Near-IR sensitization of wide band gap oxide semiconductor by axially anchored Si-naphthalocyanines
Lorena
Macor
a,
Fernando
Fungo
a,
Tomas
Tempesti
a,
Edgardo N.
Durantini
a,
Luis
Otero
*a,
Eva M.
Barea
b,
Francisco
Fabregat-Santiago
b and
Juan
Bisquert
*b
aDepartamento de Química, Universidad Nacional de Río Cuarto, Agencia Postal 3, 5800, Río Cuarto, Argentina. E-mail: lotero@exa.unrc.edu.ar
bPhotovoltaic and Optoelectronic Devices Group, Departament de Fisica, Universitat Jaume I, 12071, Castelló, Spain. E-mail: bisquert@fca.uji.es
First published on 13th March 2009
Abstract
Near-IR dye sensitized solar cells are very interesting due to their potential applications in panchromatic cells, semi-transparent windows and in tandem cells. In this work we show the utilization of axially anchored Si-naphthalocyanine dye in the spectral sensitization of TiO2 nanostructured photoelectrodes. We report the first successful evaluation of a naphthalocyanine in the production of sensitized photocurrent with maximum incident photon to current efficiency (IPCE) at λ ∼ 790 nm.
Broader contextThe continuous increment of energy demand, and the limited accessibility to traditional sources, due to both economical and environmental problems, drives the development of new materials and devices for solar energy conversion, one of the most important research areas. Solar light absorbed by organic colorants can start a process that culminates in the generation of electricity when they form part of a new kind of low cost solar energy conversion device: dye sensitized solar cells. In this frame the development of organic dyes that absorb light in the infrared could improve cell performance in the construction of smart windows. Ideally, a red/near-IR dye which is transparent over a large region of the visible spectrum would allow visible light enter to a building while it converts IR solar light into electricity. In addition to directly generating power, this also reduces indoor solar heating. In this work we show the utilization of axially anchored Si-naphthalocyanine dye in the spectral sensitization of TiO2 nanostructured photoelectrodes. We report the first successful evaluation of a naphthalocyanine in the production of sensitized photocurrent with maximum incident photon to current efficiency in the infrared region of the solar spectrum. |
Introduction
Owing to the high photoelectric conversion efficiency of low cost dye-sensitized nanocrystalline semiconductor solar cells (DSCs),1 a considerable number of organic structures have been designed, synthesized and studied as molecular sensitizers for photon harvesting.2–5 Devices based on Ru(II) polypyridyl complexes and TiO2 have attracted wide scientific and technological interest, because these cells achieve 11.2% validated solar power efficiencies and their stability data indicates, at least, 10 years of use in outdoor applications.6,7 However, the low molar extinction coefficient in the far-red/near-IR region limits further improvement in the cells. The optimal sensitizer for the dye-sensitized solar cell should be panchromatic, that is, it absorbs light in the widest possible wavelength region.8,9 For example, Bignozzi et al.10 developed a series of Os polypyridine complexes that are able to produce conversion of photons to electrons starting at ∼1100 nm. Also, tricarbocyanine clusters,11 heptamethine–cyanine12 and croconate dyes13 show extended photocurrent generation until ~900 nm. Furthermore, near-IR dyes are particularly interesting due to their potential applications in transparent solar cells for windows and tandem cells. Ideally, a red/near-IR dye which is transparent over a large region of the visible spectrum would allow visible light to enter to a building; meanwhile it converts the IR solar irradiation into electricity. In addition to directly generating power, this also reduces indoors solar heating. On the other hand, an IR dye that contains an optical window in the visible region can be used in combination with an appropriate dye to achieve panchromatic cosensitization of the oxide semiconductor, increasing the device efficiency, as has been reported by Nazeeruddin et al.14,15 using organic sensitizers. Thus, it is very important to find ways to enhance the DSCs response in the above mentioned wavelength region. Anthraquinone,16 perylene,17 squaraine,18,19 chlorophyll, porphyrins and phthalocyanines20–23 dyes, which absorb around 600–700 nm have been used for harvesting light in the red region. Although several groups have tested porphyrins and phthalocyanines24–28 as sensitizers for wide-bandgap oxide semiconductors, with different power conversion efficiencies in the IR region,29–31 there are very few antecedents for the use of naphthalocyanines as light-absorbing dyes in DSCs. Li et al.32 reported the synthesis of peripherally tetra-substituted naphthalocyanines, and presented the first evaluation of this class of dye in a photoelectrochemical solar cell via immobilization on nanocrystalline TiO2, but they found no or insignificant photocurrent generation. However, naphthalocyanines have relevant properties: they possess an intense absorption band in the near IR region (λmax ∼ 800 nm, ε ∼ 2 × 105, around 50% larger than the structurally related phthalocyanine) and excellent stability. These facts render them attractive for photovoltaic applications. Naphthalocyanines are also of interest because of the extension of the π-electron system, compared to phthalocyanines, which can effectively modify the optical spectra and other electronic properties such as redox potentials, electrical conductivity, photoconductivity and catalytic activity.Furthermore, dye anchoring onto TiO2 surface has been conducted by several methods, such as covalent bonding, electrostatic and/or hydrophobic interactions.33 Covalent attachment via a carboxyl group is probably the most widely adopted. However, the stability of this linkage is satisfactory in aqueous medium only within a certain pH range,34 and in organic solvents or aqueous/organic mixtures the strength of the linkage could be even weaker and undergoes a certain degree of dissociation. On the other hand, owing to the high affinity of the silicon-derivatives with the TiO2 semiconductor surface, and the chemical inertness of the resultant Si–O bonds, silyl anchoring groups seem to be good candidates for surface modification of TiO2. Various studies employing silyl-derivatized polypyridyl metal complexes have demonstrated the robustness of the silyl linkage on silica, SnO2, TiO2 and ITO surfaces.35,36
In this paper we report the utilization of Si–naphthalocyanine dyes 1 and 2 (Fig. 1) in the spectral sensitization of TiO2 nanostructured photoelectrodes. The dyes can be axially anchored to the semiconductor oxide surface, either via the classical carboxyl–TiO2 surface interaction (dye 2), or through a direct Si–O–Ti linkage (dye 1). This report is, as far as we are aware, the first evaluation of a naphthalocyanine dye in a DSC with successful production of observable sensitized photocurrent.
 | ||
| Fig. 1 Structures of the naphthalocyanine dyes. | ||
Results and discussion
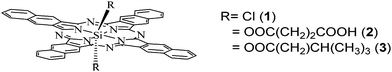
The photophysical properties of the dyes were analyzed by steady-state absorption and emission spectroscopy. The absorption spectra of 1 and 2 are shown in Fig. 2, in solution and adsorbed over FTO/TiO2 nanostructured films. As can be observed, the spectrum of the dyes in solution (Fig. 2a) features Q-bands at λmax 686 and 770 nm, whereas the B-band appears at λmax = 354 nm. Thus the dyes show strong light absorption in the UV and near IR regions of the spectrum. The sharp absorption bands in the Q-band region are assigned to monomer absorption, and the bands around 500 nm (which become stronger as the dye concentration increases) could be due to the characteristic face-to-face dimer or higher order aggregate (H-aggregate), as reported for structurally related compounds.37,38 It is well known that in going from phthalocyanine to naphthalocyanine compounds the HOMO level is destabilized, and in consequence the Q-bands become red-shifted, meanwhile the LUMO has a similar energy level. The obtained spectra are in agreement with experimental and theoretical reports,39 that showed that there is a small effect on spectra by axial substituents for silicon–naphtahlocyanines compounds. However, the Q-bands' spectral shift of around 100 nm going from phthalocyanine to naphthalocyanine could be useful for extending the response of DSCs.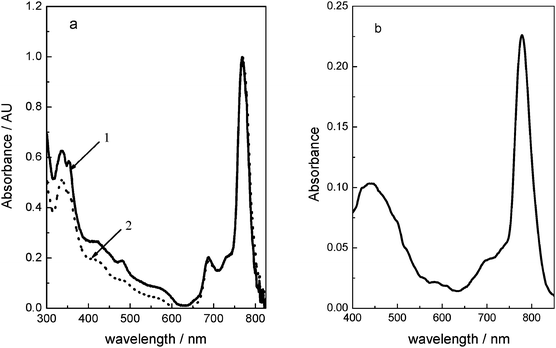 | ||
Fig. 2 Absorption spectra of naphthalocyanine dyes: (a) in DMF solution, (1) ![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ; (2) ⋯. (b) Absorption spectrum of (1) adsorbed over FTO/TiO2 electrode. ; (2) ⋯. (b) Absorption spectrum of (1) adsorbed over FTO/TiO2 electrode. | ||
On the other hand, fluorescence is observed from 1 and 2 in DMF solution (Fig. 3). Both compounds show emissions in the Q-band region for excitation in either Q or the B absorption bands. The excitation spectra of the compounds are similar to that of the absorption spectra. However, as can be observed from the excitation spectra, emission from aggregates is minimal, showing the typical quenching effect when molecular aggregates are formed in these kind of compounds. The electronic energy levels are relatively unaltered by changing the axial substituents and both compounds have a very small Stokes shift of less than 10 nm. The fluorescent quantum yield (ΦF) values are high (0.17), and similar to those observed for silicon–phthalocyanines.40 The presence of the lighter Si atom in the macrocycle leads to low rates of inter-system crossing (ISC), efficient fluorescent emission and a fairly small non-radiative decay constant.
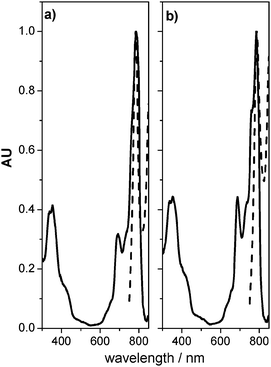 | ||
Fig. 3 (a) Excitation () and emission (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) spectra in DMF solution of 1 (a) and 2 (b) naphthalocyanine dyes. λex = 710 nm. ) spectra in DMF solution of 1 (a) and 2 (b) naphthalocyanine dyes. λex = 710 nm. | ||
Naphthalocyanine 1 and 2 adsorption onto the semiconductor produces light-gray coloration of the film. However, the amount of 2 that remains over electrode surface after solvent rinses is low (electrode absorbance below 0.07 at Q-band, less than half of that obtained with 1), indicating inefficient dye harvesting in the TiO2 semiconductor. This last fact could result from the low solubility of the dyes in DMF solution used for the adsorption procedure. Contrarily, 1 was adsorbed in larger amounts, possibly due to the already mentioned affinity of the silicon-derivatives for the TiO2 semiconductor surface. The UV–visible absorption spectrum of compound 1 adsorbed on FTO/TiO2 (Fig. 2b) shows electronic transitions (Q and B bands) broader and shifted in comparison with those in solution. This may be caused by the interaction of the dye with the polar surface of the nanostructured TiO2, as well as the possible formation of aggregates.
Fig. 4 shows the short circuit photocurrent action spectrum obtained with a sandwich cell using 1 as light absorbing dye. The incident monochromatic photon-to-photocurrent efficiencies, (IPCE)41 closely match the absorption spectrum of the organic molecule, confirming that photosensitization has successfully extended the photocurrent response of the electrodes into the IR region. The small red shift (∼5 nm) observed between the IPCE and the absorption spectrum at the Q-band could originate in the different media that surround the electrode, air and electrolyte. We observed an intense response in the red part of the spectrum with maximum IPCE of ∼17% obtained at Q-band (790 nm). There are several reasons for this value, which may be linked to the factors that determine IPCE, namely, the light harvesting efficiency of the electrode (related to the extinction coefficient and the amount of adsorbed dye), the charge injection yield (Φinj) from the excited dye to the semiconductor, and the charge collection efficiency, ηc, of the system (IPCE = LHE Φinjηc.).41 Taking into account the electrode absorbance at 790 nm the product Φinjηc for the dye at the Q-band is ∼0.4. This value is consistent with those reported for axially anchored phthalocyanines compounds by Morandeira et al.28 In our case, improvement in the IPCE (and therefore cell efficiency) could be achieved by peripheral modifications of Si–naphthalocyanines. The introduction of branched groups (such as terbutyl) allows one to obtain higher dye solubility, which precludes aggregation and could increase the effective amount of absorbed dye on the electrode.
 | ||
| Fig. 4 Photocurrent action spectrum, IPCE% (⋯) of 1 DSC. Electrode absorption spectrum () was added for comparison. | ||
In order to evaluate the energetic feasibility of the sensitization process, we estimated the oxidation potential of naphthalocyanine compounds using differential pulse voltametry (DPV) in DMF solution. However, due to the low solubility of 2, we synthesized a more soluble ester derivate of this compound (3, Fig. 1) for DPV experiments.42 The first oxidation potentials observed were 0.56 and 0.63 V vs ferrocene/ferrocinium couple used as the internal reference, for 1 and 3 respectively. With those values, the oxidation potentials of the dye-excited state (E*ox) can be estimated by subtracting the excitation energy (1.59 eV, obtained from absorption and emission spectra) from the redox potentials of the molecules in the ground state. The energy analysis shows that both processes, electron injection into TiO2 from dye-excited states and dyes reduction by I−/I3− couple are exothermic, making possible the DSC operation. It is interesting to note that Li et al.32 attributed the lack of photocurrent generation to the inability of iodide to reduce the photogenerated dye cations, consistent with the dye ground-state oxidation potential (0.25 V vs Ag/AgCl). However, the Zn–naphthalocyanine used by Li et al bears four electron-donating ether groups and, as was clearly demonstrated for porphyrins, phthalocyanines and other related dyes,43 peripheral substitution has a noticeable effect in modulating the electron donor/acceptor capabilities. Moreover, the Zn(II) central metal also contributes to HOMO destabilization, making the dye cation a weaker reductant.
Fig. 5 shows the current–voltage curve measured for 1–naphthalocyanine DSC under AM 1.5 G simulated sunlight. The cell exhibits a short circuit current, open-circuit voltage, fill factor, and power conversion efficiency of 0.38 mA cm−2, 0.47 V, 0.6, and 0.11% respectively (0.12 mA cm−2, 0.43 V, 0.6, and 0.03% for 2–naphthalocyanine DSC). To the best of our knowledge, this result represents the first report of DSCs based on naphthalocyanine sensitizers. As is shown in the photo insert Fig. 5, the complete solar cell is nearly transparent in the visible region; the light brown color is due to the iodide electrolyte. We believe that these results could start increased interest in the field of naphthalocyanine-sensitized solar cells for use as photovoltaic windows that transmit part of the visible light and harvest the rest in the UV and red/near-IR zone of the solar spectrum.
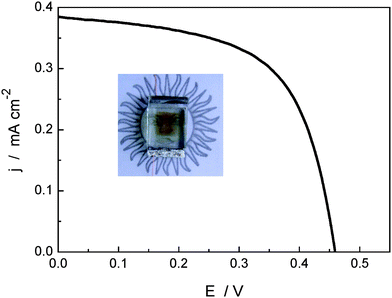 | ||
| Fig. 5 I–V curve obtained with 1 DSC under AM 1.5 G solar simulation conditions. The inserted shows a photo of the complete cell. | ||
Conclusion
The capability of axially anchored Si–naphthalocyanine for their utilization in DSCs has been demonstrated. The cells exhibit maximum incident photon to current efficiency at λ ∼ 790 nm. The electrodes absorbance and IPCE spectra show that there is little or no dye aggregation over TiO2 surface, demonstrating that Ti–O–Si axial anchoring is a promising route to the design of new IR-sensitizing dyes. The results we obtained are fairly promising and should encourage further studies on DSCs using axially anchored naphthalocyanine compounds.Experimental
Naphthalocyanine dyes
The structures of the dyes used are shown in Fig. 1. Bis(succinoyl) silicon naphthalocyanine 2 and bis(isovaleroyl) silicon naphthalocyanine 3 were synthesized following the procedure previously described with minor modifications.44 A solution of succinic acid (5 ml, 6 M) was added to 2 ml of 1 (52 mg, 0.066 mmol) in DMF. The solution was refluxed for 40 min and the reaction mixture quenched by addition of water (until complete precipitation). The precipitate was centrifuged and the solid was dried under vacuum, yielding 43 mg (66%) of 2. 1HNMR (DMSO-d6, TMS) δ (ppm) −2.10 (4H), −0.74 (4H), 8.02 (8H), 8.82 (8H), 10.14 (8H). MS-FAB [m/z] 975 [M + H]+ (974.2269 calculated for C56H34N8O8Si). Anal. calcd. C 68.98, H 3.51, N 11.49, found C 68.87, H 3.43, N 11.57. Absorption spectrum λmax (DMF) (nm) (ε, dm3mol−1cm−1) 347 (134![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 781 (327000). In a similar way, a solution of iso-valeric acid (1 ml, 6 M) was added to 2 ml of 1 (60 mg, 0.074 mmol) in DMF. The mixture was treated as described above for 2, giving 44 mg (63%) of 3. 1HNMR (CDCl3, TMS) δ (ppm) −0.66 (d, 4H, J = 5.8 Hz), −0.38 (m, 2H), 0.99 (d, 12 H, J = 5.6 Hz), 8.01 (8H), 8.67 (8H), 10.14 (8H). MS-FAB [m/z] 943 [M + H]+ (942.3098 calculated for C58H42N8O4Si). Anal. calcd. C 73.87, H 4.49, N 11.88, found C 73.76, H 4.38, N 11.97. Absorption spectrum λmax (DMF) (nm) (ε, dm3mol−1cm−1) 341 (112000), 801 (352000). Bis(chloro) silicon naphthalocyanine 1 was purchased from Aldrich (Milwaukee, WI, USA).
000), 781 (327000). In a similar way, a solution of iso-valeric acid (1 ml, 6 M) was added to 2 ml of 1 (60 mg, 0.074 mmol) in DMF. The mixture was treated as described above for 2, giving 44 mg (63%) of 3. 1HNMR (CDCl3, TMS) δ (ppm) −0.66 (d, 4H, J = 5.8 Hz), −0.38 (m, 2H), 0.99 (d, 12 H, J = 5.6 Hz), 8.01 (8H), 8.67 (8H), 10.14 (8H). MS-FAB [m/z] 943 [M + H]+ (942.3098 calculated for C58H42N8O4Si). Anal. calcd. C 73.87, H 4.49, N 11.88, found C 73.76, H 4.38, N 11.97. Absorption spectrum λmax (DMF) (nm) (ε, dm3mol−1cm−1) 341 (112000), 801 (352000). Bis(chloro) silicon naphthalocyanine 1 was purchased from Aldrich (Milwaukee, WI, USA).
Cell preparation
Colloidal paste was prepared as follows: 1:4 solution of commercial P25 TiO2 particles (Degussa) and ethanol was stirred and sonicated two times for a minute. After that we added 0.2 g of PEG and stirred overnight. To obtain a porous film of uniform thickness, the following procedure was used. The colloidal TiO2 suspension was spread onto transparent conducting glass sheets (fluorine-doped tin oxide glass, FTO) using scotch tape as a spacer. A thin film was obtained by raking off the excess suspension with a glass rod. After removing the tape and air-drying, the sample was sintered in air at 450 °C for 30 min to form a transparent TiO2 film electrode. The thickness of the film was about 7 µm. Dye-coating of the TiO2 film was carried out by soaking the film in dye-satured dimethylformamide (DMF) solution, immediately after the high-temperature annealing and while it was still warm (∼80 °C). The DSC cell was assembled using a platinized conducting glass and a 50 µm hot melt spacer. The electrolyte used was 0.5 M LiI / 0.05 M I2 in metoxipropionitrile, which was introduced into the cell by holes drilled in the platinized counter electrode.
General
Absorption and fluorescence spectra were recorded on a Shimadzu UV-2401PC spectrometer and on a Spex FluoroMax fluorometer, respectively. Spectra were recorded using 1 cm path length quartz cells at 25.0 ± 0.5 °C. The fluorescence quantum yield (ϕF) of naphthalocyanine dyes was calculated by comparing the area below the corrected emission spectrum with that of ZnPc as a fluorescence reference. A value of ϕF = 0.28 for ZnPc in DMF was calculated by comparison with the fluorescence spectrum in pyridine using ϕF = 0.30 and taking into account the refractive index of the solvents (η (DMF) = 1.431; η (DMF/water 10% [HCl] = 1.2 mM) = 1.425). The refractive indexes (η) were measured using an Atago NAR-1T (Tokyo, Japan) refractometer.Proton nuclear magnetic resonance (1HNMR) spectra were recorded on a FT-NMR Bruker 200 spectrometer at 200 MHz. FAB mass spectra were taken with ZAB-SEQ Micromass equipment.
Electrochemical characterization of the redox process for the dyes was acquired with a potentiostat–galvanostat Autolab (Electrochemical Instruments) using a Pt disc working electrode of 0.204 cm2 and a Pt counter electrode in a conventional three compartment Pyrex cell. A freshly prepared Ag/AgCl quasireference electrode was used. The working electrode was cleaned between each experiment by polishing with 0.3 µm alumina paste, followed by solvent rinses. Studies were carried out in N2-degased DMF with 0.1 M tetrabutylammonium perchlorate (TBAP, Aldrich) as the supporting electrolyte. After each experiment, ferrocene was added, and the potential axis was calibrated against the formal potential for ferrocenium/ferrocene redox.
Photocurrent action spectra were obtained by illumination of the DSC with monochromatic light obtained from a 75 W high-pressure Xe lamp (Photon Technology Instrument, PTI) and a computer controlled PTI high intensity grating monochromator. The incident light intensities at different wavelengths were measured with a Coherent Laser-Mate Q radiometer (sensitivity 1 µW).
Photocurrent and voltage were measured using a solar simulator equipped with a 1000W ozone-free Xenon lamp and AM 1.5 G filter (Oriel), where the light intensity was adjusted with an NREL-calibrated Si solar cell with a KG-5 filter to 1 sunlight intensity (100 mW cm−2).
Acknowledgements
This work was supported in part by a joint project Argentina–Spain funded by Agencia Española de Cooperación Internacional (AECI) and Universitat Jaume I. L.O. is grateful to Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET–Argentina); Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT–Argentina), Secretaría de Ciencia y Técnica de la Universidad Nacional de Río Cuarto (SECYT–UNRC). J.B. thanks financial support from Ministerio de Ciencia e Innovación of Spain under project HOPE CSD2007-00007 (Consolider-Ingenio 2010).References
- T. W. Hamann, R. A. Jensen, A. B. F. Martinson, H. Van Ryswyk and J. T. Hupp, Energy Environ. Sci., 2008, 1, 66 RSC.
- L. Otero, H. Osora, L. Li and M. A. Fox, J. Porphyrins Phthalocyanines, 1998, 2, 123 CrossRef CAS.
- M. Linke-Schaetzel, A. D. Bhise, H. Gliemann, T. Koch, T. Schimmel and T. S. Balaban, Thin Solid Films, 2004, 451, 16 CrossRef.
- D. Kuang, C. Klein, S. Ito, J. E. Moser, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2007, 17, 154 CrossRef CAS.
- L. Moreira Gonçalves, V. Bermudez, H. Aguilar Ribeiro and A. Magalhães Mendes, Energy Environ. Sci., 2008, 1, 655 RSC.
- H. Sugihara, L. P. Singh, K. Sayama, H. Arakawa, M. K. Nazeeruddin and M. Grätzel, Chem. Lett., 1998, 10, 1005 CrossRef.
- S. D. Burnside, K. Brooks, A. J. McEvoy and M. Grätzel, Chimia, 1998, 52, 557 CAS.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 8.
- J. J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martínez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS.
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fré, P. Rubino, C. Chon, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342 CrossRef CAS.
- K. Takechi, P. K. Sudeep and P. V. Kamat, J. Phys. Chem. B, 2006, 110, 16169 CrossRef.
- A. Otsuka, K. Funabiki, N. Sugiyama, H. Mase, T. Yoshida, H. Minoura and M. Matsui, Chem. Lett., 2008, 37, 176 CrossRef CAS.
- K. Takechi and P. V. Kamat, Chem. Mater., 2008, 20, 265 CrossRef CAS.
- H. Choi, S. Kim, S. O. Kang, J. Ko, M.-S. Kang, J. N. Clifford, A. Forneli, E. Palomares, M. K. Nazeeruddin and Michael Grätzel, Angew. Chem., Int. Ed., 2008, 47, 8259 CrossRef CAS.
- J.-H. Yum, S.-R. Jang, P. Walter, T. Geiger, F. Nüesch, S. Kim, J. Ko, M. Grätzel and M. K. Nazeeruddin, Chem. Commun., 2007, 4680 RSC.
- C. Li, X. Yang, R. Chen, J. Pan, H. Tian, H. Zhu, X. Wang, A. Hagfeldt and L. Sun, Sol. Energy. Mater. Sol. Cells., 2007, 91, 1863 CrossRef CAS.
- Y. Jin, J. Hua, W. Wu, X. Ma and F. Meng, Synth. Met., 2008, 158, 64 CrossRef CAS.
- J.-H. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nüesch, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc, 2007, 129, 10320 CrossRef CAS.
- J. H. Yum, S. J. Moon, R. Humphry-Baker, P. Walter, T. Geiger, F.N. üesch, M. Grätzel and M. K. Nazeeruddin, Nanotechnology, 2008, 19, 424005 CrossRef.
- C. Li, W. Wang, X. Wang, B. Zhang and Y. Cao, Chem. Lett., 2005, 34, 554 CrossRef CAS.
- A. Burke, L. Schmidt-Mende, S. Ito and M. Grätzel, Chem. Commun., 2007, 234 RSC.
- S. Barazzouk, P. V. Kamat and S. Hotchandani, J. Phys. Chem. B, 2005, 109, 716 CrossRef CAS.
- P. K. Sudeep, K. Takechi and P. V. Kamat, J. Phys. Chem. C, 2007, 111, 4884.
- J. He, G. Benko, F. Korodi, T. Polívka, B. Reiner Lomoth, B. Akermark, L. Sun, A. Hagfeldt and V. Sundstrom, J. Am. Chem. Soc., 2002, 124, 4922 CrossRef CAS.
- P. Y. Reddy, L. Giribabu, Ch. Lyness, H. J. Snaith, Ch. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J. H. Yum, K. Kalyanasundaram, M. Grätzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373 CrossRef CAS.
- M. K. Nazeeruddin, R. Humphry-Baker, M. Grätzel, D. Wöhrle, G. Schnurpfeil, G. Schneider, A. Hirth and N. Trombach, J. Porphyrins Phthalocyanines, 1999, 3, 230 CrossRef CAS.
- E. Palomares, M. V. Martínez-Díaz, S. A. Haque, T. Torres and J. R. Durrant, Chem. Commun., 2004, 2112 RSC.
- A. Morandeira, I. López-Duarte, M. V. Martínez-Díaz, B. O'Regan, Ch. Shuttle, N. A. Haji-Zainulabidin, T. Torres, E. Palomares and J. R. Durrant, J. Am. Chem. Soc., 2007, 129, 9250 CrossRef CAS.
- S. Eu, T. Katoh, T. Umeyama, Y. Matanoa and H. Imahori, Dalton Trans., 2008, 40, 5476 Search PubMed.
- J.-H. Yum, S. Jang, R. Humphry-Baker, M. Grätzel, J. J. Cid, T. Torres and Md. K. Nazeeruddin, Langmuir, 2008, 24, 5636 CrossRef CAS.
- B. C. O'Regan, I. López-Duarte, M. V. Martínez-Díaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc, 2008, 130, 2906 CrossRef CAS.
- X. Li, N. J. Long, J. N. Clifford, C. J. Campbell and J. R. Durrant, New J. Chem., 2002, 26, 1076 RSC.
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 77, 347 CrossRef CAS.
- P. Péchy, F. P. Rotzinger, M. K. Nazeeruddin, O. Kohle, S. M. Zakeeruddin, R. Humphry-Baker and M. Grätzel, J. Chem. Soc., Chem. Commun., 1995, 1093 RSC.
- P. K. Ghosh and T. G. Spiro, J. Am. Chem. Soc., 1980, 102, 5543 CrossRef CAS.
- A. K. M. Fung, B. K. W. Chiu and M. H. W. Lam, Water Res., 2003, 37, 1939 CrossRef CAS.
- J. He, G. Benko, F. Korodi, T. Polívka, R. Lomoth, B. Akermark, L. Sun, A. Hagfeldt and V. Sundstrom, J. Am. Chem. Soc., 2002, 124, 4922 CrossRef CAS.
- D. Schlettwein, T. Oekermann, T. Yoshida, M. Tochimoto and H. Minoura, J. Electroanal. Chem., 2000, 481, 42 CrossRef CAS.
- J. Andzelm, A. M. Rawlett, J. A. Orlicki, J. F. Snyder and K. K. Baldridge, J. Chem. Theory Comput., 2007, 3, 870 CrossRef CAS.
- C. A. Barker, K. S. Findlay, S. Bettington, A. S. Batsanov, I. F. Perepichka, M. R. Bryce and A. Bebí, Tetrahedron, 2006, 62, 9433 CrossRef CAS.
- P. Vlachopoulos, P. Liska, J. Augustynski and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 1216 CrossRef CAS.
- B. L. Wheeler, G. Nagasubramanian, A. J. Bard, L. A. Schechtman, D. R. Dininny and M. E. Kenney, J. Am. Chem. Soc., 1984, 106, 7404 CrossRef CAS.
- K. M. Kadish and M. M. Morrison, J. Am. Chem. Soc., 1976, 98, 3326 CrossRef CAS.
- D. M. Maree, N. Kuznetsova and T. Nyokong, J. Photochem. Photobiol., A, 2001, 140, 117 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |