Highly stable sensitizer dyes for dye-sensitized solar cells: role of the oligothiophene moiety
Ryuzi
Katoh
*a,
Akihiro
Furube
a,
Shogo
Mori
b,
Masanori
Miyashita
b,
Kenji
Sunahara
b,
Nagatoshi
Koumura
a and
Kohjiro
Hara
a
aNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: r-katoh@aist.go.jp
bDivision of Chemistry and Materials, Faculty of Textile Science and Technology, Shinshu University, 3-15-1 Tokida, Ueda, Nagano 386-8567, Japan
First published on 23rd March 2009
Abstract
The photostability of five sensitizer dyes with and without an oligothiophene moiety was studied by means of spectroscopic techniques to determine the dyes' suitability for dye-sensitized solar cells. We examined the effect of light irradiation on the dyes adsorbed on nanocrystalline TiO2 films under ambient conditions and found that the dyes containing an oligothiophene moiety showed high stability. The results of transient absorption measurements suggest that the location of holes on the oligothiophene moieties was responsible for this observed high stability.
Broader contextFor the practical application of dye-sensitized solar cells (DSSCs), long lifetime of the device is an important requirement in addition to high performance. In this context, Ru complexes have been used instead of metal-free organic dyes. Recently, it was found that some organic dyes showed higher stability, whereas the reason is unclear. To clarify the reason, we studied the photostability of five sensitizer dyes with and without an oligothiophene moiety by means of spectroscopic techniques. We examined the effect of light irradiation on the dyes adsorbed on nanocrystalline TiO2 films under ambient conditions and found that the dyes containing an oligothiophene moiety showed high stability. The results of transient absorption (TA) measurements suggest that the location of holes on the oligothiophene moieties was responsible for this observed high stability. This would be one of the strategies to design highly stable sensitizer dyes for dye-sensitized solar cells. |
Introduction
Dye-sensitized solar cells (DSSCs) based on Ru complexes are the most commonly studied type of DSSCs. Although such cells show relatively high photovoltaic conversion efficiency (η) values (η > 11%),1,2 they possess several disadvantages, such as the limited availability of ruthenium and a relatively high cost of production. To avoid such limitations, organic sensitizer dyes without metal atoms have been synthesized for use in DSSCs. Many metal-free organic dyes have been reported to produce a high conversion efficiency. For example, in 2003, Hara et al. reported high-performance DSSCs (>5%) using coumarin dyes combined with a cyano-acrylic acid group as an anchor group.3 In 2004, DSSCs using an indoline-based dye were reported to show 8% conversion efficiency.4 Recently, dyes featuring oligothiophene groups combined with carbazole moieties5 and fluorene moieties6,7 were newly synthesized, and some of them showed high conversion efficiency. Accordingly, organic-dye-based DSSCs are considered to be promising devices, though their performance is still lower than that observed for DSSCs using Ru complex dyes. Recently, researchers have sought to determine the reasons for this lower performance by studying transient photo-current and -voltage measurements of organic dye DSSCs.8For the practical application of DSSCs, long lifetime of the device is an important requirement in addition to high η. Several active species, such as electrons and dye cations, are produced in DSSCs under light irradiation. These species potentially induce degradation reactions in the device, causing η to decrease gradually with time. To sustain a high η for a long period of time, the mechanism of such degradation reactions should be clarified.
Though many factors control the lifetime of DSSCs, the stability of sensitizer dyes is an especially critical factor. Electron injection from the excited dye to the semiconductor occurs within 100 fs.9 This fast injection means that the lifetime of the dye in its excited state is short, suggesting that degradation is not likely to occur while the dye is in its excited state. After the electron injection, the dye is in a cation state until regeneration by a redox mediator occurs. The timescale of this regeneration is 100 ns–1 µs.10,11 To realize long-term stability, the sensitizer dyes must remain stable in the cation state for a long time. For example, in a 10-year operation cycle, sensitizer dyes are regenerated more than 107 times; therefore, the cation state of such sensitizer dyes must remain stable for at least 10 s. Accordingly, the molecular design of high-stability sensitizer dyes for DSSCs must be tailored to satisfy this criterion for the dyes' cation states.
Here, we evaluate the photostability of sensitizer dyes synthesized for DSSCs and measure the transient absorption (TA) spectra to understand the origin of the differences in the dyes' observed stability. Dyes adsorbed on nanocrystalline TiO2 films were irradiated by AM 1.5 light without redox mediators. Under these conditions, the lifetime of the dyes in the cation state is limited by the recombination time between an injected electron and a parent cation, which occurs in the millisecond time range.12,13 Thus, the dyes remained in the cation state for 10 s during 2 h of irradiation by AM 1.5 light, which corresponds to 10-years operation time of DSSC devices.
Experimental
Nano-porous TiO2 films were prepared by applying TiO2 paste (Nanoxide-T, Solaronix) to transparent conductive oxide glass substrates by means of doctor blade techniques. The films were annealed at 550 °C for 30 min in air. The thickness of the films was around 5 µm.The sensitizer dyes were dissolved at a concentration of 0.5 mM in a mixture of tert-butylalcohol (tBuOH) and acetonitrile (AN) (1 : 1 w/w) or a mixture of tBuOH, AN, and toluene (1 : 1 : 1 w/w/w). These solvents were used as obtained from the suppliers without further purification. Fig. 1 shows the molecular structures of the sensitizer dyes studied. The N719 dye (Solaronix SA) was used without purification. Detailed synthetic procedures for the organic dyes (NKX-2311, NKX-2587, NKX-2697 and MK-2) have been reported elsewhere.3,5,14 The semiconductor films were immersed in each dye solution at room temperature for at least 16 h. The sample specimens were then dried in air.
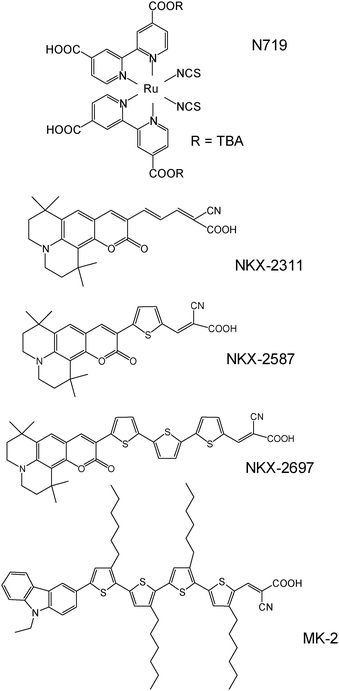 | ||
| Fig. 1 Molecular structures of the sensitizer dyes studied. | ||
The photostability of dyes adsorbed on the nanocrystalline films was evaluated by visible-light (>420 nm) irradiation from a solar simulator (WACOM WXS-80C-3) operating at AM 1.5 (100 mW cm−2) with an ultraviolet (UV) cutoff filter (HOYA, L42). Irradiation was carried out under ambient conditions. To evaluate the degradation of the dyes, absorption measurements were carried out with a spectrometer (Shimadzu, UV-3101PC) equipped with an integrating sphere at 35 and 90 min irradiation times.
For TA spectra measurements, a TA spectrometer based on an optical parametric oscillator (Spectra Physics, MOPO-SL) excited by the third harmonic of a Nd3+:YAG laser (Spectra Physics, Pro-230-10) was used. The wavelength of the laser was set to 532 nm, and the pulse duration was about 8 ns. A Xe flash lamp (Hamamatsu, L4642, 2 µs pulse duration) was used as the probe light source. The probe light was collinear with the excitation pulse, and the probe light transmitted through the sample was dispersed with a monochromator (Ritsu, MC-10N) and then detected by a Si photodiode (Hamamatsu, S-1722) for 500–950 nm or by an InGaAs photodiode (Hamamatsu, G3476-05) for 900–1600 nm. The signal from the detectors was amplified (NF Electronic Instruments, BX-31A), processed with a digital oscilloscope (Tektronix, TDS680C), and analyzed with a computer.
For TA decay measurements, a highly sensitive TA spectrometer based on a Nd3+:YAG laser (HOYA Continuum, Surelite II) was used. The repetition rate of the laser was 10 Hz. The second harmonic pulse (532 nm) was used for the excitation. A halogen lamp (100 W) was used as the probe light source, and the probe light was collinear with the excitation pulse. The light transmitted through the sample films was dispersed with a monochromator (Acton Research, SpectraPro-150) and then detected by a Si photodiode (Hamamatsu, S-1722). The photocurrent from the detector was amplified with an amplifier (NF Electronic Instruments, 5305). Signals were processed with a digital oscilloscope (Tektronix, TDS380) and analyzed with a computer. The DC component of the photocurrent from the detector was subtracted by means of an electric frequency filter (NF Electronic Instruments, FV-628B), and therefore small absorbance changes (<10−5) could be detected. The time resolution of the system was about 1 µs. The wavelength of the probe light was set at 800 nm for N719, NKX-2697, and MK-2, and at 900 nm for NKX-2311 and NKX-2587. The band width (FWHM) of the probe light was about 20 nm. The intensity of the laser pulse was measured with a pyroelectric energy meter (Ophir, PE25-SH-V2). All measurements were carried out at 295 K.
Results and discussion
Fig. 2 shows a photograph of the sample specimens of N719, NKX-2311, NKX-2587, NKX-2697, and MK-2 adsorbed on TiO2 surfaces before and after 35 min of light irradiation. The color of the NKX-2311 and NKX-2587 specimens changed dramatically, suggesting that these dyes were photochemically unstable. In contrast, no dramatic change in color was observed for the N719, NKX-2697, and MK-2 samples. | ||
| Fig. 2 A photograph of the sample specimens of N719, NKX-2311, NKX-2587, NKX-2697 and MK-2 adsorbed on nanocrystalline TiO2 films before and after 35 min of light irradiation. | ||
To investigate the degradation process in detail, we measured the absorption spectra of N719, NKX-2311, NKX-2587, NKX-2697, and MK-2 adsorbed on nanocrystalline TiO2 films before and after light irradiation (Fig. 3). For NKX-2311 and NKX-2587, the absorbance at 400–650 nm decreased sharply as a result of light irradiation. This decrease suggests that some chemical bonds of the coumarin moiety, which absorbs strongly in the visible wavelength range, were broken during the dyes' cation state lifetime. In contrast, no substantial change in absorbance was observed for N719, NKX-2697, and MK-2. For N719, the absorption peak shifted from 540 nm to 480 nm. This shift has been assigned to the detachment of an SCN− ligand from the dye.15 For NKX-2697 and MK-2, the peak positions did not change as a result of light irradiation, indicating that no photochemical reaction occurred.
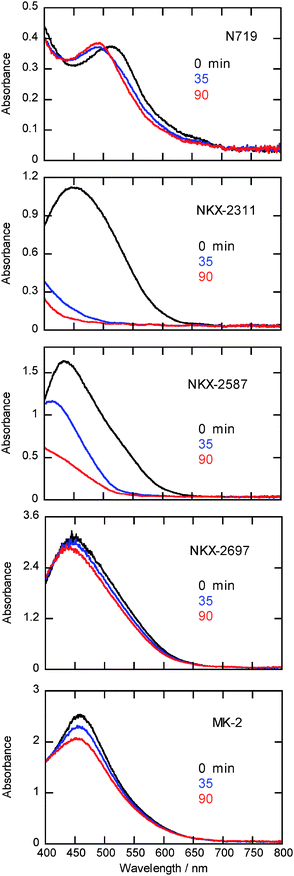 | ||
| Fig. 3 Absorption spectra of N719, NKX-2311, NKX-2587, NKX-2697 and MK-2 adsorbed on nanocrystalline TiO2 films before (black line) and after light irradiation for 35 (blue line) and 90 min (red line). | ||
Fig. 4 shows the TA spectra recorded just after excitation by a 532 nm light pulse. The characteristic features of the spectrum observed for NKX-2587 were similar to those observed for NKX-2311, as we have reported previously.16 This similarity suggests that the hole in the cation state was located on the coumarin moiety for both dyes. In contrast, the observed spectrum for NKX-2697 differed significantly from that of NKX-2311, although both dyes have a coumarin moiety. This dissimilarity clearly indicates that the location of the hole in the cation state of NKX-2697 differed from that observed for NKX-2587. The absorption spectrum of the cation state of oligothiophene moieties has been determined through the electron transfer reaction of supramolecular systems containing these moieties, and the resulting spectrum shows a characteristic absorption around 1500 nm.17 Thus, the NKX-2697 spectrum obtained here, which also displayed a peak around 1500 nm, suggests that the hole in the cation state of NKX-2697 was located on the oligothiophene moiety. For MK-2, the absorption spectrum was similar to that of NKX-2697, again suggesting that the hole was located on the oligothiophene moiety of this dye.
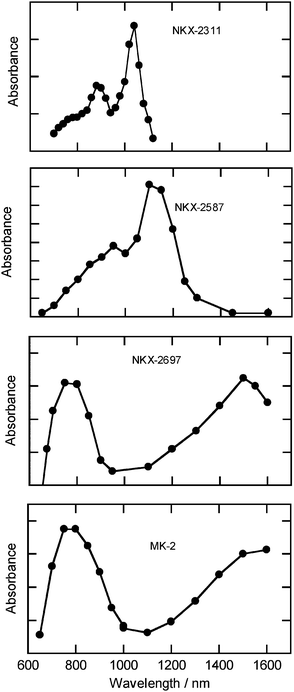 | ||
| Fig. 4 Transient absorption spectra of NKX-2311, NKX-2587, NKX-2697 and MK-2 recorded just after excitation at 532 nm. | ||
The peak position in the spectrum for the cation state of polythiophene18 (2500 nm) is at a longer wavelength than that observed for the cation state of oligothiophene moieties in supramolecular systems (∼1500 nm).17 This difference suggests that the area of the hole delocalization in polythiophene is larger than that in oligothiophene, namely, the peak position of the cation state in the near-IR region reflects the degree of delocalization of holes. As shown in Fig. 4, the peak in the near-IR region of the spectrum for the cation state of MK-2 was slightly red-shifted compared to that of NKX-2697. Accordingly, this difference in peak positions probably occurred because the area of hole delocalization over the MK-2 oligothiophene moiety was slightly larger than the delocalization over NKX-2697. This difference in delocalization area between the respective dyes' oligothiophene moieties probably can be attributed to differences in the sizes of the two oligothiophene moieties.
Organic dyes in their cation states generally have low chemical stability because they have an open-shell electronic structure. Although chemical stability varies among molecules, the possibility for degradation reactions tends to increase as the lifetime of the cation state increases. In this context, the recombination time between an injected electron and a cation strongly influences the photostability of dyes on the surface of semiconductor films. The recombination time can be evaluated by means of the time profile of the TA measurement. According to a TA study for Ru complex systems, the recombination time strongly depends on the density of electrons in TiO2nanoparticles, and the recombination time actually becomes insensitive to light intensity below the density at which one electron is produced per nanoparticle.12,13 Thus, all measurements in the current studies were carried out under weak excitation conditions (5.5 µJ cm−2). Fig. 5 shows the decay profile of TA for the dye cations. The recombination rates were evaluated as a half-life period: N719 = 1 ms, NKX-2311 = 3 ms, NKX-2587 = 3 ms, NKX-2697 = 0.2 ms, and MK-2 = 3 ms.
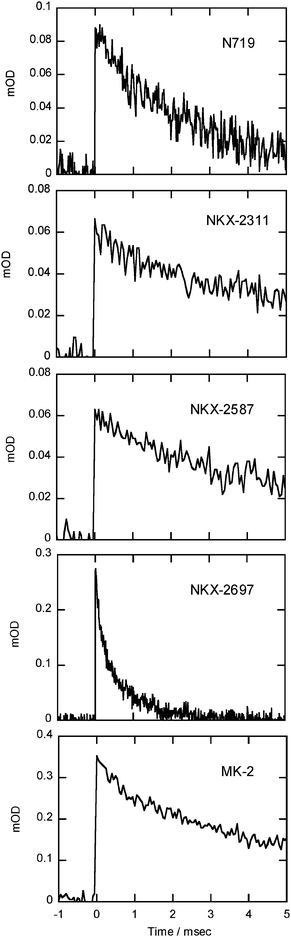 | ||
| Fig. 5 Decay profiles of transient absorption for the dye cations of N719, NKX-2311, NKX-2587, NKX-2697 and MK-2. All measurements were carried out under weak excitation conditions (5.5 µJ cm−2). | ||
Although the recombination times differed among sensitizer dyes, there was less correlation between the recombination time and the stability of a given dye under light irradiation. This result suggests that the chemical stability of the dyes was not limited by the lifetime of the cation state but instead reflected the reactivity in the cation state. Notably, the highly stable dyes NKX-2697 and MK-2 showed a similar TA spectra that indicated the presence of holes located on the oligothiophene moieties in the dyes. This result suggests that the holes located on the oligothiophene moieties were chemically stable, although the origin of the dyes' high stability is not clear.
Studies of chemical reactions in aromatic molecules in their cation states have shown that efficient chemical reactions frequently occur because a hole is localized on a specific carbon atom and forms a localized radical center.19,20 As discussed above, the holes in NKX-2697 and in MK-2 were delocalized over the oligothiophene moiety, suggesting that the radical center also was not localized on a specific atom in the molecule. Such delocalization of holes would explain the observed high stability.
Another possible degradation mechanism of the dyes adsorbed on the nanocrystalline TiO2 films is the reaction of the dyes in the ground state with singlet oxygen produced by the reaction between oxygen molecules and electrons injected from the dyes. Singlet oxygen is a reactive species that causes the degradation of organic molecules under light irradiation.21 In the present study, however, such singlet oxygen reactions were unable to occur, because the rate of singlet oxygen generation through the reaction of oxygen with electrons produced in TiO2 is around 50 ms,22,23 which is much slower than the recombination time observed in our experiments (0.2–3 ms). In other words, almost all the electrons in our experiments recombined with parent cations before singlet oxygen was produced.
Recently, some organic sensitizer dyes have been reported to show sufficiently high device stability under light-soaking conditions. In particular, organic dyes based on fluorene–dithiophene dye,6fluorene–dithienothiophene dye7 and coumarin–thiophene dye24 show high stability. Interestingly, all these dyes contain a thiophene moiety, as do the dyes we have studied here. These reported results again demonstrate the importance of the introduction of oligothiophene units in DSSC dyes to obtain high stability.
Conclusion
We examined the effect of light irradiation on organic sensitizer dyes adsorbed on nanocrystalline TiO2 films under ambient conditions and found that dyes containing oligothiophene moieties showed high stability. Using transient absorption spectroscopy, we found that holes in such dyes were located on the oligothiophene moieties. These results suggest that the holes on the oligothiophene moieties are chemically stable.References
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Han, Jpn. J. Appl. Phys., 2006, 45, L638 CrossRef CAS.
- K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597 CrossRef CAS.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218 CrossRef CAS.
- N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256 CrossRef CAS.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS.
- H. Qin, S. Wenger, M. Xu, F. Gao, X. Jing, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 9202 CrossRef CAS.
- M. Miyashita, K. Sunahara, T. Nishikawa, Y. Uemura, N. Koumura, K. Hara, A. Mori, T. Abe, E. Suzuki and S. Mori, J. Am. Chem. Soc., 2008, 130, 17874 CrossRef CAS.
- G. Benkö, J. Kallioinen, J. E. I. Korppi-Tommola, A. P. Yartsev and V. Sundström, J. Am. Chem. Soc., 2002, 124, 489 CrossRef.
- S. A. Saif, Y. Tachibana, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 1998, 102, 1745 CrossRef CAS.
- I. Montanari, J. Nelson and J. R. Durrant, J. Phys. Chem. B, 2002, 106, 12203 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538 CrossRef CAS.
- J. N. Clifford, E. Palomares, Md. K. Nazeeruddin, M. Grätzel, J. Nelson, X. Li, N. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225 CrossRef CAS.
- K. Hara, Z.-S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 489.
- H. G. Agrell, J. Lindgren and A. Hagfeldt, Sol. Energy, 2003, 75, 69.
- T. Yoshihara, R. Katoh, A. Furube, M. Murai, T. Tamaki, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 2643 CrossRef CAS.
- T. Nakamura, J. Ikemoto, M. Fujitsuka, Y. Araki, O. Ito, K. Takimiya, Y. Aso and T. Otsubo, J. Phys. Chem. B, 2005, 109, 14365 CrossRef CAS.
- R. Österbacka, C. P. An, X. M. Jiang and Z. V. Vardeny, Science, 2000, 287, 839 CrossRef CAS.
- T. Shida, E. Haselbach and T. Bally, Acc. Chem. Res., 1984, 17, 180 CrossRef CAS.
- H. D. Roth, Acc. Chem. Res., 1987, 20, 343 CrossRef CAS.
- A. A. Gorman and M. A. Rodgers, Chem. Soc. Rev., 1981, 10, 205 RSC.
- L. Xiao-e, A. N. M. Green, S. A. Haque, A. Mills and J. R. Durrant, J. Photochem. Photobiol., A, 2004, 162, 253 CrossRef.
- M. Murai, Y. Tamaki, A. Furube, K. Hara and R. Katoh, Catal. Today, 2007, 120, 214 CrossRef CAS.
- Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. B, 2008, 112, 17011 CAS.
| This journal is © The Royal Society of Chemistry 2009 |