Control of charge recombination at nanostructured quantum-dot sensitized TiO2 interfaces employing a multi-step redox cascade
Henry C.
Leventis
and
Saif A.
Haque
*
Nanostructured Materials and Devices Group, Department of Chemistry, Imperial College London, United Kingdom. E-mail: s.a.haque@imperial.ac.uk; Tel: (+44) 207 594 1886
First published on 5th August 2009
Abstract
A hybrid dye/CdS-sensitized TiO2 solar cell architecture is presented whereby the CdS nanocrystal layer is shown to perform multiple functions—retarding interfacial charge recombination and harvesting short-wavelength light, whilst acting as a redox mediator in the injection of electrons from the photo-excited dye into the mesoporous TiO2nanoparticle film.
Broader contextKey challenges to the design of more efficient solid-state dye sensitized solar cells (DSSCs) include the minimization of interfacial charge recombination losses and the improvement of the spectral overlap of the absorber layer with the solar spectrum. At present there is interest in the use of inorganic nanocrystals such as quantum dots as sensitizers in DSSCs. To improve the spectral coverage of CdS sensitized nanocrystalline TiO2 based architectures, we perform selective adsorption of dye sensitizer molecules to CdS nanocrystals. Moreover we demonstrate that the intermediate CdS layer serves as a multifunctional blocking layer that is able to absorb light and can serve as a mediator in a redox cascade system, thereby minimizing charge recombination between the photogenerated electrons and holes. |
Concerns over fossil fuel emissions are now motivating the development of alternative renewable energy sources. Photovoltaic devices based upon nanostructured molecular materials are currently attracting considerable interest for this purpose, owing to their low fabrication costs and the possibility of large area device deposition onto flexible plastic substrates. A configuration of particular interest is the dye-sensitized nanocrystalline semiconductor solar cell (DSSC).1–4 A typical dye-sensitized solar cell is based upon a mesoporous metal oxide film which is coated with a monolayer of a light harvesting dye complex. The pores of the resulting film are interpenetrated with a hole transporting matrix. Solar light to electrical energy conversion efficiencies exceeding 11% and 5% have been reported for devices employing liquid5 and solid-state organic semiconductor-based hole transporting materials (HTMs)4 respectively. Solid-state DSSCs employing molecular or polymeric hole transporting materials are particularly attractive as they offer the prospect of circumventing some of the encapsulation/sealing issues associated with liquid-based devices.4 However, the performance of such solid-state devices still remains lower than that of their liquid counterparts and significant improvements in efficiency are required before they can be considered commercially viable. Key challenges to the design of more efficient solid-state DSSCs are well known and include: (i) the minimization of interfacial charge recombination losses and (ii) the improvement of the spectral overlap of the absorber layer with the solar spectrum. One way to improve the spectral coverage is to use dye/dye3 or quantum dot/dye6 co-sensitizing combinations, where each component is tailored to harvest light from different regions of the solar spectrum. However, the success of this approach can be hindered by the decrease in sensitizing efficiency of the individual dyes upon mixing with a co-sensitizer. With regard to the minimization of interfacial charge recombination losses, several approaches have been used, with varying degrees of success. These include the use of lithium salts,7 supramolecular sensitizers8 and insulating metal oxide ‘blocking’ layers, such as MgO, SiO2 and Al2O3.9,10 In the latter approach, the insulating blocking layer is inserted between the TiO2 and the sensitizer and serves to minimize recombination losses without affecting the primary charge photogeneration yield. Moreover, to date such blocking layers have not been able to contribute to the photocurrent yield as they exhibit a wide bandgap (Eg < 3 eV). In this paper we report the use of a multifunctional blocking layer that is able to absorb light and can serve as a mediator in a redox cascade system, thereby minimizing charge recombination between the photogenerated electrons and holes. Fig. 1 shows the architecture of the photoactive layer studied in this work along with its corresponding schematic energy level diagram, with approximate redox potentials and band energies of the different components. The conduction band (CB) edge of the CdS used herein was estimated by consideration of the electronic confinement in CdS nanocrystals and the bulk CB energy.11–14 Therefore in our regime of nanocrystal size, we believe the CdS conduction band energy to lie between the TiO2 conduction band edge and the oxidation potential of the excited dye state, as shown in Fig. 1. The active layer has the following configuration: TiO2/CdS/Dye/Organic HTM; samples are studied using transient absorption spectroscopy on the µs – s timescale, and exhibit high yields of charge generation (processes 1–4 in Fig. 1a) and long-lived charge separation (processes 5–7) at the respective interfaces present in this architecture.
 | ||
| Fig. 1 (a) Shows a schematic illustration of the electron transfer processes occurring in the redox ‘cascade’ system described in the text, after the incorporation of a hole-conducting matrix consisting of spiro-OMeTAD, together with approximate redox potentials (vs. vacuum) and band energies of the different components. (b) Shows a schematic illustration of the structure of the redox ‘cascade’; also illustrated are possible electron and hole pathways following charge injection at the TiO2/CdS and N719/spiro-OMeTAD interfaces. | ||
Deposition and growth of a CdS nanocrystal layer upon the surface of mesoporous TiO2 was achieved using the ten successive ionic adsorption and reaction (SILAR) cycles.15–18 The S2− source was Na2S·9H2O (Sigma-Aldrich, 98+%), and Cd(NO3)2·4H2O (Hopkin & Williams, 99%) served as the Cd2+ source. To enable the study of charge transfer processes occurring at the two interfaces involved in this configuration, it was necessary to ensure that the N719 (cis-isothiocyanato bis(2,2′-bipyridyl-4,4′-dicarboxylato)-ruthenium(II)) dye was not able to adsorb to the TiO2 surface, but instead was forced to reside exclusively on the CdS surface. This was achieved by pre-coating the TiO2 surface with an alkyl monolayer, adsorbed to the surface by means of a phosphonic acid functionality (known to have a greater affinity for adsorption than carboxylic acid groups—the means of attachment of the N719 dye—in such systems).19 Adsorption of the alkylphoshonic acid monolayer on the TiO2 surface was achieved by immersion of the sample in a 10 mM solution of n-octylphosphonic acid (OPA, Alfa Aesar, 98%) in tetrahydrofuran (THF, 99%, BDH) for ten minutes, followed by rinsing with THF. No adsorption of the N719 dye was seen upon a bare TiO2 surface which had been treated with OPA. Fig. 2 shows the steady-state UV-visible absorption spectra obtained for TiO2/CdS and TiO2/CdS/N719 sensitized nanocrystalline films (spectra (b) and (c), respectively). Also shown is the absorption spectrum of a bare TiO2 film prior to any adsorption (spectrum (a)). Bare TiO2 films are transparent and colorless in the visible region, showing a characteristic absorption increase below ca. 400 nm due to the onset of TiO2 bandgap excitation. Introduction of the CdS layer on the nanocrystalline TiO2 film resulted in an absorption feature at ca. 450 nm–500 nm; this is consistent with previous work.6,14,15,20Sensitization of the CdS nanocrystals with the ruthenium dye complex resulted in the appearance of an additional absorption band centered at 540 nm. This feature is assigned to the MLCT transition of the N719 dye and is in agreement with previous studies.21
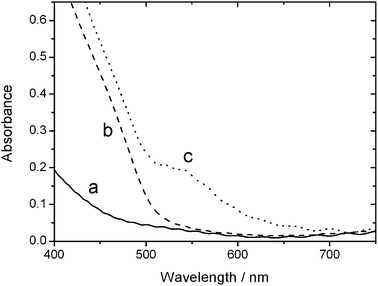 | ||
| Fig. 2 Steady-state absorption spectra of (a) TiO2, (b) CdS-sensitized TiO2 and (c) N719-sensitized CdS/TiO2 films. | ||
We consider next transient absorption spectroscopy (TAS) studies of these architectures. Fig. 3 shows the transient difference absorption spectra observed before and after the adsorption of the N719 dye to the CdS surface, measured 20 µs after pulsed laser excitation at 450 nm. In the absence of N719, a broad positive absorption band was seen at probe wavelengths between 500 nm and 1000 nm, with a gradual increase in ΔOD as the wavelength is decreased (up to ca. 600 nm), as seen in previous studies of quantum-dot sensitized TiO2 films. Such long-lived features have been assigned to the presence of photogenerated holes in the nanocrystal layer.6,14,22 Following sensitization with the N719 dye, the development of a large absorption feature centered at 815 nm is observed upon photoexcitation either at 450 nm (where both CdS nanocrystals and dye molecules are excited), or at 540 nm (where only dye molecules are excited). This feature is attributed to the transient existence of the N719+ cation,9 which can be formed in two ways: as a result of electron injection into the CdS/TiO2 layer following photo-excitation of the N719 species, or by hole injection into the N719 HOMO (highest occupied molecular orbital) after excitation of the CdS nanocrystal (reactions 2 and 3 in Fig. 1a). The latter process results from the function of the CdS nanocrystals as a light-harvesting layer.
 | ||
| Fig. 3 Transient absorption difference spectra, measured 20 µs after pulsed excitation at 450 nm, for various nanostructured films under examination: OPA/CdS/TiO2 (▲), N719/OPA/CdS/TiO2 (●), spiro-OMeTAD/N719/OPA/CdS/TiO2 (□). | ||
The transient kinetics are illustrated in Fig. 4, and it can be seen that the timescale of the decay in ΔOD to half its value at 1 µs (t½) is ca. 200 ms, retarded by over two and a half orders of magnitude relative to that seen in N719/TiO2 films.23 It is also clear that the decay in the N719+ cation population is on a significantly longer timescale in the dye-sensitized films than for the decay in the CdS-localized hole in the unsensitized arrangement (monitored at 750 nm, near the centre of the broad positive band seen in the transient spectrum). Such observations are consistent with the function of the CdS nanocrystal layer as a ‘blocking’ unit, which serves to increase the spatial separation between the dye cation and conduction band electrons, thereby resulting in a retardation of the charge recombination kinetics. Our observation of a slower charge recombination lifetime at the TiO2/CdS/dye interface is consistent with previous studies.24 It is pertinent to note, however, that in the present work we observe a significantly more pronounced retardation of interfacial recombination (by several orders of magnitude), possibly as a result of the selective adsorption of dye molecules to the CdS surface, as well as the use of TiO2 substrates as opposed to those based on SnO2.24
 | ||
| Fig. 4 Transient absorption data (pumped at 450 nm) contrasting electron recombination with dye cations in the (a) N719/TiO2 and (b) N719/OPA/CdS/TiO2 systems with the same N719 dye loading (monitored using a probe wavelength of 820 nm) with (c) the decay of ΔOD at 750 nm for an OPA/CdS/TiO2 sample; the latter is believed to provide an indication of the lifetime of CdS-localized holes. | ||
Application of the solid-state hole-conductor matrix onto the substrate was achieved by depositing 40 µl per cm2 of a 0.17 M solution of spiro-OMeTAD (Covion, observed to be pure by 1H-NMR and HPLC analysis) in chlorobenzene (Sigma-Aldrich, 99.9%), also containing 38 mM Li(CF3SO2)2N (Sigma-Aldrich, 99.9%) and 28 mM 4-tert-butylpyridine (Aldrich, 99%). The solution was allowed to penetrate the film for 30 s, after which spin coating was performed at 2000 rpm.
Fig. 5 compares the transient absorption kinetics of the TiO2 conduction band electron (probed at 950 nm) for the mesoporous films following spin-coating with spiro-OMeTAD. Optical excitation results in the appearance of two new features, shown in Fig. 3: an intense, narrow, positive band and a broad, less intense, feature, centered at 510 nm and 660 nm respectively. These are assigned to the presence of the spiro-OMeTAD+ cation,16,25 and demonstrate that hole injection has occurred into the organic semiconductor. The lifetime of conduction band electrons is seen to be extended significantly by the presence of CdS nanocrystals in the spiro-OMeTAD/N719/CdS/TiO2 arrangement, with t½ ≈ 15 ms. This is significantly longer than the value obtained for spiro-OMeTAD/N719/TiO2 films, t½ ≈ 2 ms. This increase in electron lifetime can be attributed to the presence of the adsorbed OPA monolayer upon the TiO2 surface, which serves to retard recombination at the TiO2/spiro-OMeTAD interface.
 | ||
| Fig. 5 Normalized transient absorption data monitoring TiO2 conduction band electron recombination with the spiro-OMeTAD+ cation at 950 nm, following photo-excitation at 450 nm. Data is presented for N719-sensitized films in the (a) absence and (b) presence of a CdS/OPA intermediate layer. | ||
Fig. 6 compares the current–voltage characteristics of spiro-OMeTAD/CdS/TiO2 and spiro-OMeTAD/dye/CdS/TiO2 DSSC architectures (Au was evaporated onto the HTM to complete the device). It can be seen that both the short-circuit current and the open-circuit voltage are improved by the inclusion of a CdS-adsorbed dye layer; the power conversion efficiency (PCE, under AM 1.5 illumination) increases from 0.15% to 0.25%. In its improvement of the PCE, the dye layer could be serving both to improve the spectral coverage of the device with the solar spectrum, and to increase the yields and lifetimes of interfacial charge separation in its mediation of hole transfer from CdS nanocrystals to the HTM.
 | ||
Fig. 6 Current–voltage characteristics (under AM 1.5 illumination) of solid-state DSSCs using CdS nanocrystal sensitizers, in the presence (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and absence ( ) and absence (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) of a CdS-adsorbed dye layer. ) of a CdS-adsorbed dye layer. | ||
We note that further improvements in light harvesting ability and charge-separated state lifetime can be expected by addressing the relatively poor dye loading ability of the currently employed dyes on the nanocrystal surface, and the instability of these dyes once bound (possibly by means of their thiocyanate moiety), as well as by using nanocrystal sensitizers made from narrow bandgap semiconductors such as PbS. Hence, future studies will have to employ novel dyes which bind strongly to (and are stable upon) nanocrystal surfaces, for example those bound using a thiol functional group. Such studies are currently underway and will be reported in due course.
The results presented in this communication demonstrate that CdS nanocrystals can be used as an intermediate layer in DSSCs, between the adsorbed dye and the TiO2 surface. This layer not only serves to harvest light, but is also able to mediate the electron injection from the photo-excited dye into the TiO2 conduction band. Efficient regeneration of the CdS-localized hole by an organometallic dye has been shown by means of transient absorption studies, and the regeneration of both the CdS-localized hole and dye cation species by an organic hole conductor have also been demonstrated. Finally, the rate of charge recombination between the TiO2 conduction band electron and the hole, delocalized throughout the organic matrix, has been found to be significantly extended by the presence of the CdS nanocrystal and alkylphosphonic acid monolayers, relative to that observed in conventional DSSCs. This is consistent with the function of these layers as a ‘blocking’ component which retards the interfacial recombination process by increasing the spatial separation between the recombining charges. Architectures of the type discussed in this study are currently being used in the optimization of solid-state DSSCs.
Acknowledgements
We acknowledge the Engineering and Physical Sciences Research Council (EPSRC) for their financial support and Xiaoe Li for preparing TiO2 nanoparticle pastes. S. A. H. is a Royal Society University Research Fellow.References
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. Gratzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS.
- H. J. Snaith and L. Schmidt-Mende, Adv. Mater., 2007, 19, 3187–3200 CrossRef CAS.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Y. Han, Jpn. J. Appl. Phys., Part 1, 2006, 45, L638–L640 CrossRef CAS.
- H. J. Lee, H. C. Leventis, S. J. Moon, P. Chen, S. Ito, S. A. Haque, T. Torres, F. Nüesch, T. Geiger, S. M. Zakeeruddin, M. Grätzel and M. K. Nazeeruddin, Adv. Funct. Mater., 2009, 19, 1–8.
- N. Kopidakis, K. D. Benkstein, J. van de Lagemaat and A. J. Frank, J. Phys. Chem. B, 2003, 107, 11307–11315 CrossRef CAS.
- C. A. Bignozzi, R. Argazzi and C. J. Kleverlaan, Chem. Soc. Rev., 2000, 29, 87–96 RSC.
- E. Palomares, J. N. Clifford, S. A. Haque, T. Lutz and J. R. Durrant, J. Am. Chem. Soc., 2003, 125, 475–482 CrossRef CAS.
- B. C. O'Regan, S. Scully, A. C. Mayer, E. Palomares and J. Durrant, J. Phys. Chem. B, 2005, 109, 4616–4623 CrossRef CAS.
- H. Matsumoto, T. Matsunaga, T. Sakata, H. Mori and H. Yoneyama, Langmuir, 1995, 11, 4283–4287 CrossRef CAS.
- R. Dewitt and A. Kirschdemesmaeker, J. Electrochem. Soc., 1983, 130, 1995–1998 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CAS.
- Y. Tachibana, K. Umekita, Y. Otsuka and S. Kuwabata, J. Phys. Chem. C, 2009, 113, 6852–6858 CrossRef CAS.
- R. Vogel, P. Hoyer and H. Weller, J. Phys. Chem., 1994, 98, 3183–3188 CrossRef CAS.
- R. Plass, S. Pelet, J. Krueger, M. Gratzel and U. Bach, J. Phys. Chem. B, 2002, 106, 7578–7580 CrossRef CAS.
- V. P. Tolstoy, Russ. Chem. Rev., 2006, 75, 161–175 Search PubMed.
- Y. L. Lee and Y. S. Lo, Adv. Funct. Mater., 2009, 19, 1–6.
- S. Pawsey, K. Yach and L. Reven, Langmuir, 2002, 18, 5205–5212 CrossRef CAS.
- Y. Tachibana, H. Y. Akiyama, Y. Ohtsuka, T. Torimoto and S. Kuwabata, Chem. Lett., 2007, 36, 88–89 CrossRef CAS.
- M. K. Nazeeruddin and M. Gratzel, in Photofunctional Transition Metals Complexes, Editon edn., 2007, vol. 123, pp. 113–175 Search PubMed.
- H. J. Lee, J. H. Yum, H. C. Leventis, S. M. Zakeeruddin, S. A. Haque, P. Chen, S. I. Seok, M. Grazel and M. K. Nazeeruddin, J. Phys. Chem. C, 2008, 112, 11600–11608 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Gratzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538–547 CrossRef CAS.
- C. Nasr, S. Hotchandani, W. Y. Kim, R. H. Schmehl and P. V. Kamat, J. Phys. Chem. B, 1997, 101, 7480–7487 CrossRef CAS.
- U. Bach, Y. Tachibana, J. E. Moser, S. A. Haque, J. R. Durrant, M. Gratzel and D. R. Klug, J. Am. Chem. Soc., 1999, 121, 7445–7446 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |