. Even though their Raman spectra are similar it is possible to discriminate between different cancer and non-cancer cell lines using advanced statistical treatments of their Raman spectra. The combined features of chemical identification under dynamic conditions with high enough spatial resolution to follow events at the sub-cellular level without recourse to labelling therefore offer great potential for future imaging in cell biology. |
Introduction
Quantitative imaging and analysis of single eukaryotic cells and their responses to the stimulus of individual signalling pathways in the cell's organisational structure is a challenging analytical goal. However, there are currently no established systems for image analysis at the sub-cellular level that yield non-biased chemical information. To be able to perturb and image whole cells, whilst simultaneously deriving interconnected chemical information from processes and organelles in the cell, rather than from isolated individual processes, could revolutionise quantitative measurement in systems biology, developmental biology, medicine and tissue engineering, all of which are priorities in clinical research. Current instruments and methods which life scientists can call upon to visualise particular processes and structures at the cellular level comprise confocal fluorescence microscopy together with fluorescence labelling techniques, atomic force microscopy and optical tweezer measurements. More recently there has been a surge of interest in the combination of spectroscopic methods with microscopy, particularly infrared spectroscopy, for the study of isolated cancer cells and tissues. Whilst these approaches have been successful at the cellular level, the limited spatial resolution available is not sufficient to be able to resolve sub-cellular species because the resolution limit obtainable using conventional optical microscopy and an infrared source is only a few microns. Furthermore, studies are normally performed on multiple cells grown or fixed on a surface which leaves a question about the influence of surface adhesion and cell to cell interactions upon the biochemical state of the cells studied.Given the application of such techniques in biology over the last ten years in parallel with the application of optical tweezers for single cell trapping and force measurements in bio-polymers, it is perhaps surprising the combination of these techniques in the form of Raman tweezers has only emerged as a practical tool for single cell studies in the last five years. Being able to isolate a single cell using optical tweezers and to then interrogate the cell using Raman spectroscopy brings the potential advantages of higher spatial resolution, spectroscopic identification of cellular components and freedom from intercellular and surface adhesion effects at the cellular level.
Raman spectroscopy relies upon the detection of inelastically scattered light from the sample as a result of illumination of the sample by a laser beam. Such Raman scattered light is shifted in wavelength from the pump beam wavelength either to longer wavelengths (Stokes shift, lower frequency) or shorter wavelengths (anti-Stokes shift, higher frequency) and appears as sharp spectral lines that are dispersed using a spectrometer and detected using a CCD array. Stokes shifts are more commonly observed than anti-Stokes shifts and the position of Raman lines yields information about vibrational and rotational energies of molecular bonds and chemical species in which those bonds are present. In the context of biological studies this enables chemical identification of specific components in cells, tissues and organisms.
An optical trap is created when a laser beam is brought to a tight focus by use of a high numerical aperture (NA) lens. Thus conventional single trap optical tweezers utilise inverted microscopes with a trapping laser beam passed through the microscope objective. As such, this configuration lends itself to Raman spectroscopy either by using the trapping beam as the Raman pump beam in a single beam configuration or by using a separate laser beam as the pump in a two beam system. The combination of Raman spectroscopy with optical tweezers in this way allows simultaneous trapping and analysis of a wide range of micron size particles, including biological cells, bacteria and organelles such as mitochondria.1–4 Optical trapping allows levitation of the cells above the substrate, which reduces fluorescence effects as well as Brownian motion of the un-trapped cells. Trapping in this way also eliminates the need to immobilise cells to substrates, which could alter the cells’ microenvironment.5 Conversely, Raman tweezers can be used to study the effect of the environment on a single isolated cell as elegantly demonstrated by Singh et al.6 who used Raman tweezers to detect hyperosmotic stress in trapped single yeast cells. Raman tweezers have therefore found applications in the life sciences, and more recently in bio-medicine. In this review we consider first the techniques and instrumentation required to perform spectroscopic measurements on single cells with Raman tweezers. This is followed by a review of applications of the technique for eukaryotic cell analysis, demonstrating that the Raman tweezer technique offers the unique feature of facilitating spectroscopic measurements on isolated cells in an environment free from extrinsic influences such as cell to cell interactions, cell to cell signalling and chemo-taxis. It is easy to envisage how Raman tweezers could be configured for the very purpose of studying such interactions at the microscopic scale.
Perhaps one of the reasons that the application of Raman tweezers to single live cell analysis has been adopted slowly is the early perception that it was not possible to trap cells as large as 20–30 µm, which is typical for eukaryotic cells, and that the trapping laser would damage the trapped cell through heating and photochemical effects, consequently making interpretation of cell biochemistry difficult to derive from acquired Raman spectra. However these effects, which are also discussed in this review, tend to be wavelength dependent so by careful choice of trapping laser wavelength, power and trapping time they can be minimised, leading to a rapid emergence of the technique for single cell analysis. Similarly dual beam approaches using optical fibre traps as discussed later in this review can mitigate such heating effects.
Practical requirements for Raman tweezers
Optical trapping
The principal pioneer of optical laser trapping (also known as optical tweezers) is considered to be Arthur Ashkin. In 1970 he successfully used radiation pressure from a continuous laser source to trap micron-sized particles in stable optical potential wells.7 This principle was later used to trap atoms and dielectric particles8,9 and has since become popular not only to manipulate micron-sized particles, bacteria and cells10–13 but also to measure forces in the piconewton (pN) range.An optical trap is created when a laser beam is brought to a tight focus onto a spherical dielectric particle by use of a high NA objective lens. If the refractive index of the particle is greater than the refractive index of the medium in which the particle is suspended in, then the particle near the focal point is trapped by an optical force, which is due to the transfer of momentum from the incident photons. The net trapping force is commonly considered as resulting from two component forces: the scattering force, which acts in the direction of light propagation, and the gradient force, which acts in the direction of the spatial light gradient. However, both the scattering force and the gradient force originate from the same physical phenomenon. For ease of discussion herein we will use the traditional approach and consider the net force to be composed by the scattering and the gradient force. Fig. 1 illustrates the effect of the gradient force on a dielectric particle due to a pair of rays of the focused laser beam. The gradient force pulls the particle towards the focus of the laser.
 | ||
| Fig. 1 Optical trapping of a dielectric sphere, showing refraction of rays a and b. The sum of Fa and Fb gives the restoring force, Fgrad. For stable traps in which gradient force > scattering force the particle is trapped near the focal point. The graded shaded area represents the intensity gradient of the focused laser. | ||
In an optical trap the gradient force is created when fluctuating dipoles in the particle, caused by the laser irradiation, interact with the heterogeneous electric field at the focal point. For optical trapping of a dielectric particle, the gradient force pulling the particle towards the focal point must be greater than the scattering component of the force pushing the particle in the opposite direction (away from the focal point). Equilibrium is reached just beyond the focal point. If the particle moves laterally, away from the centre of the trap, a restoring force will bring the particle back into the centre. For small displacements of <150 nm the trap behaves like a Hookean spring and the force imparted on the particle is directly proportional to the displacement. The proportionality constant, termed the spring constant, is proportional to the intensity of the light. This characteristic of the optical trap allows it to be used to apply and to measure forces up to ca. 100 pN, such as those involved in the unfolding of RNA and the compliance of cellular membranes.14,15
Although the forces experienced by spherical particles can be computed using the Mie scattering theory (for particles much bigger than the trapping wavelength) and Rayleigh scattering theory (when the wavelength is much bigger than the particle),16 and some progress has been made in the theoretical determination of the forces for particles of size similar to that of the wavelength used, in practice the forces applied are determined empirically.
In Raman tweezers the optical trap is merely used to select and hold cells in place to enable the Raman spectrum to be acquired and hence we will not discuss further the use of optical tweezers to apply and to measure forces. For further information on force measurements the reader should refer to the comprehensive review by Svoboda and Block17 and for information on the practical aspects of designing and building optical traps, refer to the very useful review on optical trapping published by Neuman and Block.18
Applications of optical trapping
In the context of this review Ashkin and Dziedzic demonstrated one of the first published applications of optical tweezers in biology; by trapping and manipulating bacteria and viruses.10 This was followed, in short succession, by further work concerning laser trapping and manipulation of bio-particles (mainly micro-organisms),11–13 which in many ways presaged the development of the technique in biology and the techniques described here.In addition to simply isolating a single cell, optical tweezers have the potential to investigate the physical properties of cells, and assess the link between cell elasticity, membrane elasticity and disease state.15 In particular this has been used to study red blood cells19 and also to relate elastic properties of cancer cells to their malignancy. Guck et al. used the same principles of optical trapping to design an “optical stretcher” to show that malignant breast cells were more easily deformed than normal cells, and that deformability increased with an increase in the invasiveness of the cells. The greater deformation for the malignant cells is thought to be due to the fact that the cells need to deform so they can pass through the surrounding tissue matrix to the circulatory system.20 This method could be used as a microfluidic cell-sorting and diagnostic technique based on membrane deformability.21 As of yet this is the only research published in this area. For further applications of optical tweezers in biology refer to ref. 22.
Raman spectroscopy
The essential feature of a laser source for Raman tweezers, or indeed any other form of Raman spectroscopy, is that it emits coherent monochromatic light of high intensity. Depending on the wavelength and power required, there are a range of lasers available.23 In the UV/visible region these include Nd∶YAG lasers (532 nm—i.e. frequency doubled 1064 nm), argon ion lasers (514, 488, 457 nm and frequency doubled lines at 257 and 244 nm) and krypton ion lasers (752, 647, 406 nm) lasers. In addition to the 1064 nm Nd∶YAG laser, diode lasers, Ti∶sapphire and Nd∶YVO4 (914 nm) provide wavelengths in the near infrared region between 690 and 1000 nm. For tweezer applications typical powers used range from 5 mW to a few W, a range which is also suitable for Raman spectroscopy.Continuous-wave (CW) lasers that emit a beam with a Gaussian beam intensity profile are generally used for Raman tweezer applications. The choice of laser wavelength has several implications. Firstly, the Raman scattering efficiency is inversely proportional to the fourth power of the wavelength.24 Using a long wavelength will result in less Raman light reaching the detector than a shorter wavelength thus reducing signal-to-noise ratio (SNR). Secondly, potential photodamage is linked to wavelength (see below). The use of longer wavelengths however can reduce competing fluorescence effects.24 The response of charged-coupled devices (CCD), which are used as detectors, also depends on wavelength; for a typical front illuminated silicon CCD, maximum quantum efficiency (approx. 50%) occurs at between 600 and 800 nm although the quantum efficiency remains above 30% down to a wavelength of 200 nm. At the red end the CCD response is only 10% at 1000 nm and falls to near 0% at 1100 nm thus restricting its use in the near infrared although recent CCD detectors may extend further into this region.
Optical requirements
In conjunction with an high quality inverted microscope high quality optics are required to direct laser light to the sample and to direct Raman light from the sample to the detector. Optical components are also required to remove Rayleigh scatter of the Raman pump laser line and other unwanted laser radiation that is present in the Raman tweezer system. Some optics commonly employed in a typical Raman tweezers set up, used for live cell analysis by the authors (at the Rutherford Appleton Laboratory, Raman Tweezers Facility, Oxford, UK) are shown schematically in Fig. 2 and 3.25 | ||
| Fig. 2 A typical back-scattering single-beam Raman tweezers configuration showing mirrors (M); edge filter (EF); dichroic mirror (DM); dichroic filter (DF) and spectrometer. By permission from the Journal of Biomedical Optics.25 | ||
 | ||
| Fig. 3 A typical forward-scattering double-beam Raman tweezers configuration showing mirrors (M); edge filter (EF); dichroic mirror (DM); bandpass filter (BP); Raman differentiator (RD) and spectrometer. By permission from the Journal of Biomedical Optics.25 | ||
In the set-ups shown in Fig. 2 and 3 dichroic filters and mirrors are used which selectively transmit desired wavelengths whilst reflecting others. A reflective dichroic edge filter at the base of the microscope will transmit laser light but reflect Raman scattered light. A Raman differentiator is a dichroic mirror with a shallow transition, which transmits 514.5 nm light and reflects 525–630 nm Raman light at a 30° angle. An edge filter is so called because the transition from reflection to transmission occurs in a couple of nanometres at an angle of 45°. In the configurations shown in Fig. 2 and 3 the edge filter was used to clean up any remaining stray Rayleigh scatter.
Spectrometer and detector
The vast majority of Raman spectrometers utilise grating spectrometers and CCD detectors to process the Raman signal. Raman light enters the spectrometer via an entrance slit, where it is dispersed into separate wavelengths by a grating. Spectral resolution is determined by a combination of the size of the entrance slit and the diffraction grating groove density.26 The latter is defined as the number of grooves per unit length. A grating is also defined by the blaze wavelength, which is the wavelength at which it is at its most efficient. This defines the wavelength range over which the spectrometer can be operated. Reducing the entrance slit and/or increasing the grating groove density will increase spectral resolution. The latter has the downside of reducing the spectral wavelength range, whilst reducing slit width will reduce light throughput which will ultimately degrade the signal to noise ratio (SNR), assuming that the principal source of noise derives from the detector as dark current. Dark current is a residual thermally generated signal from the CCD and is temperature dependent. Consequently, CCDs are often cooled thermoelectrically or by using liquid nitrogen to reduce the amount of dark current generated by the detector, thus reducing noise to increase the SNR. Most CCDs are now cooled thermoelectrically. However liquid nitrogen cooled CCDs are still available.The output spectrum will of course be a combination of signal from the sample as well as noise. The source of limiting noise is an important consideration in Raman spectroscopy because the high levels of noise can severely reduce spectral quality and instrument precision. The observed spectral noise can be subdivided into two sources; additive (background) noise and multiplicative noise. Additive noise is always present, regardless of the presence of a sample, and comprises dark current as well as noise arising from the blank (when no sample is present). Additive noise is independent of the signal strength, whilst multiplicative noise increases proportionately with signal strength.
Shot noise is due to the quantum nature of matter and photons, and its magnitude is proportional to the square root of the signal.27 Therefore, increasing integration time or summing spectral readouts will increase the SNR. Johnson noise occurs as a result of the thermal motion of electrons in resistive elements of electric circuits, and is independent of signal magnitude.27 Flicker noise is largely associated with the laser source and is dependent on the signal magnitude.26 Therefore, if flicker noise is the dominant noise contribution no improvement can be made by increasing integration time or summing spectral readouts.
Raman tweezer configurations
Including the configurations shown in Fig. 2 and 3 there are four possible Raman tweezers configurations as shown in Fig. 4. These configurations are based on an inverted microscope. | ||
| Fig. 4 Possible Raman tweezers configurations in an inverted microscope. The graded shaded areas represent the laser intensity gradient of the optical trap and the arrows the direction of propagation of the excitation beam and the collected Raman light. | ||
Configurations 2 and 4 in which different lasers are used for trapping and excitation have some advantages over single beam setups. One such advantage includes more freedom to manipulate the trapped cell with increased sensitivities, because the cell and the focus of the laser can be aligned properly allowing for optimisation of the signal. Furthermore, such configurations allow lasers to be selected taking into account the need to minimise damage to cells caused by the trapping laser, Raman scatter efficiency at the excitation wavelength and the quantum efficiency of the CCD detector.
Trapping powers can vary from a few mW to over 1 W in the specimen plane, though more commonly a few to a few hundred mW are commonly used. The excitation power used is usually a few to a few tens of mW, i.e. when the excitation laser is not the trapping laser.
Photodamage considerations
The use of high powered, high intensity lasers required for Raman tweezers leads to local intensities exceeding MW cm−2.33 Lasers are known to activate photochemical reactions within cells.34 Therefore, the potential for cell damage (photodamage) is a concern when designing Raman tweezer systems. Laser photodamage of cells can include: photochemical damage (e.g. oxidation, DNA damage, cell metabolism); photothermal (heat induced damage); bubble formation; acoustic wave formation and photomechanical stress.35 Photothermal and photochemical effects are discussed in the following text.Photothermal damage
Research by Liu et al.36,37 determined that the temperature increase for a trapped Chinese hamster ovary (CHO) cell was between 1–1.15 °C per 100 mW power rise for a 1064 nm Nd∶YAG operating at powers of up to 400 mW. Theoretical calculations by Block et al.38 and Ramser et al.39 suggest laser induced temperature increases are in the order of 1 kelvin per watt (K W−1) for wavelengths between 488 and 830 nm. These small temperature rise values have allowed researchers to assume that thermal damage from a continuous wave laser source can be neglected over the relatively short periods required for trapping and analysis.40 However, Berns41 advises that for laser powers of 300 mW or greater, the effect of temperature should not be dismissed.Intuitively it might seem that photothermal damage would be related to the absorbance of the trapped object and the efficiency of subsequent photothermal conversion to heat, i.e. low absorbance produces little photothermal damage. However in a study performed by Peterman et al. on Brownian motion of polystyrene beads and silica beads in both water and glycerol33 the role of the suspending medium was explored. From experimental data, temperature increases at the laser focal point of 7.7 ± 0.1 K W−1 for 500 nm silica beads and 8.1 ± 2.1 K W−1 for 444 nm silica beads were observed. These researchers found that the most significant contribution to heating was not direct absorption by the beads, but rather absorption of laser light in the water, causing indirect heating of the beads. They also observed that the heating effect is increased as the distance between cover slip, on which the beads suspension was placed, and the particle is increased. These results have potentially important implications when designing Raman tweezer systems.
Photochemical damage
Researchers have postulated two likely mechanisms of photochemical damage in cells: single photon absorption resulting in formation of reactive species (e.g. singlet oxidation), and two-photon absorption.34,37,40,42,43 Neuman et al.40 found strong evidence for the role of oxygen in photodamage by exposing trapped E. coli bacteria to aerobic and anaerobic conditions and measuring the LD50 of the cell population. A three to six fold increase in LD50 was found when oxygen was removed. Using the same cell types Neumann found a near-linear relationship between sensitivity and laser power, which suggested the role of single-photon absorbance in photodamage. Xie42 also postulated the role of reactive species and photon absorption in the photochemical damage. Xie used the Raman peak at 1606 cm−1, assigned to aromatic amino acids, to monitor photodamage in yeast cells, and found that there was a reduction in this peak’s intensity with increased exposure to laser light at 690 nm. The author suggested that this reduction could be due to oxidation of the aromatic ring by singlet oxidation, or by a two-photon process inducing photodynamic reactions.Relationship between photodamage and wavelength
Both Neuman et al.40 and Liang et al.44 carried out studies to determine the effect of the use of different laser wavelengths on the viability of cells. Liang measured the cloning efficiency of CHO cells exposed to different wavelengths, whilst Neuman measured the LD50 of E. coli bacteria. When plotted on the same graph there is general agreement between the two, which suggests that the photodamage mechanisms are similar for the two cells (Fig. 5). The results for E. coli demonstrate that the most damaging wavelengths occur at 870 and 930 nm, whilst the least damaging occurs at 970 and 830 nm.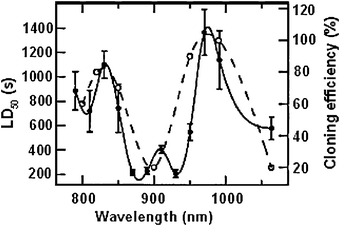 | ||
| Fig. 5 Relationship between wavelength and cell photodamage for E. coli (solid line and left axis) and CHO cells (dashed line and right axis). The higher the LD50 and % cloning the less damage the laser causes for a given wavenumber. Figure reproduced from Neuman et al.40 with permission from the Biophysical Society. | ||
The results summarised in Fig. 5 are limited however, as wavelengths of less than 800 nm were not studied. A study by Puppels et al.45 showed that photodamage occurred for human lymphocytes and chromosomes using a confocal Raman microspectrometer wavelength of 514.5 nm at powers as low as 5 mW at the sample focus. However such damage was not observed for DNA solutions and histoneprotein solutions at powers of 25 mW. It was also noted that no damage to cells and chromosomes occurred when a laser wavelength of 660 nm was used. It was thought by the authors therefore that the most plausible mechanism of damage was likely to be photochemical oxidation of DNA bases and amino acids, and not multiphoton or heat induced damage.
Applications of Raman tweezers in biology and bio-medicine
Cancer studies
Given the success of the application of infrared vibrational spectroscopy to discriminate between cancer and non-cancerous cells46–49 it is perhaps not surprising that there has been significant work done with Raman tweezers in this area. Thus early work by Zheng et al.50 describes the potential of Raman tweezers in cancer diagnosis. These authors report the use of Raman tweezers to analyse 200 normal and 200 cancerous colorectal cells.50 Their research applied artificial neural networks (ANNs) to PCA reduced data (PCA—principal component analysis) to classify obtained spectra. A description of ANN can be found in ref. 50. A training set of 320 total spectra was generated and blind-tested by the remaining 80 spectra. Sensitivities and specificities of 86.3% each were obtained. A double-blind test gave sensitivities and specificities of 85% and 92.5%, respectively. These terms are commonly used clinical parameters to describe the performance of diagnostic models. Sensitivity is a measure of the probability of the diagnostic model giving a positive cancer test for patients who have the disease. Specificity is a measure of the probability of the diagnostic model giving a negative cancer test for patients who do not have the disease. High values approaching 100% indicate good diagnostic models, and are desirable for robust clinical tools.Chen et al.51 also obtained Raman spectra from optically trapped cancerous and normal colorectal epithelial cells. The aim of this research was to develop diagnostic techniques that can be used to investigate intact cells. Cell suspensions were obtained from tissue of patients suffering from colorectal adenocarcinomas. Assessment of the Raman spectra suggested that cancerous cells contained higher amounts of nucleic acids and proteins, which were consistent with other Raman studies.52,53 This work was progressed by generating diagnostic models, which by blind testing gave sensitivity and specificity results of 82.5% and 92.5%, respectively.
The requirement to improve the accuracy of diagnosis in prostate cancer is pressing given the high rate of false positives that are evident with the prostate specific antigen test (PSA) which leads to unnecessary biopsies. As mentioned in the introduction of this review, Guck20 utilised an optical stretcher to study the change in elastic properties of mammalian cells that occur as cells become more cancer-like. The hypothesis that cells become less elastic as they progress to a more invasive and hence metastatic form has been tested for prostate cancer cell lines by Correia Faria et al.54 using atomic force microscopy (AFM). The same group has recently applied Raman tweezers in both forward and back-scattering modes to enable discrimination between prostate cancer cells (PC-3) and bladder cell lines (MGH-U1) using PCA techniques for statistical analysis,25 which is a necessary precursor study to the development of a simple urine test. In this study, the effect of differing cell sizes between PC-3 and MGH-U1 was eliminated as a reason for effective discrimination. Furthermore, through loading plot analysis at different wavelengths, it was tentatively suggested that the principal reason that discrimination can be obtained is that nucleic acid and proteins are present in relatively higher amounts in MGH-U1 than PC-3 cells. A typical Raman spectrum obtained for a trapped cancer cell is shown in Fig. 6.55
 | ||
| Fig. 6 A Raman spectrum of a single trapped PC-3 cancer cell using the back-scattering Raman configuration and pump wavelength of 514.5 nm.55 | ||
As can be seen from this example the Raman spectrum is rich in chemical species information, for example the amide I and amide II bands can be seen at approximately 1650 cm−1 and 1570 cm−1, respectively. The lipid region around 1200 cm−1 to 1400 cm−1 (CH2 wagging) is dominant and a protein associated phenylalanine peak at 1002 cm−1 is clearly observed. The band at 860 cm−1 can also be attributed to proteins and weaker bands below 800 cm−1 can be attributed to the presence of nucleic acids.31
Deng et al. carried out preliminary Raman tweezer studies on normal and cancerous colorectal cells.56 Initial results suggested that the intensity of the band ratio, 1002/1300, was 1.08 for normal and 0.85 for cancerous cells. Consultation of ref. 56 indicates that the 1002 cm−1 is a peak due to proteins and that the peak at 1300 cm−1 is due to lipids, suggesting that these non-cancerous cells may contain more proteins, but less lipids. Hamden et al.28 analysed the virus-infected cancerous haematopoietic cell line , BCBL-1 and BC-1 and the normal non-infected BJAB cell line . Analysis of the spectra showed increases in the intensity of proteins and nucleic acid moieties for infected cells. Subsequent PCA showed significant separation between cancerous and normal cells, with the cancerous cells displaying a large data spread. This observation demonstrated the heterogeneity nature of cancerous cells in relation to normal cells.
A short study into the potential of Raman tweezers to distinguish between the normal brain cell, astrocyte, and its cancerous equivalent, astrocytoma, was carried out by Banerjee and Zhang.57 These researchers found an increase in spectral intensity of protein and lipid moieties for cancerous compared with normal cells. This research shows promise, with the potential for more comprehensive studies in the future.
Erythrocyte and lymphocytes studies
Raman tweezers have found particular useful applications for the study of erythrocytes (red blood cells) and lymphocytes (white blood cells). This technique could have potential as a diagnostic tool for haematological malignancies such as leukaemia and lymphomas. Erythrocytes and lymphocytes differ primarily from epithelial cells in that they are a smaller size (~7 µm), allowing for more effective trapping. Erythrocytes also differ in that they contain haemoglobin, which influences their optical properties and thus trapping efficiency, and they differ in their morphology, as they are disc shaped, compared with the more rounded epithelial cell.Raman spectra of normal and transformed (neoplastic) lymphocytes cells were obtained by Chan et al.58 Spectra showed lower DNA band intensities for neoplastic than normal cells, in contrast to FTIR (Fourier transform infrared) spectra, which showed higher DNA intensities (the FTIR study was cited in ref. 58—no original FTIR work was done in this paper). A possible explanation for this discrepancy is as follows. The neoplastic cells have increased transcription and replication activities. This requires an open configuration of the chromatin resulting in lower DNA concentrations in the Raman probe volume compared with normal cells, which have a more compact chromatin configuration. However, for FTIR the more compact chromatin could result in lower DNA intensities for normal cells compared with neoplastic cells. This is because the highly compact chromatin in the normal cells would make the nucleus virtually opaque to IR light, resulting in lower DNA signals. This theory was postulated by Mohlenhoff et al. to explain the absence of DNA signals in the spectra of pyknotic cells.59
Further research by Chan et al.demonstrated that highly reproducible Raman spectra could be obtained by trapping live leukaemia cells obtained from patients suffering from the disease.60 The authors suggest that this high reproducibility is likely due to the consistent trapping of the nucleus, and that due to the high N/C ratio of these cells, contributions from non-nucleic material are likely to be minimal. Chan was also able to demonstrate that healthy and diseased live cells could be separated using PCA. Supervised classification by PC-LDA (LDA—linear discriminant analysis) gave average sensitivities of 95% and 90% for normal and diseased cells, respectively. Whilst this is a useful study using real clinical samples, the number of different patients used in the study is unclear. For a full clinical study a large cohort of patients would be required to ensure results are statistically and clinically significant.
Multiple trapping, in which cells are trapped using more than one beam, was investigated by Cojoc et al.30 In this study red blood cells (RBCs) were trapped at multiple locations by the use of a 1064 nm Nd∶YAG laser, split into multiple beams by a diffractive beamsplitter. The sample was probed with an Ar-ion laser at 514.5 nm. The focus of this paper was directed towards the development of instrumentation and practical considerations. However the authors report that multiple trapping has three distinct advantages. Firstly, cell stability allows for spatial mapping of the cell. Secondly, the RBC can be trapped in a horizontal position, allowing for lateral movement of the cell and thus excitation from different regions and finally, beam power is delocalised over the cell, reducing potential photodamage.
There is a considerable need for dynamic studies and in this respect Deng et al.56 investigated the effect of alcohol on RBC using Raman tweezers. Spectra of cells exposed to 20% alcohol were recorded as a function of time. Results showed that band intensities assigned to haemoglobin groups depleted with increased alcohol exposure as was to be expected. Similarly, Ramser et al.39 developed a dual-beam Raman tweezers system to study erythrocytes. Raman spectra were obtained from RBC using three different excitation wavelengths; 446, 514.5 and 568.2 nm. The 514.5 nm laser gave the highest spectral quality, with the 446 nm line suffering from a large fluorescence background. A time series experiment was carried out, which involved recording five consecutive spectra, of 30 seconds each, for both the 568.2 and 514.5 nm lines. The quality of the spectra from the 568.2 nm excitation wavelength made it difficult to ascertain if any spectral changes had taken place, whilst for the 514.5 nm line fluorescence increased with time. Despite the limitations of the spectra, frequency shifts for peaks in the 1500 to 1650 cm−1 region can be observed, which are sensitive to photo-dissociation of oxygen from haem groups.
The influence of multi-stacking of RBCs in a laser trap on signal strength and signal-to-noise ration (SNR) was investigated by Jess et al.61 Results showed an improvement in both signal strength and SNR when the number of cells vertically stacked in the trap increased from one to three. However, this methodology of stacking cells might prove problematic when larger epithelial cells are considered.
Micro-organisms
Raman tweezers have also been applied to identify micro-organisms. A Raman tweezer study was carried out by Xie et al.,29 who sorted micro-organisms by use of Raman tweezers. Xie and co-workers analysed live and dead yeast cells, and then compared the spectra against known spectra of live and dead cells. Once the cell status was determined, they were transported in the laser trap via a micro-chamber to a sorting chamber where the cells adhered to a substrate. Validation of this method was determined by staining cells with eosin, which only stains dead cells. For six sorted live and dead cells the accuracy of this method was 100%. However this study was only carried out on six cells. A bigger study would be necessary to confirm these findings. Within the same study the researchers sorted micron size particles in spoiled milk, and by doing so were able to discriminate bacteria from food particles.29 In this publication the researchers have demonstrated the use of Raman tweezers as an alternative to flow cytometry for the sorting of micro-organisms.Raman spectra of trapped E. coli bacteria and yeast using a 785 nm laser were obtained by Xie and Li.5 Spectra of trapped E. coli cells displayed significantly better SNR and reduced background contributions compared with spectra of cells immobilised onto a glass substrate. This is a good example of the benefits of obtaining Raman spectra of trapped cells rather than immobilised cells. In the same publication a study of E. coli cells trapped at different temperatures found significant reduction in nucleic acid bands above 60 °C. Further work by this group showed that Raman tweezers could be used to discriminate single bacterial cells in various growth conditions.62 Results showed that both unsupervised and supervised chemometric methods could be used to classify different bacterial types.
Creely et al.31 applied Raman tweezers using a wavelength of 785 nm to obtain spectra of yeast cells. Results over a trapping period of 2 hours showed minimal cell damage with no observable change in protein confirmations. Further studies in this field by the same group looked at observing the production of ethanol and glycerol by trapped yeast cells exposed to a glucose (hyperosmotic) environment, as well as changes in biochemical compositions during the lag and G1 phase of the growth and cell cycles.63
Chan et al.64 obtained Raman spectra of trapped bacterial spores, polystyrene and glass beads. A concentration analysis of a mixture of these particles showed that, by sorting using Raman spectra, a good correlation was achieved in comparison with the known composition. The spectra were of quite poor quality, but this paper outlines an important proof of principle: that Raman tweezers could be used to sort mixtures of unknown composition.
Studies of organelles and sub-cellular structures
So far this review has concentrated on the application of Raman tweezers to single cells. However there has been some significant work in recent years concerned with Raman analysis of sub-cellular components that have been excised from the parent cell. As stated earlier Puppels et al.45 had developed a non-tweezer confocal Raman system for studying chromosomes and this is a theme that has also been applied latterly using Raman tweezers. Thus Ojeda et al.65 have applied Raman tweezers for spectral analysis of chromosomes as an alternative to cytogenetic techniques based on staining methods. The method utilised a generalised discriminate analysis (GDA, a supervised classification model) to compare spectra obtained from human chromosomes 1, 2 and 3 after G-banding for positive identification and it was shown that it is possible to segregate these and identify human chromosomes without chromatin staining. These authors postulated that the technique could in future be applied to chromosome identification and more detailed chromosomal structure than with current staining methods. This claim has caused some controversy largely over the validity of the evaluation of Raman spectra using the GDA technique. Bak and Jørgensen66 questioned the robustness of the experimental method employed in terms of proven reproducibility and the variance in the spectra. Furthermore, these authors felt that it had not been shown that the GDA model used could generalise to other cases in the absence of a cross-validation procedure. Ojeda et al.67 in a subsequent letter defended their original paper robustly reiterating its value in demonstrating the potential of the Raman tweezers technique. Whatever outcome, this exchange of letters highlights potential pitfalls that could arise using advanced chemometric techniques such as PCA, LDA and GDA to extract meaningful differences between spectra. It is essential that the sources of variance upon which the models operate are indeed in the original data and not derived from experimental variability.In a more clear-cut application of near-infrared Raman tweezers Tang et al.4 have applied the technique to an investigation of single trapped mitochondria isolated from rat liver, heart muscle, and kidney tissue. They recorded Raman peaks corresponding to lipids, proteins and nucleic acids and hypothesised that differences in the mitochondria extracted from different tissues were due to differences in the content of these components. For example, differences between spectra from liver mitochondria and heart mitochondria might arise from differences in the lipid composition and molecular weight of proteins. Studying the decrease in the 1602 cm−1 Raman peak when mitochondria were subjected to 100 µM Ca2+ in KClbuffer solution enabled these authors to study swelling of the mitochondriai.e. a measure of the bio-activity of individual mitochondria. This is an important result as it indicated the Raman tweezers could be developed as a non-invasive technique to study dynamic bioactivity of cells and cell components towards for example drugs and toxins.
Using a less conventional configuration a Raman tweezer study into primary human keratinocyte (PHK) cells was carried out by Jess et al.68 The cell was trapped by two fibre laser beams propagating in opposite directions directed from either side of the microscope stage, whilst the cell was interrogated with a laser from below the stage, and perpendicular to the trapping beams. Using this configuration the trapped cell was stable enough to obtain spectra from the nucleus, cytoplasm and membrane components. Furthermore the resulting spectra were of sufficient quality to afford separation between spectra obtained from the nucleus, cytoplasm and membrane using PCA.
Summary and future potential
At the outset of this review we stated that “there are currently no established systems for image analysis at the sub-cellular level that yield non-biased chemical information.” Based on the work reported in this review it is evident that the future development of Raman tweezers could make a major contribution to achieving the goal of providing such information as both static and dynamic measurements can be made on single isolated cells and organelles. At present work on sub-cellular species using tweezers requires the excision of the species from the cell but it is reasonable to expect that this will be achieved within the cell provided that the particular species to be studied can be correctly identified, imaged and trapped in the cellular medium.To illustrate the possibilities we have to go back to an early but ingenious optical trapping experiment by Liang et al.69 In this work a Nd∶YAG laser at a wavelength of 1.06 µm was focused into a phase contrast microscope to form an optical trap in which mitotic chromosomes were manipulated in PTK2 cells (from the kidney of the male rat kangaroo; Potorous tridactylis). The trapped chromosomes were seen to initiate movement of the meta-phase plate. These experiments demonstrated that forces could be applied directly to sub-cellular components using optical tweezers and it is reasonable to assume that these types of experiments could be combined with Raman identification in first steps towards gaining dynamic chemical information of trapped sub-cellular components.
Conventional Raman tweezer apparatus employ a trapping beam of Gaussian radial intensity profile i.e. a TM00 mode beam that normally require spherical symmetry of the trapped object for efficient trapping. There is however great potential in the use of Laguerre–Gaussian (LG) mode laser beams70 to trap irregularly shaped objects and cells with minimum interaction between the beam and the cell. Essentially an LG mode beam produces a radially symmetric doughnut shaped beam in which the object is trapped in the centre of the doughnut beam thus minimising interaction with the beam itself and avoiding photodamage. By using a second probe laser of shorter wavelength tightly focused beam may offer improvements for improved mapping of the cell and monitoring of cellular processes, e.g. taking place in organelles.
A further but significant advantage that has been reported by Cormack et al.71 is the reduction in background fluorescence from optical components in the Raman system when using an LG and holey Gaussian trapping beams when compared to a Gaussian beam. To achieve this reduction the optical Raman signal is collected from the dark space in the LG annulus where the trapped object (in this case a polymer sphere) is partly located. Combined with the use of multiple traps that have been reported30,72 and notwithstanding the caveats in Cojac et al.,30 microfluidic platforms that allow the trapping and interrogation and Raman imaging of many cells simultaneously promise to provide an extremely powerful diagnostic tool for eukaryotic cell biology. Furthermore the combination of Raman spectroscopy with combined AFM and tweezer systems will undoubtedly open up the possibility of studying dynamic biochemical changes in isolated cells when subjected to physical as well as chemical stress.
References
- C. Xie, M. A. Dinno and Y.-Q. Li, Opt. Lett., 2002, 27, 249–251 Search PubMed.
- J. W. Chan, A. P. Esposito, C. E. Talley, C. W. Hollars, S. M. Lane and T. Huser, Anal. Chem., 2004, 76, 599–603 CrossRef CAS.
- C. Xie, C. Goodman, M. A. Dinno and Y.-Q. Li, Opt. Express, 2004, 12, 6208–6214 CrossRef.
- H. Tang, H. Yao, G. Wang, Y. Wang, Y.-Q. Li and M. Feng, Opt. Express, 2007, 15, 12708–12716 CrossRef.
- C. Xie and Y.-Q. Li, J. Appl. Phys., 2003, 93, 2982–2986 CrossRef CAS.
- G. P. Singh, C. M. Creely, G. Volpe, H. Grotsch and D. Petrov, Anal. Chem., 2005, 77, 2564–2568 CrossRef CAS.
- A. Ashkin, Phys. Rev. Lett., 1970, 24, 156–159 CrossRef CAS.
- A. Ashkin, J. M. Dziedzic, J. E. Bjorkholm and S. Chu, Opt. Lett., 1986, 11, 288–290 Search PubMed.
- S. Chu, J. E. Bjorkholm, A. Ashkin and A. Cable, Phys. Rev. Lett., 1986, 57, 314–317 CrossRef CAS.
- A. Ashkin and J. M. Dziedzic, Science, 1987, 235, 1517–1520 CrossRef CAS.
- A. Ashkin, Gravitational Space Biol., 1991, 4, 133–146 Search PubMed.
- A. Ashkin and J. M. Dziedzic, Ber. Bunsen-Ges., 1989, 93, 254–260.
- A. Ashkin, J. M. Dziedzic and T. Yamane, Nature, 1987, 330, 769–771 CrossRef CAS.
- H. Changbong and D. Thirumalai, Biophys. J., 2007, 92, 731–743.
- J. Dai and M. P. Sheetz, in Methods in Cell Biology, Volume 55—Laser Tweezers in Cell Biology, ed. M. P. Sheetz, Academic Press, London, 1998, ch. 9, pp. 157–171 Search PubMed.
- A. Ashkin, Biophys. J., 1992, 61, 569–582 Search PubMed.
- K. Svoboda and S. M. Block, Annu. Rev. Biophys. Biomol. Struct., 1994, 23, 247–285 CrossRef CAS.
- K. C. Neuman and S. M. Block, Rev. Sci. Instrum., 2004, 75, 2787–2809 CrossRef CAS.
- J. Sleep, D. Wilson, R. Simmons and W. Gratzer, Biophys. J., 1999, 77, 3085–3095 CrossRef CAS.
- J. Guck, S. Schinkinger, B. Lincoln, F. Wottawah, S. Ebert, M. Romeyke, D. Lenz, H. M. Erickson, R. Ananthakrishnan, D. Mitchell, J. Käs, S. Ulvick and C. Bilby, Biophys. J., 2005, 88, 3689–3698 CrossRef CAS.
- S. Suresh, Acta Biomater., 2007, 3, 413–438 CrossRef.
- Methods in Cell Biology, Volume 55—Laser Tweezers in Cell Biology, ed. M. P. Sheetz, Academic Press, London, 1998 Search PubMed.
- R. L. McCreery, in Modern Techniques in Raman spectroscopy, ed. J. J. Laserna, Wiley, Chichester, 1996, pp. 41–72 Search PubMed.
- E. Smith and G. Dent, Modern Raman Spectrsocopy—A Practical Approach, Wiley, London, 2005, ch. 1, pp. 8–12 Search PubMed.
- T. J. Harvey, C. Hughes, A. D. Ward, E. Gazi, E. Correia Faria, R. Snook, M. Brown, N. W. Clarke and P. Gardner, Proceedings of the XXI International Conference on Raman Spectroscoscopy, ed. R. Withnal and B. Z. Chowdhry, IM Publications, Chichester, 2008, p. 1015 Search PubMed; T. J. Harvey, E. Correia Faria, A. Henderson, E. Gazi, A. D. Ward, N. W. Clarke, R. D. Snook and P. Gardner, J. Biomed. Opt., 2008, 13, 064004–064011 Search PubMed.
- M. J. Pelletier, in Analytical Applications of Raman Spectroscopy, ed. M. J. Pelletier, Blackwell Science, Michigan, 1999, ch. 2, p. 75 Search PubMed.
- K. A. Rubinson and J. F. Rubinson, Contemporary Instrumental Analysis, Prentice Hall, New Jersey, 2000, pp. 759–762 Search PubMed.
- K. E. Hamden, B. A. Bryan, P. W. Ford, C. Xie, Y.-Q. Li and S. M. Akula, J. Virol. Methods, 2005, 129, 145–151 CrossRef CAS.
- C. Xie, C. De and Y.-Q. Li, Opt. Lett., 2005, 30, 1800–1802.
- D. Cojoc, E. Ferrari, V. Garbin and E. Di Fabrizio, Proc. SPIE, 2005, 5930, 1–11.
- C. M. Creely, G. P. Singh and D. Petrov, Opt. Commun., 2005, 245, 465–470 CrossRef CAS.
- R. Thurn and W. Kiefer, Appl. Opt., 1984, 24, 1515–1519 Search PubMed.
- E. J. G. Peterman, F. Gittes and C. F. Schmidt, Biophys. J., 2003, 84, 1308–1316 CrossRef CAS.
- P. P. Calmettes and M. W. Berns, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 7197–7199 CrossRef CAS.
- D. O. Lapotko and V. P. Zharov, Lasers Surg. Med., 2005, 36, 22–30 CrossRef.
- Y. Liu, D. K. Cheng, G. J. Sonek, M. W. Berns, C. F. Chapman and B. J. Tromberg, Biophys. J., 2005, 68, 2137–2144.
- Y. Liu, G. J. Sonek, M. W. Berns and B. J. Tromberg, Biophys. J., 1996, 71, 2158–2167 CrossRef CAS.
- S. M. Block, in Noninvasive Techniques in Cell Biology, ed. S. Grinstein and K. Foskett, Wiley-Liss, New York, 1990, pp. 375–401 Search PubMed.
- K. Ramser, K. Logg, M. Goksör, J. Enger, M. Käll and D. Hanstorp, J. Biomed. Opt., 2004, 9, 593–600 CrossRef.
- K. Neuman, E. H. Chadd, G. F. Liou, K. Bergman and S. M. Block, Biophys. J., 1999, 77, 2856–2863 CrossRef CAS.
- M. W. Berns, in Methods in Cell Biolog, Volume 82—Laser Manipulation of Cells and Tissues, ed. M. W. Berns and K. O. Greulich, Academic Press, London, 2007, pp. 455–466 Search PubMed.
- C. Xie, PhD Thesis, East Carolina University, 2004.
- K. König, H. Liang, M. W. Berns and B. J. Tromberg, Opt. Lett., 1996, 21, 1090–1092 Search PubMed.
- H. Liang, K. T. Vu, P. Krishnan, T. C. Trang, D. Shin, S. Kimel and M. W. Berns, Biophys. J., 1996, 70, 1529–1533 CrossRef CAS.
- G. J. Puppels, J. H. F. Olminkhof, G. M. J. Segers-Nolten, C. Otto, F. F. M. de Mul and J. Greve, Exp. Cell Res., 1991, 195, 361–367 CrossRef CAS.
- B. R. Wood, M. A. Quinn, F. R. Burden and D. McNaughton, Biospectroscopy, 1996, 2, 143–153 CrossRef CAS.
- P. Lasch, A. Pacifico and M. Diem, Biopolymers, 2002, 67, 335–338 CrossRef.
- E. Gazi, J. Dwyer, P. Gardner, A. Ghanbari-Siakhali, A. P. Wade, J. Myan, N. P. Lockyer, J. C. Vickerman, N. W. Clarke, J. H. Shanks, C. Hart and M. Brown, J. Pathol., 2003, 201, 99–108 CrossRef.
- T. J. Harvey, A. Henderson, E. Gazi, N. W. Clarke, M. Brown, E. Correia Faria, R. D. Snook and P. Gardner, Analyst, 2007, 132, 292–295 RSC.
- F. Zheng, Y. Qin and K. Chen, J. Biomed. Opt., 2007, 12, 034002–034008 CrossRef.
- K. Chen, Y. Qin, F. Zheng, M. Sun and D. Shi, Opt. Lett., 2006, 31, 2015–2017 CrossRef.
- K. M. Omberg, J. C. Osborn, S. L. Zhang, J. P. Freyer, J. R. Mourant and J. R. Schoonover, Appl. Spectrosc., 2002, 56, 813–819 CrossRef CAS.
- A. Taleb, J. Diamond, J. J. McGarvey, R. Beattie, C. Toland and P. Hamilton, J. Phys. Chem. B, 2006, 110, 19625–19631 CrossRef CAS.
- E. Correia Faria, N. Ma, E. Gazi, P. Gardner, M. Brown, N. W. Clarke and R. D. Snook, Analyst, 2008, 133, 1498–1500 RSC.
- T. J. Harvey, PhD Thesis, University of Manchester, 2008.
- J. Deng, Q. Wei, Y. Wang and Y.-Q. Li, Proc. SPIE, 2006, 5630, 308–315.
- H. N. Banerjee and L. Zhang, Mol. Cell. Biochem., 2007, 295, 237–240 CrossRef CAS.
- J. W. Chan, D. S. Taylor, T. Zwerdling, S. M. Lane, K. Ihara and T. Huser, Biophys. J., 2006, 90, 648–656 CAS.
- B. Mohlenhoff, M. Romeo, M. Diem and B. R. Wood, Biophys. J., 2005, 88, 3635–3640 CrossRef CAS.
- J. W. Chan, D. S. Taylor, S. M. Lane, T. Zwerdling, J. Tuscano and T. Huser, Anal. Chem., 2008, 80, 2180–2187 CrossRef CAS.
- P. R. T. Jess, V. Garcés-Chávez, A. C. Riches, C. S. Herrington C S and K. Dholakia, J. Raman Spectrosc., 2007, 38, 1082–1088 CrossRef CAS.
- C. Xie, J. Mace, M. A. Dinno, Y.-Q. Li, W. Tang, R. J. Newton and P. Gemperline, Anal. Chem., 2005, 77, 4390–4397 CrossRef CAS.
- G. P. Singh, C. Creely, G. Volpe, H. Grotsch and D. Petrov, Proc. SPIE, 2005, 5930, 1–11.
- J. W. Chan, A. P. Esposito, C. E. Talley, C. W. Hollars, S. M. Lane and T. Huser, Anal. Chem., 2004, 76, 599–603 CrossRef CAS.
- J. F. Ojeda, C. Xie, Y.-Q. Li, F. E. Bertrand, J. Willey and T. J. McConnell, Opt. Express, 2006, 14, 5385–5393 CrossRef CAS.
- J. Bak and T. M. Jørgensen, Opt. Express, 2007, 15, 5997–5999 CrossRef.
- J. F. Ojeda, C. Xie, Y.-Q. Li, F. E. Bertrand, J. Willey and T. J. McConnell, Opt. Express, 2007, 15, 6000–6002 CrossRef.
- P. R. T. Jess, V. Garcés-Chávez, D. Smith, M. Mazilu, L. Paterson, A. Riches, C. S. Herrington, W. Sibbett and K. Dholakia, Opt. Express, 2006, 14, 5779–5791 CrossRef.
- H. Liang, W. H. Wright, W. Hei and M. W. Berns, Exp. Cell Res., 1991, 197, 21–35 CrossRef CAS.
- N. B. Simpson, L. Allen and M. J. Padgett, J. Mod. Opt., 1996, 43, 2485–2491.
- I. G. Cormack, M. Mazilu, K. Dholakia and C. S. Herrington, Appl. Phys. Lett., 2007, 91, 023903-1–3.
- R. A. Flynn, A. L. Birkbeck, M. Gross, M. Ozcan, B. Shao, M. M. Wang and S. C. Esener, Sens. Actuators, B, 2002, 87, 239–243 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |