Hydrogen production from renewable sources: biomass and photocatalytic opportunities
R. M.
Navarro
,
M. C.
Sánchez-Sánchez
,
M. C.
Alvarez-Galvan
,
F. del
Valle
and
J. L. G.
Fierro
*
Instituto de Catálisis y Petroleoquímica, CSIC, Cantoblanco, E-28049, Madrid, Spain. E-mail: jlgfierro@icp.csic.es; Fax: +34 915 854 769
First published on 22nd October 2008
Abstract
The demand for hydrogen over the coming decade is expected to grow for both traditional uses (ammonia, methanol, refinery) and running fuel cells. At least in the near future, this thirst for hydrogen will be quenched primarily through the reforming of fossil fuels. However, reforming fossil fuels emits huge amounts of carbon dioxide. One approach to reduce carbon dioxide emissions, which is considered first in this review, is to apply reforming methods to alternative renewable materials. Such materials might be derived from plant crops, agricultural residues, woody biomass, etc. Clean biomass is a proven source of renewable energy that is already used for generating heat, electricity, and liquid transportation fuels. Clean biomass and biomass-derived precursors such as ethanol and sugars are appropriate precursors for producing hydrogen through different conversion strategies. Virtually no net greenhouse gas emissions result because a natural cycle is maintained, in which carbon is extracted from the atmosphere during plant growth and released during hydrogen production. The second option explored here is hydrogen production from water splitting by means of the photons in the visible spectrum. The sun provides silent and precious energy that is distributed fairly evenly all over the earth. However, its tremendous potential as a clean, safe and economical energy source cannot be exploited unless it is accumulated or converted into more useful forms of energy. Finally, this review discusses the use of semiconductors, more specifically CdS and CdS-based semiconductors, which are able to absorb photons in the visible region of the spectrum. The energy stored within a semiconductor as electronic energy (electrons and holes) can be used to split water molecules by simultaneous reactions into H2 and O2. This conversion of solar energy into a clean fuel (H2) is perhaps the greatest challenge for scientists in the 21st century.
Rufino M. Navarro is a Tenured Scientist at the Institute of Catalysis and Petrochemistry of the National Council of Scientific Research (CSIC). His research focuses on heterogeneous catalysis applied to clean energy production: reforming of hydrocarbon, hydrogen technologies, renewable energies, thermochemical cycles and water splitting technologies. |
Maria Cruz Sánchez is a PhD candidate in the Institute of Catalysis and Petrochemistry of the National Council for Scientific Research (CSIC). Her research activities are concentrated in the field of hydrogen production from bioalcohols. |
Consuelo Alvarez-Galvan received her doctoral degree from the University of La Laguna and then moved to the Institute of Catalysis and Petrochemistry of the National Council for Scientific Research (CSIC) where she is as a Ramon y Cajal postdoctoral scientist. Her research interests include hydrocarbon reforming, thermochemical cycles, and catalytic combustion. |
Fabian del Valle is a PhD candidate in the Institute of Catalysis and Petrochemistry of the National Council for Scientific Research (CSIC). His research activities are in the field of water splitting on semiconductor surfaces. |
Jose L.G. Fierro is Research Professor at Institute of Catalysis and Petrochemistry of the National Council of Scientific Research (CSIC). His research interests include hydrogen technologies, renewable energy, natural gas conversion, chemical technology, solid state chemistry, environmental chemistry, and commodity and bulk chemicals production. |
Broader contextHydrogen is a versatile energy carrier that is usually produced from steam reforming of carbon-containing sources with the coproduction of huge amounts of CO2. To take full advantage of the environmental benefits of hydrogen, low carbon emitting, low polluting, low cost hydrogen production systems are needed. This review details the fundamentals of and describes some major achievements in the production of hydrogen from two renewable resources: biomass and water splitting with visible light. Particular emphasis is placed on biomass gasification under near- and supercritical water conditions, and also in semiconductors which split water under ambient temperature and pressure upon irradiation with light of wavelength in the visible spectrum. |
1. Introduction
Hydrogen (H2) is the most common element on earth, but it does not occur to a significant extent in elemental form. It is mostly present in water, biomass and hydrocarbons. Hydrogen gas is currently produced from a variety of primary sources, such as natural gas, naphtha, heavy oil, methanol, biomass, wastes, coal, solar, wind and nuclear.1–4 It is a clean energy fuel because the chemical energy stored in the H–H bond is released when it combines with oxygen, yielding only water as a reaction product. Accordingly, a future energy infrastructure based on hydrogen has been perceived as an ideal long-term solution to energy-related environmental problems.5–7 There is no doubt that hydrogen has the potential to provide a clean and affordable energy supply that can minimize our dependence on oil and therefore enhance the global economy and reduce environmental pollution.It is generally understood that the renewable energy-based processes of hydrogen production (solar photochemical and photobiological water decomposition, electrolysis of water coupled with photovoltaic cells or wind turbines, etc.) are unlikely to involve significant reductions in hydrogen costs over the next ten-to-twenty years. Industry generates some 48 million metric tons of hydrogen globally each year from fossil fuel. Almost half of this hydrogen goes into making ammonia, while refineries use the second largest volume of hydrogen for chemical processes such as removing sulfur from gasoline and converting heavy hydrocarbons into gasoline and diesel fuel. Food producers add a small percentage of hydrogen to some edible oils through a catalytic hydrogenation process.
The demand for hydrogen is expected to grow over the next ten years, for both traditional uses, such as making ammonia, and running fuel cells. At least in the near future, this thirst for hydrogen will be quenched primarily through the use of fossil fuels. To make hydrogen, industry uses Steam Methane Reforming (SMR), which is the most widely used and most economical process for producing hydrogen.1–4 Although SMR is a complex process involving many different catalytic steps, as long as natural gas (or CH4) and hydrocarbon fuels remain at a low or even moderate price, SMR will continue to be the choice technology for the mass production of H2. Following several decades of improvements in catalyst technology, substantial improvements have been introduced over the years. The SMR process also produces carbon monoxide and carbon dioxide, the primary greenhouse gas. Although this approach still generates pollution, these gases are released in a potentially more manageable way, as opposed to the case of millions of automobile engines.
Nonetheless, shedding the habit of fossil fuel entirely is the only way a wholesale shift to hydrogen will work in the long-term. One approach to this goal is to apply steam-reforming methods to alternative renewable materials. Such materials might be derived from crops. Not only do these biomass-conversion schemes turn waste into a valuable product, but, researchers say, there is another benefit: any carbon dioxide released in the processes could be soaked up by planting new crops to provide the required biomass. A biomass strategy of hydrogen generation could be a useful intermediate step between the current fossil fuel method and the dream of efficient water splitting. Still, any realistic contender for hydrogen generation must first replace the reforming of fossil fuel as the cheapest and most efficient process.
Although hydrogen production and storage/distribution infrastructures are commercially available in chemical and refining industries around the world, existing conversion and storage technologies are too expensive for widespread use in energy schemes. The development of hydrogen production as a realistic, viable energy option will require an unprecedented level of sustained and coordinated activities and, most importantly, the development of new concepts for the distributed production of hydrogen. Not only fossil fuel burning, which contributes to the greenhouse gas pool, but also the eventual depletion of the world's fossil-fuel reserves are threatening sustainable development.8,9 Abundant, clean, and carbon-neutral hydrogen is widely believed to be the ultimate mobile energy carrier replacing gasoline, kerosene and diesel in internal combustion engines. The present status of hydrogen production from less costly and abundant biomass for producing low-cost hydrogen without net carbon emissions is reviewed.10–14 In addition, this review includes advances in the fully renewable conversion of solar energy into hydrogen via the water splitting process assisted by photo-semiconductor catalysts.
2. Hydrogen production from carbon-neutral precursors
Hydrogen is currently produced almost entirely from natural gas, liquid hydrocarbons and coal. All these C-containing sources release massive amounts of CO2 into the atmosphere during the production of hydrogen. Thus, renewable biomass, a product of photosynthesis, is an attractive alternative to fossil feedstocks, as it can be considered a CO2 neutral precursor. Photosynthesis is the biological process that converts light energy into chemical energy and stores it in carbohydrates as “nCO2 + nH2O → [CH2O]n + nO2”, and fixes atmospheric carbon into biomass. At the dawn of the 21st century, a combination of economic, technological, resource and political developments is driving the emergence of a new carbohydrate economy.15,16Biomass can be converted into hydrogen and other useful products through several thermochemical processes, such as liquefaction, pyrolysis and gasification. Among these, pyrolysis and gasification have received considerable attention in recent years. In addition, there are other biochemical processes for the conversion of biomass into hydrogen. A brief account of biological H2 production and the future challenges in this field are also featured in this review.
2.1 Thermal pyrolysis
Pyrolysis is the thermal decomposition of solid biomass resources at a temperature of 650–800 K at 1–5 bar in the absence of air to yield liquid oils, solid charcoal and gaseous compounds. Pyrolysis can be classified into slow pyrolysis and fast pyrolysis (Fig. 1). Although most pyrolysis processes are designed for biofuel production, hydrogen can be produced directly through fast or flash pyrolysis if both high temperature and sufficient volatile phase residence time are provided for as follows (eqn 1):| CxHyOz + heat → H2 + CO + CH4 + HCs | (1) |
| CH4 + HCs + H2O → CO + H2 | (2) |
| CO + H2O → CO2 + H2 | (3) |
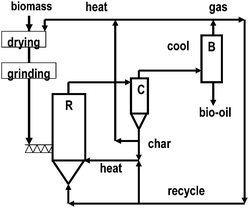 | ||
| Fig. 1 Sketch diagram of a fast pyrolysis plant. (R), reactor; (C), cyclone; (B), bio-oil. Adapted from ref. 21. | ||
Lower process temperatures and longer vapour residence times favour the production of charcoal. High temperatures and longer residence times increase biomass conversion to gas, and moderate temperatures and short vapour residence times are optimum for producing liquid. Thus, fast pyrolysis for liquid production is currently of particular interest because liquids can be stored and transported more easily and at a lower cost than solid biomass.17 Besides the gaseous products, the bio-oil products can also be processed for hydrogen production.18 In general maximum hydrogen yield at about 90% is obtained with steam reforming over Ni-based catalysts and additional water–gas shift reaction. Pyrolysis reaction rates are accelerated by some chlorides and carbonates.19 Since it is difficult to gasify tar, extensive studies on the catalytic effect of inexpensive dolomite and CaO on the decomposition of HCs in tar have been carried out.20 The beneficial effects of other catalysts, such as Ni,21 Y-type zeolite,22 carbonates,23 and metal oxides24 have also been investigated. Virtually any form of biomass can be considered for fast pyrolysis. While most work has been carried out on wood because of its consistency between tests, nearly one hundred different biomass types have been employed, ranging from agricultural wastes such as straw, olive pits and nut shells to energy crops, forestry wastes and solid wastes.
2.2 Steam/oxygen gasification
Biomass is also gasified at temperatures above 1000 K in the presence of oxygen and/or water. Under these conditions, biomass undergoes partial oxidation and/or steam reforming reactions yielding gas and char product. The char is subsequently reduced to form H2, CO, CO2 and CH4. This conversion process can be expressed as:| CxHyOz + H2O + O2 → H2 + COx + CH4 + HCs + char | (4) |
Most biomass gasification processes employ air as gasifying agent, which results in a low calorific value gas stream (3–5 MJ m−3). This gas can be used after cleaning in gas-fired engines or gas turbines. For gas turbines connected to a steam turbine, medium calorific value gas (10–15 MJ m−3) is more favourable than low calorific gas. Steam injection into the gas turbine combustion chamber requires at least medium calorific value gas. The production of methanol or hydrogen via biomass gasification or the use of producer gas in low-temperature fuel cells also require either gasifiers operating with highly-enriched oxygen or gasifiers using steam as a gasification medium to generate the necessary medium calorific value raw gas with high hydrogen content.
The gasification of wood and wood-type residues and waste in fixed bed or fluidised bed gasifiers with subsequent burning of the gas for heat production is the state-of-the-art. Significantly greater technical problems are posed by the gasification of straw and other solid agricultural feedstocks, which mostly have higher concentrations of chlorine, alkali, nitrogen and sulfur. The gasification of herbaceous biomass is still at an early stage of research and development. Intensified development efforts involving gasification technologies for herbaceous biomass feedstocks are desirable, as the potential supply of this group of fuels is comparatively large. Thorough gas cleaning and the adaptation of the gas from biomass gasification to the specific requirements of the gas utilisation systems are the prerequisites for gas use in gas-fired engines, gas turbines and fuel cells.
One of the major issues in biomass gasification is the tar formation that occurs during the process. The unwanted tar polymerizes to a more complex structure, which is not favourable for hydrogen production through steam reforming. Currently, three methods are available for minimising tar formation: (i) proper gasifier design; (ii) incorporation of catalysts; and (iii) control of operating variables. Regarding method (iii), the operating parameters, such as gasifying agent, temperature and residence time, are key factors in the formation and decomposition of tar. Tar can be thermally cracked at temperatures above 1273 K.25 In the type (ii) method, the use of additives (such as dolomite, olivine and even char), also facilitates tar reduction.26 Dolomite is particularly suited because 100% tar elimination can be achieved with this additive.27 Catalysts also reduce tar content, but are particularly effective for improving gas product quality and conversion. Dolomite-loaded nickel catalysts and alkaline metal oxides are widely used as gasification catalysts.
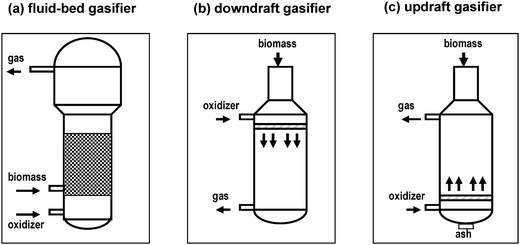 | ||
| Fig. 2 Sketch pictures of biomass reactors: (a), fluidized-bed gasifier; (b), downdraft gasifier; and (c), updraft gasifier (ref. 28). | ||
In the fluidized-bed reactor (Fig. 2(a)) the biomass, which is previously reduced to a fine powder, air, steam, or oxygen enters through the bottom of the gasifier. A high linear velocity of the gas stream forces the fine particles of biomass upward through a bed of silica beads. Pyrolysis and char gasification take place in this process. This type of gasifier is suitable for large-scale applications and has a medium tar yield of around 10 g Nm−3. In the downdraft gasifier (Fig. 2(b)), the air or oxygen and biomass particles enter through the top of the reactor as a fine powder and flow downward, and the gas exits through the bottom of the reactor. The product gas contains the lowest concentration of particulates and tars of nearly 1 g Nm−3, which is a much lower level than in fluidized-bed reactor, because most of the tars are combusted. The flame temperature in this reactor is 1200–1600 K. This reactor configuration is ideal when clean gas is needed. The main disadvantage of this gasifier reactor is its low overall thermal efficiency, as well as the difficulty in handling ash content. In the updraft gasifier (Fig. 2(c)), biomass enters through the top and air/oxygen/steam flow upward from the bottom and the gas exits through the top. This reactor primarily forms tars at a very high level (in the order of 100 g Nm−3). The principal advantages of an updraft gasifier include: it is a mature technology for heat production, it can be used for small-scale applications, and it can handle feeds with high moisture content. On the other hand, the tar yield of this gasifier is very high and it has slagging potential.
Absorption enhanced steam reforming (AER) is another technology that has been explored recently for the continuous gasification of biomass and the production of a H2-rich gas stream.35,36 In the AER process, the CO2 produced during steam gasification is separated from the reactor by an adsorbent, so that the resulting product gas contains a high H2 concentration and low concentration of carbon oxides. CO2 absorption not only shifts the equilibrium towards the desired product, but also delivers heat for the endothermic reactions. A gas with increased CO2 concentration is produced along the regeneration step, which simplifies CO2 separation. The concept of the AER process is outlined in Fig. 3. The two fluidised bed reactors are coupled in such a manner that the sorbent bed material circulates between the AER gasifier (CO2 absorption) and the combustor (CO2 desorption). A nearly nitrogen-free product gas with a caloric value of 12–14 MJ Nm−3 (dry) is produced in the AER gasifier.
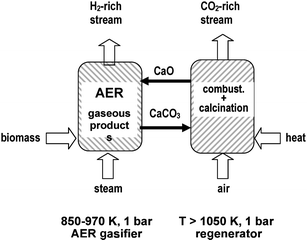 | ||
| Fig. 3 Coupling of two fluidized-bed reactors for the continuous production of an H2-rich gas flow from biomass. The adsorbent bed material circulates between the AER reactor (absorption) and the regenerator (desorption). | ||
AER is being used for the unpressurised steam gasification of biomass (eqn 5). Through simultaneous CO2 absorption (in the example with CaO) (eqn 7), the equilibrium of the homogeneous WGS reaction (eqn 6) is shifted towards H2 and CO2 and all the parallel gasification/reforming reactions are also influenced in favour of the desired products. Accordingly, a H2-rich product gas results with reduced CO and CO2 concentration. Eqn 4 represents the idealized sum reaction for AER gasification. For the sake of simplicity, the formation of methane coke and tars is ignored.
| CHxOy + (1−y)H2O → CO + (0.5x +1−y)H2 (ΔH > 0) | (5) |
| CO + H2O → CO2 + H2 (ΔH < 0) | (6) |
| CaO + CO2 → CaCO3 (ΔH > 0) | (7) |
| CHxOy + (1−y)H2O + CaO → CO + (0.5x + 2−y)H2 | (8) |
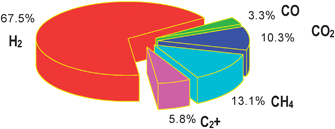 | ||
| Fig. 4 Typical gas composition of a 100 kW gasifier operated in the Absorption Enhanced Reforming (AER) mode. | ||
Sorbent enhanced reforming technology is still at the experimental stage, and shows promise for low cost H2 production. Critical issues in this methodology are sorbent lifetime and system design.
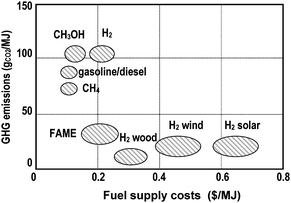 | ||
| Fig. 5 Specific greenhouse gas emissions of the supply chain and use of fuel with respect to fuel costs (without taxes) from ref. 68. | ||
Hydrogen costs delivered to the end user will generally be higher than the costs of current fossil fuel options. Especially hydrogen based on renewable sources will increase fuel costs compared with conventional fuels but at a very low specific CO2 level. Higher efficiencies of fuel cell systems, costs for CO2 capture and storage – necessary for fossil-based hydrogen – as well as bonus points for less specific carbon dioxide emissions will modify Fig. 5.
2.3 Biomass gasification in supercritical water
Supercritical water (SCW) is obtained at pressures above 221 bar and temperatures above 647 K. In the absence of oxygen, biomass is converted under supercritical water conditions into fuel gases (eqn 9), which are easily separated from the water phase by cooling to ambient temperature.41,42| CxHyOz + (2x−z)H2O → xCO2 + (2x−z + y/2)H2 | (9) |
Other reactor types include capillaries and tubular steel reactors. Quartz capillaries have also been used as batch microreactors. This reactor configuration allows for inexpensive and high-speed testing, with the further possibility of visual observation. A drawback is that the pressure inside the capillary cannot be directly measured; it is calculated from the temperature and the sample plus reactor volume. For continuous operation, tubular steel reactors are often used. Other types of reactor, such as the stirred tank reactor can be used in principle, but to date this configuration has not yet been applied. To maintain the same capacity, the volume of a stirred tank reactor should be larger than the tubular reactor. The biomass concentration is lower in a stirred tank reactor due to the fast dilution to reactor outlet concentration.
2.3.2.1 Chemistry. The mechanism of hydrothermal degradation of biomass is complex. Reactions involving cellulose and glucose were studied by Minowa's group in either the absence or presence of catalysts.45 They used an autoclave as a reactor and ran the reaction in hot water within the 473 to 623 K temperature range. In all cases, the reaction products were gases, aqueous phase, oily material and solid residue. In the absence of catalyst, cellulose was slightly decomposed over 473 K to yield sugars, which are water-soluble products. As no gases, oil or char formation was produced, it could be inferred that hydrolysis is the primary step for the gasification reaction.45 At 523 K, cellulose was decomposed to form gases, oil, char, sugars and other non-sugar compounds. Over 573 K, cellulose conversion was complete; sugars and oil decomposed while char production increased. Finally, char is mainly obtained with a yield ca. 60% on a carbon basis, with 15% of non-sugar water-soluble products and 10% of gas, mainly CO2, with very small amounts of CO. From these results, a simple reaction scheme was proposed (Fig. 6). Additional experiments using glucose as starting feedstock concluded that hydrolysis is the first step in cellulose conversion.46 For this feedstock, the product distribution of gas, oil and char at different reaction temperatures was essentially the same as that for cellulose. The degradation scheme for glucose and cellulose is therefore the same.
The onset temperature of cellulose degradation in the presence of nickel catalysts is similar to that found for catalyst-free operation, but the nickel phase gasifies the water-soluble products into a CO-free mixture containing CO2, H2 and CH4. Oil and char are also produced, but their yields were very low. The change of gas composition revealed that CO2 and H2 are primary products, although a minor proportion of CH4 is later formed via methanation.
2.3.2.2 Gasification under subcritical conditions. Catalytic gasification at lower temperatures (ca. 720 K) has been studied by several groups with the objective to produce either H2 or a methane-rich gas with a medium calorific value. At temperatures below 723 K, the catalytic effects of the reactor walls are minimized, and the product distribution depends on the catalyst incorporated. For a slurry of 10% wood sawdust, Waldner47 reported conversion to gases of about 21% at 409 °C in the absence of a catalyst. For cellulose, Minowa et al.45 found only about 10% carbon conversion to gases at 623 K and 60 min residence time. In addition, 67% of the feed carbon was recovered as char, 5% as bio-oil and 13% dissolved in the aqueous phase.
In USA, Modell et al.41 were the first to demonstrate that wood could be gasified in supercritical water without the formation of char and tars at low conversions. Elliott et al.48–51 at the Pacific Northwest National Laboratory developed a process to gasify biomass under subcritical conditions (623 K, 20 MPa) using a variety of catalysts. Rhodium, ruthenium and nickel phases deposited on ZrO2 (monoclinic), α-Al2O3, TiO2 (rutile) and carbon. In general, the product gas consisted of more than 50% vol% CH4, 40–50 vol% CO2, less than 10 vol% H2 and light hydrocarbons at trace levels. Sealock et al.52 reported CH4 yield of 0.22 g CH4/g wood with 33 vol% CH4 using a stirred batch autoclave provided with a stainless steel liner operated at 623 K and 34 MPa for 150 min with a Ni catalyst. The Ni phase sintered rapidly although it could be stabilized by adding a second metal.53 The presence of inorganic salts and/or when N- and S-compounds were present resulted in catalyst deactivation.
In Japan, Osada et al.54 investigated several supported noble metal catalysts for the gasification of lignin and propyl phenols.54–57 They suggested that the metal catalyses the decomposition of lignin to lower molecular weight products, i.e. alkylated phenols, and also causes the gasification of these phenolics. Only the first decomposition step was affected by the water density, however higher water densities enhanced the decomposition of lignin but did not influence the gasification of 4-propyl phenol. The catalyst became deactivated in the presence of sulfur compounds, although the rate of deactivation did not depend significantly on the type of the sulfur compound. They also pointed out that sulfur most likely blocks the sites responsible for the C–C bond scission and for the methanation but not for the water gas-shift reaction nor for the decomposition of C1 compounds such as formaldehyde. Minowa et al.58 carried out a study on cellulose gasification at 473–673 K using nickel catalysts in a stirred autoclave. The gas yield was found to be a strong function of the amount of nickel catalyst. These authors proposed a simplified mechanism involving water-soluble intermediates that can either polymerize to oil and further to char or react to form gases.59–61 Park and Tomiyasu62 gasified cellulose, several polymers and model compounds on unsupported RuO2 catalyst at 723 K and 44 MPa. Experiments with deuterated water enabled them to propose a redox mechanism including formation of CO and H2O in a first step (eqn 10), and H2 production in a second (eqn 11):
| (−CH2O)n + nRuO2 → nH2O + nCO + nRuO | (10) |
| nRuO + nH2O → nRuO2 + nH2 | (11) |
In Europe, Waldner and Vogel67 gasified spruce sawdust slurries with feed concentrations up to 30 wt% around 673 K in a batch reactor. A number of catalysts were screened for their activity and selectivity. From the catalysts, Raney nickel, 1%Ru/TiO2 and 2%Ru/C were then tested for their long-term stability in a continuous test setup. A mixture of five organic compounds representing hydrolyzed wood was used as a feed. The Raney nickel catalyst sintered after a short time whereas 1%Ru/TiO2 was not active enough. On the contrary, the 2%Ru/TiO2 catalyst was hydrothermally stable for more than 200 h on-stream at 673 K and 30 MPa. Dosing 8 ppm Na2SO4 in the feed resulted in slow catalyst deactivation. This deactivation could be explained by an irreversible bonding of sulfate anion to the surface ruthenium, although sulfate might not be the actual catalyst poison under reaction conditions as it may be reduced to a sulfide species.67
Most studies in the field of high temperature SCW biomass gasification to hydrogen have been conducted in USA, Europe and Asia. A brief account of the activities carried out by all these groups in SCW has been given recently by Peterson et al.68
In USA, Antal et al.44 investigated supercritical gasification of wet biomass and glucose. They reported that complete gasification of glucose can occur at 873 K 34.5 MPa and a 30 s residence time, and also that Inconel walls of the reactor strongly catalyse the water gas-shift reaction. It was shown that wood sawdust, dry sewage sludge or other particulate biomass could be mixed with a corn starch to form a viscous gel.69 At the critical pressure of water (22 MPa) this paste vaporizes without the formation of char. A packed bed of carbon catalyst at 923 K causes the tarry vapors to react with water to produce H2, CO2, some methane and only traces of CO, but this catalyst deactivated after several hours on-stream. More recently, Hong and Spritzer70 at General Atomics designed a continuous reactor with a thermal sleeve inside the pressure vessel to study SCW oxidation under 23.5 MPa pressure for several feedstocks such as wood, corn starch, coal and solid wastes. According to this concept, external heating was avoided because the heat required for running the reaction was released by the partial oxidation. In their preliminary experiments, ethanol was added to the feed with the objective of enhancing the heating. They reported yields in a directly heated system similar to Antal's yields in an indirectly heated system, after compensating for heating differences. Based on these works, General Atomics completed pilot-scale testing and concluded that by using negative cost waste streams they will be able to produce H2 at cost of about $3/GJ.
In Europe, the earliest work carried out by Schneider et al.71 and Kruse et al.72 on SCW gasification of a range of feedstocks demonstrated complete gasification to H2 at 898 K and 25 MPa in both batch and continuous tubular-flow reactors. They found that potassium compounds such as KOH and K2CO3 drastically increased the yield of hydrogen. Based on these and other studies a pilot plant was built at Forschungszentrum, Karlsruhe. This pilot, which was designed for a continuous flow capacity of 100 kg h−1, maximum temperature of 930 K and 28 MPa, has been in operation since 2003. Using feedstocks of 9–25 wt% ethanol, pyroligneous acid and corn silage, very high yields of H2 were obtained. Notwithstanding, the plant showed some plugging of the preheaters even at low reaction times (3.5 h). As indicated, most of the SCW works conducted at FZK employed alkali salts as catalysts because they catalyse the water gas-shift reaction.71–74 While the mechanism by which K-salts increase H2 yield is still unclear, there is an important body of current agreement that the interaction of alkali salt solutions with the reactor walls under SCW conditions is a potential source of hydrogen. First, the chromium layer of stainless steel is solubilised by alkali, then the Cr (and/or Fe) atoms located in sub-surface layers react with water molecules to yield additional H2. However, recent SCW experiments conducted in sealed quartz tubes examined the influence of KOH and NaOH without the complication of the metal reactor walls. Using this experimental setup Kersten et al.75 reported that NaOH increased the H2 yield from 9.9% to 17–21% during the gasification of 17% glucose at 873 K under 30 MPa and 60 s residence time. Indeed, detailed studies are required to get a clearer understanding of the catalytic effect of alkali salts during SCW gasification.
At the University of Twente, Potic et al.76 have constructed small sealed quartz capillaries (id = 1 mm) with an objective of avoiding the catalytic effect of metal surfaces. Kersten et al.75 investigated in detail the SCW gasification of glucose, glycerol and pine-wood in these reactors. They found that, in the absence of catalyst, complete gasification was only achieved at concentrations 2 wt%, however incorporation of a Ru/TiO2 catalyst allowed glucose solutions of up to 17 wt% to be gasified. To demonstrate the catalytic effect of the reactor walls when using conventional autoclave reactors, Inconel (a typical alloy of SCW metal reactors) powder was added and found to have a dramatic effect, increasing the gasification of a 5% glucose solution.
In China, the work by Guo et al.77 was basically focused on the use of different reactor configurations including a miniature plant with a movable-piston feed pump to handle slurry feeds. They used a great variety of model compounds such as glucose, cellulose, lignin and xylan as well as feedstocks like sawdust, rice straw, rice shell, peanut shell, corn stalk, corn cob and wheat stalk under typical reaction conditions of 873 K, 25 MPa pressure and 10 wt% feed. The gasification efficiency achieved was very high, often above 80% conversion, together with high hydrogen yields for every feedstock, except lignin which was the most difficult compound to gasify.
The cost of hydrogen production from supercritical water gasification of wet biomass has been analyzed by Demirbas.79 Cost analysis of hydrogen produced via gasification of biomass in SCW has been made at a series of temperatures, pressures and different resident times. For all explored gasification conditions, analysis revealed that the cost H2 produced via SCW gasification is several times higher than the current price of hydrogen from steam methane reforming. In addition, an evaluation of SCW gasification and biomethanation in terms of process cost, energy efficiency, and CO2 emissions has been made by Matsumura.80 Gasification of 1 t/d (dry) of water hyacinth was selected as a model case. Assumptions were made that the system should be energetically independent, that no environmentally harmful material should be released, and that carbon dioxide should be removed from the product gas. Energy efficiency, carbon dioxide payback time, and price of the product gas were chosen as indices for energy, environmental, and economic evaluation, respectively. Under these assumptions, supercritical water gasification appeared to be more advantageous over biomethanation, but the cost of the product gas was still 1.86 times more expensive than city gas (in Tokyo), although some improvement in efficiency of supercritical water gasification could be obtained by proper design of the heat exchanger. Finally, utilization of fermentation sludge made biomethanation much more advantageous.
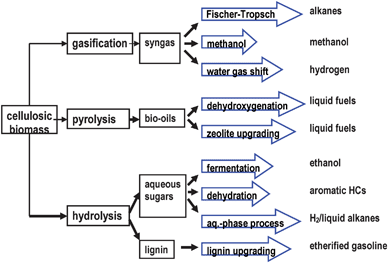
2.4 Reforming of biomass-derived products
| C6H12O6 → 2C2H5OH + 2CO2 | (12) |
| CH3CH2OH + 3H2O → 6H2 + 2CO2 (ΔH0 = + 173.3 kJ mol−1) | (13) |
| CH3CH2OH + 1.5O2 → 3H2 + 2CO2 (ΔH0 = − 552.0 kJ mol−1) | (14) |
| CH3CH2OH + xO2 + (3−2x)H2O → (6−2x)H2 + 2CO2 (0 < x < 0.5) (ΔH0 = ((3−2x)/3)*207.7 – (x/1.5)* 545.2 kJ mol−1) | (15) |
2.4.1.1 Steam reforming. The overall steam reforming reaction of ethanol can be represented by eqn 13. This reaction is usually conducted at temperatures in the 823–1073 K range in the presence of a catalyst. Ethanol-reforming reactions involve several reaction pathways (dehydration, decomposition, dehydrogenation, coking) depending on the catalysts and reaction conditions81 (Fig. 7). Therefore, the choice of catalyst is essential in the reforming process. Reactions to avoid are those that lead to C and C2H4 products that are precursors of carbon deposition on catalyst surfaces. Consistent with this, catalysts for the steam reforming of ethanol to produce H2 selectively must be able to: (i) dehydrogenate ethanol; (ii) break the carbon–carbon bonds of surface intermediates to produce CO and CH4; and (iii) reform these C1 products to generate hydrogen. On the basis of the influence of the nature of both the metal and support on the catalytic characteristics of supported metals, the choice of these elements is a key factor in developing supported catalysts that will fulfill the above requirements. Different oxide catalysts,82 metal-based catalysts (Ni, Co, Ni/Cu,)83–85 and noble metal-based catalysts (Pt, Pd, Rh)86–89 have proven to be active in the ethanol-reforming reaction.
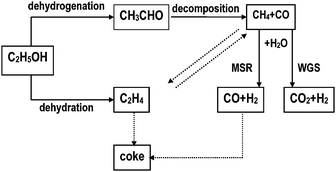 | ||
| Fig. 7 Reaction mechanism for steam reforming of ethanol from ref. 4. | ||
Both the metallic function and the acid–base property of the catalyst play major roles in the reforming reaction of ethanol. This is illustrated by the Cu/Ni/K/γ-Al2O3 catalyst, which exhibits acceptable activity, stability and hydrogen selectivity at relatively low temperature (573 K) and atmospheric pressure.83 In this catalyst, the copper is the active phase and nickel promotes C–C bond rupture, increasing hydrogen selectivity, while the potassium neutralizes the acidic sites of the γ-alumina substrate and improves the general performance of the catalyst. The nature of the support influences the catalytic performance of the supported catalyst for the steam reforming of ethanol, since it affects the dispersion and stability of the metal and may participate in the reaction. Lanthanum oxide is a particularly suitable support for the metallic function of ethanol-reforming catalysts. The Ni/La2O3 or Ni–La2O3/Al2O3 catalysts have high activity and long-term stability for hydrogen production.90,91 A 20% Ni/La2O3/Al2O3 catalyst has good stability at 1023 K for reaction times over 150 h, with only a small reduction in ethanol conversion from 95% to 90%, while hydrogen selectivity remains essentially unchanged. These results indicate the uniqueness of the Ni–La2O3 system in terms of its protracted stability. The unusual stability of the Ni–La2O3 catalyst has been attributed to the scavenging of coke deposition on the Ni surface by lanthanum oxycarbonate species existing on top of the Ni particles.87 The effects of basic additives (K, Mg, Ca, Ce) that favour water adsorption and OH surface mobility in Al2O3 supports, to lower the rate of coke deposition on catalyst surfaces have also been investigated on nickel-based catalysts.92,93 Coke formation on bare and Ce-, K- or Mg-modified catalysts does occur, but at orders of magnitude lower than that in alumina-supported Ni catalysts.
Cobalt-based catalysts have also been studied in the ethanol steam reforming reaction. Llorca et al.85 performed the reaction between ethanol and water in the 573–723 K temperature range at atmospheric pressure over supported cobalt catalysts. The ZnO-supported cobalt catalyst had a very high catalytic performance. Using an EtOH)/H2O = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 (molar ratio) mixture, total conversion of ethanol and high values of H2 and CO2 were obtained, in the absence of deactivation. Complete EtOH conversion was also reached on the Co-free ZnO substrate, but the yields of H2 and CO2 alone were found to be substantially lower. The decomposition of EtOH into acetone occurs to a large extent on Co/ZnO catalysts. Since this reaction results from consecutive reactions, such as dehydrogenation and aldol condensation, activity tests conducted at low contact times have indicated that the reforming reaction is relatively fast, while EtOH decomposition to acetone via aldol condensation of acetaldehyde is supressed. In addition, the Co/ZnO catalyst accumulates a considerable amount of carbon throughout the reaction, which causes the deactivation of the cobalt catalyst.
:13 (molar ratio) mixture, total conversion of ethanol and high values of H2 and CO2 were obtained, in the absence of deactivation. Complete EtOH conversion was also reached on the Co-free ZnO substrate, but the yields of H2 and CO2 alone were found to be substantially lower. The decomposition of EtOH into acetone occurs to a large extent on Co/ZnO catalysts. Since this reaction results from consecutive reactions, such as dehydrogenation and aldol condensation, activity tests conducted at low contact times have indicated that the reforming reaction is relatively fast, while EtOH decomposition to acetone via aldol condensation of acetaldehyde is supressed. In addition, the Co/ZnO catalyst accumulates a considerable amount of carbon throughout the reaction, which causes the deactivation of the cobalt catalyst.
Noble metals supported on porous oxide substrates (A2O3, SiO2, CeO2, TiO2 and MgO)86,89,94–100 are highly active in the steam reforming of ethanol to COx and H2. The support plays a significant role in the steam reforming of ethanol over noble metals. When CeO2/ZrO2, which has oxygen storage capability, is used as the support for noble metals, ethylene formation is not observed and the order of activity at higher temperature is Pt ≈ Rh > Pd.86 Alumina-supported catalysts are very active at low temperatures in the dehydration of ethanol to ethylene. At higher temperatures, ethanol is converted into H2, CO, CO2 and CH4, with an activity order of metals as follows: Rh > Pd > Ni = Pt.85 Auprêtre et al.99 studied the effect of both the metal and the support in the steam reforming of ethanol. They found that the hydrogen yield on alumina-supported metal catalysts at 973 K decreased in the following order: Rh > Pd > Pt > Ru. They concluded that the high activity of the metals in ethanol steam reforming and their poor efficiency in the water gas-shift reaction would provide active and selective catalysts for ethanol reforming. Auprêtre et al.99 also reported that the H2 yield on Rh/CeO2 was higher than that on Rh/Al2O3 at 873 K. It was concluded that a metal–ceria interaction affects the absorption–decomposition of ethanol to CH4 and CO products and their subsequent reforming reactions with steam. On the Rh/Al2O3 catalyst, Cavallaro et al.100 reported that ethanol is firstly converted to ethylene by dehydration on the Al2O3 surface, or to acetaldehyde by dehydrogenation on Rh particles. The acetaldehyde undergoes decarbonylation on the rhodium surfaces to form methane and CO, while ethylene is also steam reformed on metal particles to C1 (very fast reactions). Liguras et al.87 also found that among the low-loaded catalysts, Rh was significantly more active and selective towards hydrogen formation than Ru, Pt and Pd. The catalytic performance of Rh was greatly improved by the increase of metal loading. In addition, the 5 wt% Rh/Al2O3 recorded good stability at 923 K and high H2 selectivity (up to 95%) without carbon formation as demonstrated by long-term activity tests.100
2.4.1.2 Catalytic partial oxidation. The partial oxidation of ethanol (POE, eqn 14) has been investigated with less intensity than in the case of steam reforming. Partial oxidation is a very interesting process for H2 production because the catalysts used can be run autothermally, thereby eliminating the need for external heat. POE is much faster than the catalytic steam reforming that allows fast start-up and short response times to variations in H2 production. It is a slightly endothermic reaction, so alone it will not produce the heat necessary for autothermal operation. Therefore, a small part of ethanol must be combusted to generate the heat for the operation in the 973–1273 K range necessary for the reaction to be sustained. It is emphasised that the pure POE process is not indicated for bio-ethanol reforming since bio-ethanol is an ethanol–water mixture in which the removal of all the water has a significant cost. Therefore, the processes for bio-ethanol partial oxidation are usually combined with steam reforming in autothermal schemes with the stoichiometry shown in eqn 16. Additionally, adding water to the reaction stream is very useful since catalyst stability is improved while coke formation is minimized. In addition, as the H atoms from H2O can also be converted into H2, complete conversion of the ethanol and water from this reaction could generate 5H2 per C2H5OH, which gives a maximum H2 selectivity of 5/3 or 167%.
| C2H5OH + 2 H2O + ½O2 → 2CO2 + 5H2 (ΔH0298K = −68.2 kJ mol−1) | (16) |
| C6H14O6 + 6H2O → 6CO2 + 13H2 (ΔH0298K = +443.5 kJ mol−1) | (17) |
The reaction pathway of APR involves cleavage of C–H, C–C, and O–H bonds of sugar molecules to form adsorbed species on the catalyst surface. Adsorbed CO must be removed by the WGS reaction to form CO2 and additional H2. Undesired parallel reactions also occur and proceed via C–O bond splitting followed by hydrogenation to yield alcohols or even acids. Thus, good catalysts for the production of H2 by APR reactions must be highly active for C–C bond cleavage and also capable of removing adsorbed CO by the WGS reaction, but must not facilitate C–O bond cleavage and the hydrogenation of carbon oxides. H2 selectivity depends on the feed sugar, catalyst, and reaction conditions. As a general trend, H2 selectivity decreases upon increasing molecular size of the feed molecule.
Kinetic studies were conducted for the APR of ethylene glycol (a probe molecule for sorbitol) over silica-supported Pd, Ni, Pt, Ir, Ru, and Rh catalysts at moderate temperatures (483–498 K) and moderate pressure (22 bar). The catalytic activity for APR of ethylene glycol, as measured by the rate of CO2 formation per surface atom at 483 K followed the order: Pt ∼ Ni > Ru > Rh ∼ Pd > Ir.109 Silica-supported Ni, Ru, and Rh catalysts recorded low selectivity for H2 production and high selectivity for alkane production. In addition, the Ni/SiO2 catalyst became rapidly deactivated at 498 K. On the other hand, Pt/SiO2 and Pd/SiO2 catalysts exhibited higher selectivity for the production of H2, with lower rates of alkane production. It was also found that both the activity and selectivity of Pt-based monometallic catalysts can be enhanced by depositing Pt phase on TiO2, carbon, and Al2O3 substrates110 or by adding Ni, Co, or Fe to a monometallic Pt/Al2O3 catalyst.111 Alumina-supported PtNi and PtCo catalysts with Pt/Co or Pt/Ni atomic ratios ranging from 1:1 to 1:9 had the highest turnover frequencies for H2 production (moles of H2 per mole of surface site measured by CO adsorption) with values of 2.8–5.2 min−1 for APR of ethylene glycol solutions at 483 K, compared to a value of 1.9 min−1 for the monometallic Pt/Al2O3 under similar reaction conditions.
Nickel catalysts are also active for APR reactions; however, they have low selectivity and stability. The H2 selectivity of Ni-based catalysts can be enhanced by adding Sn to the Ni catalyst, whereas its stability can be improved by using bulk Ni catalysts, for example, Raney Ni.112 The rates of H2 production by APR of ethylene glycol over a SnNi catalyst with Ni/Sn atomic ratios up to 14:1 are comparable to a 3 wt% Pt/Al2O3 catalyst, based on reactor volume. The incorporation of Sn to Raney Ni catalysts markedly decreases the rate of methane formation from reactions of COx with H2, while maintaining the high rates of C–C cleavage necessary for the production of H2. Nonetheless, the reactor must operate at near the bubble-point pressure of the feed and moderate space times to achieve high H2 selectivities over Raney SnNi catalysts. Remarkably, these Raney SnNi catalysts are stable for more than 250 h time-on-stream.112
2.5 Biological hydrogen production
Biological H2 production processes are becoming important because they can use renewable energy resources, and they usually operate at ambient temperature and atmospheric pressure. A number of microorganisms possess enzymes that can produce H2 from water when they are exposed to an outside energy source, such as sunlight. Specific ways in which microorganisms can produce H2 have recently been summarized by Das et al.113 There are three main processes for biological H2 production: (i) biophotolysis of water using green algae and blue-green algae (cyanobacteria); (ii) photofermentation; and (iii) dark fermentation.The direct biophotolysis (type (i) process) of H2 production is a biological process that uses solar energy and photosynthetic algae systems to convert water into chemical energy:
| 2H2O + photons → 2H2 + O2 | (18) |
The two photosynthetic systems responsible for the photosynthesis process are photosystem I (PSI), which produces reductant for CO2, and photosystem II (PSII) which splits water to evolve O2. The two photons obtained from the splitting of water can either reduce CO2 by PSI or form H2 in the presence of hydrogenase. Due to the lack of hydrogenase in plants, only CO2 reduction takes place. By contrast, green algae and cyanobacteria (blue-green algae) contain hydrogenase and thus have the ability to produce H2.114 In these organisms, electrons are generated when PSII absorbs light energy, which is then transferred to ferredoxin. A reversible hydrogenase accepts electrons directly from the reduced ferredoxin to generate H2 in the presence of hydrogenase:
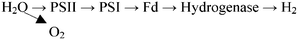 | (19) |
Type (ii) biological H2 production process is photofermentation. Purple non-sulfur bacteria produce hydrogen mainly due to the presence of nitrogenase under oxygen-deficient conditions using light energy and reduced compounds (organic acids). The reaction is as follows:
| C6H12O6 + 12H2O + photons → 12H2 + 6CO2 | (20) |
Dark fermentation (type (iii) process) is a ubiquitous phenomenon under anoxic or anaerobic conditions. The oxidation of the substrate by bacteria generates electrons, which need to be disposed of in order to maintain electrical neutrality. Under aerobic conditions, O2 serves as the electron acceptor, whereas under anaerobic or anoxic conditions other compounds, such as protons, act as the electron acceptor and are reduced to molecular H2.116 Carbohydrates, mainly glucose, are the preferred carbon sources for this process, which predominantly give rise to acetic and butyric acids together with H2 evolution:117
| C6H12O6 + 2H2O → 2CH3COOH + 2CO2 + 4H2 | (21) |
| C6H12O6 + 2H2O → CH3CH2COOH + 2CO2 + 2H2 | (22) |
A challenging problem in establishing biohydrogen as a source of energy is the renewable and environmentally-friendly generation of large quantities of H2 gas. However, two major aspects need vital attention, viz., a suitable renewable biomass/wastewater and ideal microbial consortia that can convert this biomass efficiently to H2. Comparative studies on the available processes indicate that biohydrogen production requires greater improvement on the process, mainly with respect to H2 yield from the cheaper raw materials. The future of biological H2 production depends not only on research developments, i.e., the improvement in efficiency through genetically engineered microorganisms and/or the development of bioreactors, but also on economic considerations.
3. The solar option
Of the various renewable energy sources, by far the largest resource is provided by the sun. More energy from sunlight strikes the earth in 1 h (4.3 × 1020 J) than all of the energy currently consumed on the planet in 1 yr (4.1 × 1020 J in 2001).118 Yet, in 2001, only <0.1% of electricity and <1.5% of fuels (mostly from biomass) were provided by a solar source.118,119 Against the backdrop of the daunting carbon-neutral energy needs of our global future, the large gap between our present use of solar energy and its enormous undeveloped potential defines a compelling imperative for science and technology in the 21st century. Indeed, the solar energy striking the earth exceeds what can possibly be consumed by any technologically-advanced society. However, to make a material contribution to the primary energy supply, solar energy must be captured, converted, and stored to overcome the diurnal cycle and the intermittency of the terrestrial solar resource. Undoubtedly, the most attractive method for this energy conversion and storage is in the form of H–H bonds.Within this scenario, the conversion of solar energy into hydrogen via the water splitting process assisted by photo-semiconductor catalysts is one of the most interesting ways of achieving clean and renewable energy systems.120,121
3.1 Basic principles of water splitting
Among the methods for H2 generation outside the C-cycle, photocatalytic hydrogen production via water splitting under visible light irradiation attracts the greatest attention for its potential to use the abundance of solar energy (the maximum direct insolation frequently reaches ca. 700 W m−2 in the sunbelt regions) and water. Thermodynamically, the overall water splitting reaction is an uphill reaction with a highly positive change in Gibbs free energy (ΔG0 = + 237.2 kJ mol−1):| H2O(l) → H2(g) + ½O2(g) (ΔG0 = + 237.2 kJ mol−1) | (23) |
Fig. 8 shows a sketch diagram of the basic principle of overall water splitting on a solid photocatalyst. Under irradiation with an energy equivalent to or greater than the bandgap (Eg) of the semiconductor photocatalyst, the electrons (e−) of the valence band are excited into the conduction band (CB) while the holes (h+) are left in VB. Electrons and holes that migrate to the surface of the semiconductor without recombination can respectively reduce and oxidize the water molecules adsorbed on the semiconductor surface. To achieve overall water splitting, the bottom of the CB must be located at a more negative potential than the reduction potential of H+/H2 (0 V vs. NHE at pH = 0), while the top of the VB must be positioned more positively than the oxidation potential of H2O/O2 (1.23 V vs. NHE). Therefore, according to this theoretical value, it can drive water splitting only if the photon energy is equal or superior to 1.23 eV. This energy is equivalent to the energy of a photon with a wavelength of around 1010 nm, indicating that visible light is energetically sufficient for the decomposition of water. However, the activation energy barrier in the charge transfer reaction between the water molecules and the semiconductor surface requires photon energy greater than the bandgap of the semiconductor to drive overall water splitting at a measurable reaction rate. The potential of the band structure is precisely the thermodynamic requirement. Other factors such as charge separation, mobility and the lifetime of photogenerated electrons and holes also have an effect on the photocatalytic properties of semiconductors. The generation and separation of photoexcited carriers (electrons and holes) strongly depend on both the presence of co-catalysts on the surface of the photocatalysts and the latter's structural and electronic properties. In general, high crystallinity has a positive effect on activity since the density of defects, which act as recombination centres of electrons and holes, decreases when crystallinity increases. Co-catalysts are usually loaded onto the photocatalysts to assist the redox reactions taking place on their surfaces.122,123 The co-catalysts are typically a noble metal (e.g. Pt, Rh) or metal oxide (e.g. NiO, RuO2) loaded on the surface as nanoparticles to reduce the electron–hole recombination and to produce active sites that reduce the activation energy for gas evolution.
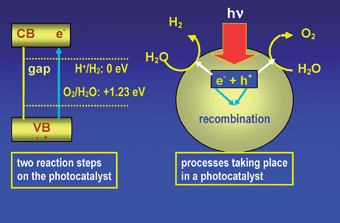 | ||
| Fig. 8 Sketch diagram of the basic principle of overall water splitting on a solid photocatalyst. Irradiation with an energy equivalent to or greater than the bandgap (Eg) of the semiconductor photocatalyst, the electrons (e−) of the valence band are excited into the conduction band (CB) while the holes (h+) are left in VB. | ||
3.2 Photocatalysts for water splitting
Several types of semiconductors, over 130 materials including oxides, nitrides, sulfides⋯124–141 have been reported to act as efficient photocatalysts for hydrogen evolution via water splitting. Among these photocatalysts, the higher quantum yields (QY) are reported for Ba-doped Sr2Nb2O7 (50% QY, pure water, UV)135 and NiO/NaTaO3 (56% QY, pure water at 270 nm).129 Unfortunately, these exciting developments have only a limited value for practical, large-scale hydrogen manufacture because UV light accounts for only about 3–4% of solar radiation energy. Therefore, regarding the use of solar energy, it is essential to develop photocatalysts that split water efficiently under visible light (λ > 400 nm). The materials for visible light-driven photocatalysts had been quite limited. However, many oxides, sulfides, oxynitrides and oxysulfides have recently been found to be active for H2 and O2 evolution under visible light irradiation.136–140 So far, the maximum quantum efficiency over visible light-driven photocatalysts achieves only a few percent at wavelengths as long as 500 nm (Cr/Rh–GaN/ZnO 2.5% QY, pure water, visible light).141 This value is still far from the QE of 30% marked as the initial starting point for practical application.142 Hence the development of new photocatalyst materials is still a major issue. There are several common approaches adopted in the search for photocatalyst for splitting water under visible light irradiation: (i) find new materials, (ii) tune the band gap energy by modifying cations or anions of UV-active photocatalysts with substitutional doping and, (iii) fabricate multi-component photocatalyst by forming solid solutions. The following sections will review the above approaches for the development of active visible light photocatalysts for water splitting.The second main strategy followed to improve the visible light response of TiO2 is related to the doping of anions such as N,149 S150 or C151 as substitutes for oxygen in the TiO2 lattice. For these anion doped TiO2 photocatalysts, the mixing of the p states of doped anion (S, N or C) with the O 2p states was reported to shift the valence band edge upwards to narrow the band gap energy of TiO2. However, these materials do not record activity for pure water splitting, although the conduction and valence bands have enough potential for the reduction and oxidation of water, due to the large over-potential for H2 and O2 evolution on the surface of these photocatalysts.
The development of visible light-driven photocatalysts by doping metal ions into SrTiO3 is also reported.152 A survey of dopants for SrTiO3 revealed that the doping of Rh or the co-doping of Cr3+–Ta5+ or Cr3+–Sb5+ were effective in making SrTiO3 visible light-responsive. Rh-doped SrTiO3 produced H2 from an aqueous methanol solution, while Cr–Ta-codoped-SrTiO3 was effective for O2 evolution from aqueous AgNO3 solutions. These doping photocatalysts that show activities for only half reactions can be used to construct Z-scheme photocatalysts for overall water splitting.127
Oxynitrides such as TaON, LaTiO2N and (Ga1−xZnx)(N1−xOx) have evolved as promising water-splitting catalysts that can operate under visible light and without sacrificial reagents. Among these, the most promising formulation was the solid solution of GaN and ZnO ((Ga1−xZnx)(N1−xOx)). Density functional calculations indicated that the bottom of the conduction band of the GaN–ZnO solid solution was mainly composed of 4s and 4p orbitals of Ga, while the top of the valence band consisted of N 2p orbitals, followed by Zn 3d orbitals. The presence of Zn 3d and N 2p electrons in the upper valence band might provide p–d repulsion for the valence band resulting in a narrowing of the band gap. H2 and O2 were found to evolve steadily and stoichiometrically from solid solution photocatalysts with an average quantum efficiency of 0.14% in the range of 300–480 nm.141 Different transition metal oxides have been examined as cocatalysts to promote activity of (Ga1−xZnx)(N1−xOx) solid solutions. Among the various materials examined, the largest improvement in activity was obtained when (Ga1−xZnx)(N1−xOx) was loaded with a mixed oxide of Rh and Cr. The quantum efficiency of the Rh–Cr loaded (Ga1−xZnx)(N1−xOx) photocatalyst for overall water splitting reaches ca. 2.5% at 420–440 nm.154
3.2.4.1 CdS-based photocatalysts. Among the available sulfide semiconductors, nanosized CdS is an interesting photocatalyst material, since it has a narrow band gap (2.4 eV) and a suitable conduction band potential to effectively reduce H+.159–165 However, the photocatalytic properties of CdS are limited due to its photocorrosion under visible light irradiation.166 Although photocorrosion can be significantly reduced by adding sacrificial reagents, life cycle assessment studies associated with the processes of water splitting using Cd-based photocatalysts must be performed in order to evaluate the potential environmental impacts associated with Cd. In spite of the drawbacks associated with CdS, considerable efforts are still being made to improve its photocatalytic properties. The steps reported in the literature to improve the activity of CdS include: (i) changes in the structural characteristics of CdS, (ii) combination of CdS with different elements or semiconductors to form mixed photocatalysts with different band gap size and, (iii) addition of small amounts of metals or transition metal oxides (cocatalysts) to CdS.
Combination of CdS with CdO and ZnO. Changes in the photoactivity of CdS can be achieved by combining the CdS semiconductor with other semiconductors with different energy levels (ZnS,131 ZnO,167 TiO2,168 AgGaS2169…). It is claimed that photogenerated electrons in these composite systems move from CdS to the attached semiconductors, while photogenerated holes remain in CdS. This charge-carrier separation stops charge recombination, therefore improving the photocatalytic activity of CdS. The optoelectronic properties of ZnO and CdO make these materials interesting as semiconductors for combining with CdS.152,171 CdO has a band gap in the interval 2.2–2.4 eV with high transmittance and very low resistance, while ZnO has a wide band gap (3.2 eV) that may improve the photoabsorption ability of CdS. In spite of the absence of studies in the literature on the effect of the presence of CdO and ZnO on the activity of CdS under visible light, there are interesting results showing the increase in CdS activity for samples mixed with CdO and ZnO.172 The CdS–CdO–ZnO catalyst prepared by sequential precipitation of CdS and CdO and ZnO showed that the activity of CdS in the hydrogen produced under visible light from aqueous solutions containing SO32− + S2− as sacrificial reagents increases with the addition of CdO and ZnO to CdS (Fig. 9). Taking into account the absence of significant changes in the structure of CdS with the addition of both oxides, the observed differences in activity of CdS–CdO–ZnO mixtures with respect to bare CdS are related to a better charge separation associated with the diffusion of photoelectrons generated in CdS toward surrounding CdO and ZnO.
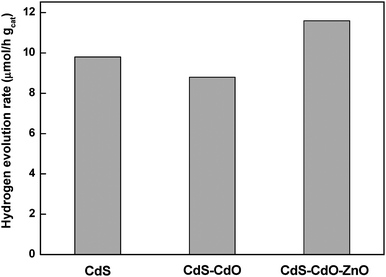 | ||
| Fig. 9 Hydrogen evolution rate from aqueous solution containing Na2S + Na2SO3 under visible light irradiation over CdS, CdS–CdO and CdS–CdO–ZnO catalysts (catalyst: 0.1 g, reactant solution 150 ml (0.1M Na2S +0.04M Na2SO3, 150 W Xe lamp). | ||
The effect of thermal treatments under inert atmosphere on crystallinity and the phase transformations of precipitated CdS–CdO–ZnO catalyst was also studied.172Fig. 10 shows hydrogen evolution under visible light irradiation over CdS–CdO–ZnO catalysts treated at 573, 773 and 923 K. Hydrogen production was found to increase with annealing temperature, with a decrease in activity for the sample treated at temperatures higher than 773 K. The physicochemical characterization of thermally treated CdS–CdO–ZnO shows that heating induces significant changes at structural and chemical levels. Fig. 11 shows that the catalyst has crystal structures of CdS that gradually change from a cubic structure to a more crystalline hexagonal phase with the increase in annealing temperature. The evolution of CdS phase in the CdS–CdO–ZnO mixture during thermal treatment is similar to the evolution observed in the bare CdS sample, suggesting that neither the presence of CdO nor that of ZnO modifies the thermal evolution of the CdS phase. Changes in structure after thermal treatment have a bearing on the photophysical properties of CdS–CdO–ZnO samples, recording an increase in band gap size from 2.24 to 2.33 eV for the sample annealed at temperatures higher than 773 K, associated with changes in its order degree. When comparing the photophysical properties of thermally treated CdS–CdO–ZnO samples with photocatalytic hydrogen production results, no direct correlation was found between visible light absorption and photoactivity. From this observation, it is inferred that the effective photo-utilization of electrons and holes, rather than photoabsorption ability, plays a key role in the catalytic activity of CdS–CdO–ZnO mixtures. The differences in activity are possibly related to the reduction in CdS surface exposure and contact between CdS and CdO/ZnO, as a consequence of the thermal crystalline growth that decreases the generation of photoelectrons in CdS and their diffusion toward surrounding CdO/ZnO nanoparticles, thereby increasing the probability of electron–hole recombination.
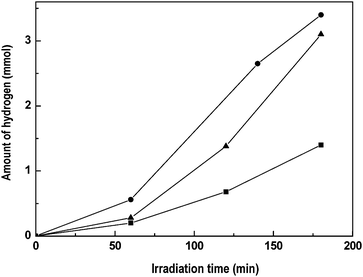 | ||
| Fig. 10 Hydrogen evolution from aqueous solution containing Na2S + Na2SO3 under visible light irradiation over CdS–CdO–ZnO catalysts annealed at different temperatures: (■) 573 K, (●) 773 K and (▲) 973 K (catalyst: 0.1 g, reactant solution 150 ml (0.1M Na2S + 0.04 M Na2SO3, 150 W Xe lamp). | ||
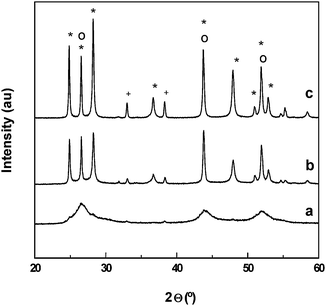 | ||
| Fig. 11 XRD patterns of CdS–CdO–ZnO catalysts treated under inert flow at different temperatures; (a) 573 K, (b) 773 K and (c) 973 K ((*) hexagonal CdS, (o) cubic CdS (+) CdO). | ||
As mentioned previously, the third way to enhance the activity of CdS photocatalyst is through the addition of small amounts of metals (cocatalyst) such as Pt, Rh and Ru.170,171 Within this scenario, we have investigated the structural changes in CdS–ZnO–CdO mixtures associated with the photodeposition of metal cocatalysts (Pt and Ru).172 The photodeposition of Pt or Ru on the CdS–ZnO–CdO mixture changes the crystalline size of CdS with hexagonal phase as consequence of the photoetching phenomena on the CdS structures caused by the light irradiation used for photodeposition.173 The absorption properties of CdS–ZnO–CdO mixture after the photodeposition of Pt or Ru showed only a slight blue-shift in the adsorption edge with respect to unloaded photocatalyst, consistent with the observed reduction in the crystalline degree of CdS after the photodeposition of noble metals. Hydrogen production on Ru- and Pt/CdS–ZnO–CdO catalysts markedly depends on the type of noble metal incorporated, with the Ru-based catalyst recording a higher level of hydrogen production than that obtained over its Pt counterpart (Fig. 12).
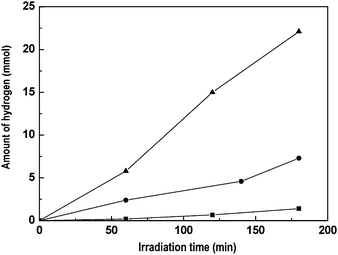 | ||
| Fig. 12 Hydrogen evolution from aqueous solution containing Na2S + Na2SO3 under visible light irradiation over CdS–CdO–ZnO with Pt and Ru cocatalysts: (■) bare CdS–CdO–ZnO, (●) Pt/CdS–CdO–ZnO and (▲) Ru/CdS–CdO–ZnO (catalyst: 0.1 g, reactant solution 150 ml (0.1M Na2S + 0.04 M Na2SO3, 150 W Xe lamp). | ||
The levels of hydrogen production over the noble metal catalysts contrast with the low rate obtained over the noble-metal-free CdS–ZnO–CdO counterpart. In particular, ruthenium strongly enhances the rate of hydrogen production by a factor of almost 50 times with respect to the CdS–ZnO–CdO substrate. The enhancement of photocatalytic activity has been explained in terms of a photoelectrochemical mechanism in which the electrons generated by irradiation of CdS are transferred to the loaded metal particles, decreasing electron–hole recombination. The XRD results showed similar changes in the CdS–ZnO–CdO substrate after Pt and Ru deposition, thereby indicating that the observed differences in activity are related to the state and surface concentration of noble metal entities after photodeposition onto the CdS–ZnO–CdO substrate. Accordingly, the better activity observed for Ru catalysts was associated with the presence of Ru oxide entities of high intrinsic activity with good surface coordination with cadmium sulfide particles that enhance the electronic transfer between both phases, thereby increasing the efficiency of water splitting by decreasing the probability of electron–hole recombination.
CdS–ZnS solid solutions. The incorporation of elements in the structure of CdS making solid solutions is a powerful strategy for improving the photocatalytic properties of CdS.174 Solid solution photocatalysts have advantages over doped photocatalysts as solid solution allows for controlling the potentials of the conduction and valence bands, and the photogenerated electrons and holes are able to move in the continuous valence and conduction bands instead of the discrete donor levels seen in the doped photocatalysts. Zn is interesting as a semiconductor for combining with CdS. CdS and ZnS form a continuous series of solid solutions (Cd1−xZnxS) where metal atoms are mutually substituted in the same crystal lattice.175–177 The Cd1−xZnxS solid-solution has a higher band gap than CdS, making the material attractive for solar applications. Photophysical and photocatalytic properties of Cd1−xZnxS solid solutions with different Zn concentration (0.2 < x < 0.35) prepared by co-precipitation were investigated.178
X-ray diffraction patterns of Cd1−xZnxS samples (Fig. 13) displayed reflections corresponding to hexagonal CdS with higher hexagonal lattice parameters than those observed for pure CdS hexagonal phase, indicating the formation of solid Cd1−xZnxS solutions in all samples. The successive shift in lattice spacing indicated the higher substitution degree of Zn into CdS structure with the increase in Zn concentration. X-ray line broadening analysis revealed that the average crystalline size of Cd1−xZnxS solid solution varies with Zn concentration, with the highest crystalline size being obtained in the sample with a concentration of Zn equal to 0.3. UV–Vis absorption spectra of Cd1−xZnxS samples showed a blue shift of the absorption edge with the increase in Zn concentration. The energy band gap, determined from the adsorption onset, increases from 2.49 to 2.68 when Zn concentration in solid solution increases from 0.2 to 0.3. The changes in optical and structural characteristics of Cd1−xZnxS solid solutions have had an influence on their photocatalytic activities (Fig. 14). The photocatalytic activity of samples increases gradually when the Zn concentration increases from 0.2 to 0.3. The change in activity for H2 production for these samples arises mainly from the modification of the energy level of the conduction band as the concentration of Zn increased. The activity of the solid solution decreases when the Zn concentration increases from 0.3 to 0.35 because it may be affected by a decrease in the number of available photons with the widening of the band gap and/or the decrease in the particle size of the solid solution.
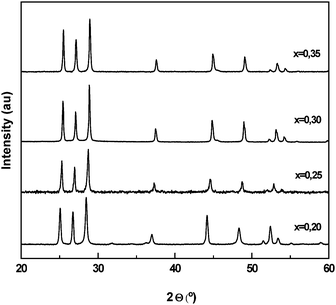 | ||
| Fig. 13 XRD patterns of Cd1−xZnxS with different Zn concentration (x = 0.2, 0.25, 0.30 and 0.35). | ||
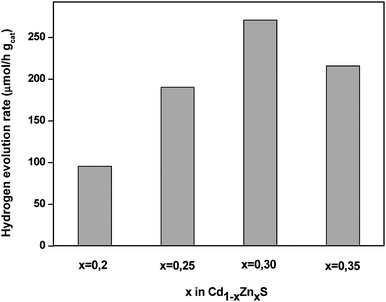 | ||
| Fig. 14 Hydrogen evolution rate from aqueous solution containing Na2S + Na2SO3 under visible light irradiation over Cd1−xZnxS with different Zn concentration (x = 0.2, 0.25, 0.30 and 0.35) (catalyst 0.1 g, reactant solution 150 ml (0.1M Na2S + 0.04M Na2SO3, 150 W Xe lamp). | ||
Taking into account the studies performed on CdS165,179 that revealed the importance of the structural characteristics (crystalline phase, crystalline size and geometrical surface area) in the control of band structure and in the concentration and mobility of photocatalyst charges, studies have been conducted on the influence of structural changes induced by thermal treatments on the photophysical properties of Cd1− xZnxS solid solutions (x = 0.2).180 The Cd0.8Zn0.2S solid solution subjected to thermal treatment under inert flow from 873 to 1023 K increases its crystalline size and slightly increases the substitution degree of Zn into CdS structure after heating at 1023 K. The structural changes induced by thermal treatments influence the UV–Vis absorption spectra of solid solution, with an increase being observed in the energy band gap of the solid solution (from 2.41 to 2.53 eV) with the increase in annealing temperature. The photocatalytic activity of Cd0.8Zn0.2S solid solution increases with treatment temperature, with a remarkable improvement in activity for the sample treated at 1023 K (Fig. 15). The combination of photoactivity results with structural characterization indicated that the improvement in photoactivity of the solid solution mainly arises from the increase in its crystalline size and absorbance derived from thermal treatments. The good crystallinity of phases with few crystal defects decreases the possibility of photoelectron–hole recombination, thereby leading to higher activity. In addition to these factors, the increase in the substitution degree of Zn into CdS structure observed for the sample treated at 1023 K is indicated as being responsible for the remarkable improvement in activity observed for this sample.
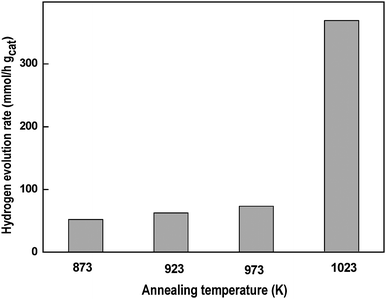 | ||
| Fig. 15 Hydrogen evolution rate from aqueous solution containing Na2S + Na2SO3 under visible light irradiation over Cd0.8Zn0.2S treated under inert flow at different temperature (873, 923, 973 and 1023 K) (catalyst: 0.1 g, reactant solution 150 ml (0.1M Na2S + 0.04M Na2SO3, 150 W Xe lamp). | ||
3.2.4.2 Other metal sulfide-based photocatalysts. ZnS is the other major metal sulfide investigated for photochemical water splitting. Combinations of AgInS2 and ZnS produce a series of (AgIn)xZn2(1−x)S2 solid solutions that crystallize in the cubic zinc blende or hexagonal wurtzite structure.140 The optical adsorption of these materials can be adjusted between 400 and 800 nm depending on composition. (AgIn)xZn2(1−x)S2 solid solutions showed photocatalytic activities for H2 evolution from aqueous solutions containing sacrificial reagents, SO32− and S2−, under visible light irradiation. Pt loaded (AgIn)0.22Zn1.56S2 with a 2.3 eV band gap, showed the highest activity for H2 evolution and the apparent quantum yield at 420 nm was 20%. Several ternary indium sulfides also show photochemical activity for water splitting under visible light. However these systems involve smaller quantities of H2 under visible light. For example, a quantum efficiency of 3.7% at 420 nm was reported for the Na14In17Cu3S35 photocatalyst.181
Other metal sulfides (In2S3, SnS2, HgS, Tl2S, PdS, CoS, Fe2S3…) have also been tested without showing photoactivity under visible light because of their small band gaps (>2 eV).
4. Conclusions
The major source of the world's energy supply currently comes from fossil fuels, but they will decline in availability over the coming decades. The large-scale production of hydrogen from natural gas and other available hydrocarbons through catalytic reforming processes remains the cheapest source of hydrogen. Even when the cheapest production method is used (steam methane reforming), some authors estimate that the cost of hydrogen production is still four times that of gasoline for the equivalent amount of energy.182 However, H2 production involving reforming technologies produces massive amounts of CO2, which has an impact on global warming. One way to reduce CO2 emissions is to apply reforming methods to alternative renewable precursors. Biomass precursors derived from plant crops, agricultural residues, woody biomass, etc., are being used for generating heat, electricity, and liquid transportation fuels (ethanol, sugars). Clean biomass precursors are converted into a gas mixture from which hydrogen is extracted. Virtually no net greenhouse gas emissions result because a natural cycle is maintained, in which carbon is extracted from the atmosphere during plant growth and is released during hydrogen production, although the time constants of the carbon cycle are different.There is a need to develop non-conventional processes for H2 production outside the C-cycle. Photonic energy technology offers the most promising outlook for the future because the water splitting reaction on semiconductor surfaces can generate potentially large quantities of clean, concentrated energy in H–H chemical bonds. Since the pioneering work by Fujishima and Honda,124 numerous attempts have been made to develop efficient photocatalysts under visible light. Consequently, more than 130 photocatalytic materials have been described as catalysts for photochemical water splitting under visible light. In spite of these developments, current results still record low efficiencies for light-to-hydrogen conversion. So far, the maximum quantum efficiency over visible light-driven photocatalysts achieves only a few percent at wavelengths as long as 500 nm (Cr/Rh–GaN/ZnO, 2.5% QE pure water, visible light.159 This value is still far from the QE of 30% marked as the initial starting point for practical application.142 Therefore, more efficient photocatalytic materials with a band gap as narrow as 2 eV (corresponding to 600 nm) need to be developed. This goal can quite possibly be achieved if sufficient knowledge is accumulated about the factors that determine the photoactivity of materials: molecular reaction mechanism, composition, structure, particle size, defect density, surface structure and charge transfer between semiconductor and cocatalysts. The molecular mechanisms and reaction kinetics of water reduction and oxidation on the semiconductor surface have yet to be elucidated in sufficient detail and should be investigated as a way of refining the materials to maximize efficiency. Finding new photocatalytic materials with a unique structure and phase is still likely to be the key to success. High throughput screening or combinational chemistry approaches, as well as a more rational search based on fundamental calculations/predictions, would be useful. The control of syntheses of materials for customising the crystallinity, electronic structure and morphology of catalysts at nanometric scale presents significant opportunities for improving water splitting photocatalysts, as these properties have a major impact on photoactivity. Taking into account the advances made in UV photocatalysts from the pioneering work of Fujisima and Honda in 1972 through to the present today, technically and economically viable visible light photocatalysts for water splitting could become available in the near future.
Acknowledgements
We are grateful to many of our colleagues for stimulating discussions and to our research sponsors CICyT and CAM (Spain) under grants ENE2007-07345-C03-01/ALT and S-0505/EN/0404, respectively. One of the authors (MCA) would also like to acknowledge financial support from the Ministry of Science and Education (Spain) through the R&C Program.References
- M. A. Peña, J. P. Gomez and J. L. G. Fierro, Appl. Catal., A, 1996, 144, 7 CrossRef CAS.
- J. A. Armor, Appl. Catal., A, 1998, 176, 159.
- D. L. Trimm and Z. I. Onsan, Catal. Rev. Sci. Eng., 2001, 43, 31 CrossRef CAS.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952 CrossRef CAS.
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2002, 27, 731 CrossRef CAS.
- J. M. Ogden, in Testimony to the Committee on Science; US House of Representatives; Washington DC, 2003 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS.
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981 CrossRef CAS.
- A. E. Farrell, R. J. Plevin, B. T. Turner, A. D. Jones, M. O'Hare and D. M. Kammen, Science, 2006, 311, 506 CrossRef CAS.
- T. A. Milne, C. C. Elam, R. J. Evans, Hydrogen from Biomass, Report for the International Energy Agency, No. IEA/H2/TR-02/001, 2002 Search PubMed.
- P. C. Hallenbeck and J. R. Benemann, Int. J. Hydrogen Energy, 2002, 27, 1185 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964 CrossRef CAS.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993 CrossRef CAS.
- J. R. Salge, B. J. Dreyer, P. J. Dauenhauer and L. D. Schmidt, Science, 2006, 314, 801 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef.
- D. Morris, J. Sci. Food Agric., 2006, 86, 1743 CrossRef CAS.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87 CrossRef CAS.
- T. A. Milne, N. Abatzoglou, R. J. Evans, NREL Technical Report, NREL/TP-570-25357, National Renewable Energy Laboratory, Golden, CO, 1998 Search PubMed.
- J. Corella, M. P. Aznar, J. Gil and M. A. Caballero, Energy Fuels, 1993, 13, 122.
- D. Sutton, B. Kelleher and J. Ross, Fuel Process. Tech., 2001, 73, 155 Search PubMed.
- A. Bauen, in Encyclopedia of Energy, ed. C. J. Cleveland, Elsevier, Amsterdam, vol. 1, 2004 Search PubMed.
- K. Tomishige, M. Asadullah and K. Kunimori, Catal. Today, 2004, 89, 389 CrossRef CAS.
- S. Rapagna, N. Jand, A. Kiennemann and P. U. Foscolo, Biomass Bioenergy, 2000, 19, 187 CrossRef CAS.
- K. Polychronopoulou, J. L. G. Fierro and A. Efstathiou, J. Catal., 2004, 228, 417 CrossRef CAS.
- E. G. Baker, L. K. Mudge and M. D. Brown, Ind. Eng. Chem. Res., 1987, 26, 1335 CrossRef CAS.
- L. K. Mudge, E. G. Baker, D. H. Mitchell and M. D. Brown, J. Sol. Energy Eng., 1985, 107, 89.
- J. M. Encinar, F. J. Beltran, A. Ramiro and J. F. Gonzalez, Fuel Process. Technol., 1998, 55, 219 CrossRef CAS.
- D. Dayton, NREL Technical Report No. NREL/TP-510-32815, National Renewable Energy Laboratory, Golden, CO, 2002. Search PubMed.
- W. B. Hausseman, Int. J. Hydrogen Energy, 1994, 19, 413 CrossRef.
- S. Lin, M. Harada, Y. Suzuki and H. Hatano, Fuel, 2004, 83, 869 CrossRef CAS.
- G. P. Curran, J. T. Clancey, D. A. Scarpillo, C. E. Fink and E. Gorin, Chem. Eng. Prog., 1996, 62, 80 Search PubMed.
- D. C. McCoy, G. P. Curran and J. D. Sudbury, Automot. Ind., 1976, 33 Search PubMed.
- S. Lin, Y. Suzuki, H. Hatano and M. Harada, Energy Fuels, 2001, 15, 339 CrossRef CAS.
- J. Wang and T. Takarada, Energy Fuels, 2001, 15, 356 CrossRef CAS.
- J. Hufton, W. Waldron, S. Weigel, M. Rao, S. Nataraj, S. Sircar, in Proceedings of the 2000 Hydrogen Program Review, NREL/CP-570-28890, 2000 Search PubMed.
- U. Zuberbühler, M. Specht, T. Marquard-Möllenstedt, in Proc. 14th European Biomass Conference and Exhibition, Biomass for Energy, Industry and Climate Protection, October 17–21, Paris, 2005 Search PubMed.
- B. Puchner, E. Höftberger, R. Rauch, H. Hofbauer, in Proc. 14th European Biomass Conference & Exhibition, Biomass for Energy, Industry and Climate Protection, October 17–21, Paris, 2005 Search PubMed.
- http://www.aer-gas.de/ .
- P. L. Spath, D. C. Dayton, NREL Technical Report No. NREL/TP-510-34929, National Renewable Energy Laboratory, Golden, CO, 2003 Search PubMed.
- B. Höhlein, Th. Grube and D. Stolten, German J. BWK, 2004, 56, 1–2 Search PubMed.
- M. Modell, in Fundamentals of Thermochemical Biomass Conversion, ed. R. P. Overend, T. A. Milne and L. K. Mudge, Elsevier Applied Science, New York, 1985, pp. 95 Search PubMed.
- M. Modell, J. Electrochem. Soc., 1980, 127(3), C139.
- M. Sasaki, Z. Fang, Y. Fukushima, T. Adschiri and K. Arai, Ind. Eng. Chem. Res., 2000, 39(8), 2883 CrossRef CAS.
- S. Manarungson, W. S. Mok and M. J. Antal, Jr., Abstr. Pap. Am. Chem. Soc., 1990, Search PubMed Paper CELL-107.
- T. Minowa, F. Zhen and T. Ogi, J. Supercrit. Fluids, 1998, 13, 253 CrossRef CAS.
- D. C. Elliott, L. J. Sealock and E. G. Baker, Ind. Eng. Chem. Res., 1993, 32, 1542 CrossRef CAS.
- M. H. Waldner and F. Vogel, Ind. Eng. Chem. Res., 2005, 44(13), 4543 CrossRef CAS.
- R. Butner, L. Sealock, Jr. and D. Elliott, Biotechnol. Bioeng. Symp., 1986, 15(15), 3 Search PubMed.
- D. Elliott, L. Sealock, Jr. and E. Baker, Ind. Eng. Chem. Res., 1993, 32(8), 1542 CrossRef CAS.
- D. Elliott, L. Sealock, Jr. and E. Baker, Ind. Eng. Chem. Res., 1994, 33(3), 558 CrossRef CAS.
- D. Elliott and L. Sealock, Jr., Chem. Eng. Res. Dev., 1996, 74(5), 563 Search PubMed.
- L. Sealock, Jr. and D. Elliott, US Pat. 5 019 135, 1991 Search PubMed.
- D. Elliott and T. Hart, US Pat. 6 152 975, 2000 Search PubMed.
- M. Osada, T. Sato, M. Watanabe, T. Adschiri and K. Arai, Energy Fuels, 2004, 18(2), 327 CrossRef CAS.
- M. Osada, N. Hiyoshi, O. Sato, K. Arai and M. Shirai, Energy Fuels, 2008, 22(2), 845 CrossRef CAS.
- M. Osada, O. Sato, K. Arai and M. Shirai, Energy Fuels, 2006, 20(6), 2337 CrossRef CAS.
- M. Osada, N. Hiyoshi, O. Sato, K. Arai and M. Shirai, Energy Fuels, 2007, 21(3), 1400 CrossRef CAS.
- Y. Usui, T. Minowa, S. Inoue and T. Ogi, Chem. Lett., 2000, 29(10), 937.
- T. Minowa, T. Ogi, Y. Dote and S. Yokoyama, Renew. Energy, 1994, 5(5–8), 813 CrossRef CAS.
- T. Minowa and T. Ogi, Catal. Today, 1998, 45(1–4), 411 CrossRef CAS.
- T. Minowa and S. Inoue, Renew. Energy, 1999, 16(1–4), 1114 CrossRef CAS.
- K. Park and H. Tomiyasu, Chem. Commun., 2003, 694 RSC.
- Y. Izumizaki, K. Park, Y. Tachibana, H. Tomiyasu and Y. Fujii, Prog. Nucl. Energy, 2005, 47(1–4), 544 CrossRef CAS.
- M. Watanabe, H. Inomata and K. Arai, Biomass Bioenergy, 2002, 22(5), 405 CrossRef CAS.
- M. Watanabe, M. Osada, H. Inomata, K. Arai and Kruse, Appl. Catal., A, 2003, 245(2), 333 CAS.
- M. Watanabe, T. Iida, Y. Aizawa, H. Ura, H. Inomata and K. Arai, Green Chem., 2003, 5(5), 539 RSC.
- M. H. Waldner, F. Krumeich and F. Vogel, J. Supercrit. Fluids, 2007, 43(1), 91 CrossRef CAS.
- A. A. Peterson, F. Vogel, R. P. Lachance, M. Fröling, M. J. Antal, Jr. and J. W. Tester, Energy Environ. Sci., 2008, 1, 32 RSC.
- X. Xu and M. Antal. Jr., Environ. Prog., 1999, 17(4), 215.
- G. Hong and M. Spritzer, Supercritical Water Partial Oxidation, Proceedings of the 2002 US DOE Hydrogen Program Review, 2002, NREL/CP-610–32405 Search PubMed.
- H. Schmieder, J. Abeln, N. Boukis, E. Dinjus, A. Kruse, M. Kluth, G. Petrich, E. Sadri and M. Schacht, J. Supercrit. Fluids, 2000, 17(2), 145 CrossRef CAS.
- A. Kruse, D. Meier, P. Rimbretch and M. Schacht, Ind. Eng. Chem. Res., 2000, 39(12), 4842 CrossRef CAS.
- A. Kruse and E. Dinjus, Z. Phys. Chem., 2005, 219(3), 341 CrossRef CAS.
- A. Kruse and M. Faquir, Chem. Eng. Technol., 2007, 30(6), 749 CrossRef CAS.
- S. Kersten, B. Potic, W. Prins and W. van Swaaij, Ind. Eng. Chem. Res., 2006, 45(12), 4169 CrossRef CAS.
- B. Potic, S. Kersten, W. Prins and W. van Swaaij, Ind. Eng. Chem. Res., 2004, 43(16), 4580 CrossRef CAS.
- L. Guo, Y. Lu, X. Zhang, C. Ji, Y. Guan and A. Pei, Catal. Today, 2007, 129(3–4), 275 CrossRef CAS.
- Calzavara, C. Joussot-Dubien, G. Boissonnet and S. Sarrades, Energy Convers. Manage., 2005, 46, 615 CrossRef CAS.
- A. Demirbas, Energy Sources, 2005, 27, 1409 CrossRef CAS.
- Y. Matsumura, Energy Convers. Manage., 2002, 9–12, 1301 CrossRef.
- F. Frusteri, S. Freni, L. Spadaro, V. Chiodo, G. Bonura, S. Donato and S. Cavallaro, Catal. Commun., 2004, 5, 611 CrossRef CAS.
- J. Llorca, P. R. de la Piscina, J. Sales and N. Homs, Chem. Commun., 2001, 641 RSC.
- A. N. Fatsikostas, D. I. Kondarides and X. E. Verykios, Chem. Commun., 2001, 851 RSC.
- F. J. Marino, M. Jobbagy, G. Baronetti and M. Laborde, Stud. Surf. Sci. Catal., 2000, 130, 2147.
- J. Llorca, N. Homs, J. Sales and P. R. de la Piscina, J. Catal., 2002, 209, 306 CrossRef CAS.
- J. P. Breen, R. Burch and H. M. Coleman, Appl. Catal., B, 2002, 39, 65 CrossRef CAS.
- D. K. Liguras, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 43(4), 345 CrossRef CAS.
- V. Fierro, O. Akdim and C. Mirodatos, Green Chem., 2003, 5(1), 20 RSC.
- R. N. Navarro, M. C. Alvarez-Galván, M. C. Sanchez-Sanchez, F. Rosa and J. L. G. Fierro, Appl. Catal., B, 2005, 55(4), 229 CrossRef CAS.
- A. N. Fatsikostas, D. I. Kondarides and X. E. Verykios, Catal. Today, 2002, 75, 145 CrossRef CAS.
- M. C. Sánchez-Sánchez, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2007, 129, 336 CrossRef CAS.
- F. Frusteri, A. Freni, V. Chiodo and L. Spadaro, Appl. Catal., A, 2004, 270, 1 CrossRef CAS.
- M. C. Sánchez-Sanchez, R. M. Navarro and J. L. G. Fierro, Int. J. Hydrogen Energy, 2007, 32, 1462 CrossRef CAS.
- S. Cavallaro and S. Freni, Int. J. Hydrogen Energy, 1996, 21, 465 CrossRef CAS.
- S. Freni, J. Power Sources, 2001, 94, 14 CrossRef CAS.
- S. Cavallaro, Energy Fuels, 2000, 14, 1195 CrossRef CAS.
- S. Freni, S. Cavallaro, N. Mondello, L. Spadaro and F. Frusteri, J. Power Sources, 2002, 108, 53 CrossRef CAS.
- P. Y. Sheng, A. Yee, G. A. Bowmaker and H. Idriss, J. Catal., 2002, 208, 393 CrossRef CAS.
- G. Auprêtre, C. Descorme and D. Duprez, Catal. Commun., 2002, 3, 263 CrossRef CAS.
- S. Cavallaro, V. Chiodo, S. Freni, N. Mondello and F. Frusteri, Appl. Catal., A, 2003, 249, 119 CrossRef CAS.
- L. V. Mattos and F. B. Noronha, J. Catal., 2003, 233, 453.
- J. R. Salge, G. A. Deluga and L. D. Schmidt, J. Catal., 2005, 235, 69 CrossRef CAS.
- Y. Yee, S. J. Morrison and H. Idriss, Catal. Today, 2000, 63, 327 CrossRef.
- L. V. Mattos and F. B. Noronha, J. Power Sources, 2000, 152, 50.
- C. Wheeler, A. Jhalani, E. J. Klein, S. Tummala and L. D. Schmidt, J. Catal., 2004, 223, 191 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 300, 2075.
- G. W. Huber and J. A. Dumesic, Catal. Today, 2006, 111, 119 CrossRef CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549 CrossRef CAS.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2003, 43, 1 CrossRef.
- J. W. Shabaker, G. W. Huber, R. R. Davda, R. D. Cortright and J. A. Dumesic, Catal. Lett., 2003, 88, 1 CrossRef CAS.
- G. W. Huber, J. W. Shabaker, S. T. Evans and J. A. Dumesic, Appl. Catal., B, 2006, 62, 226 CrossRef CAS.
- J. W. Shabaker, D. A. Simonetti, R. D. Cortright and J. A. Dumesic, J. Catal., 2005, 231, 67 CrossRef CAS.
- D. Das, N. Khanna and T. N. Veziroglu, Chem. Ind. Chem. Eng., 2008, 14(2), 57 Search PubMed.
- M. Ni, D. Y. C. Leung, M. K. H. Leung and K. Sumathy, Fuel Process. Tech., 2006, 87, 461 Search PubMed.
- M. L. Ghirardi, P. W. King, M. C. Posewitz, P. C. Maness, A. Fedorov, K. Kim, J. Cohen, K. Schulten and M. Seibert, Biochem. Soc. Trans., 2005, 33, 70 CrossRef CAS.
- D. B. Levin, L. Pitt and M. Love, Int. J. Hydrogen Energy, 2004, 29, 173 CrossRef CAS.
- K. Nath and D. Das, Appl. Microbiol. Biotechnol., 2005, 68, 533 CrossRef CAS.
- Energy Information Administration, Annual Energy Outlook, US Department of Energy, Washington, DC, 2005 Search PubMed.
- United Nations Development Program, World Energy Assessment Report: Energy and the Challenge of Sustainability, United Nations, New York, 2003 Search PubMed.
- M. Anpo, in Metal Oxides: Chemistry and Applications, ed. J. L. G. Fierro, Chap. 19, CRC Press, FL, 2006, pp. 595 Search PubMed.
- J. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. Kamat, J. Phys. Chem. B, 2001, 105, 11439 CrossRef CAS.
- S. Sakthivel, M. V. Shankar, M. Palanichamy, B. Arabindoo, D. W. Bahnemann and V. Murgesan, Water Res., 3004, 38, 301.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 3829 RSC.
- M. A. Gondal, A. Hameed and Z. H. Yamani, Energy Sources, 2005, 27, 1151 CrossRef CAS.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS.
- A. Kudo, K. Omorin and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- N. Kakuta, K. H. Park, M. F. Finlayson, A. Ueno, A. J. Bard, A. Campion, M. A. Fox, S. E. Webber and J. M. White, J. Phys. Chem., 1985, 89, 732 CrossRef CAS.
- A. Kudo and M. Sekizawa, Catal. Lett., 1999, 58, 241 CrossRef CAS.
- A. Kudo, I. Tsuji and H. Kato, Chem. Commun., 2002, 17, 1958 Search PubMed.
- G. Hitoki, T. Tataka, J. N. Kondo, M. Hara, K. Kobayashi and K. Domen, J. Phys. Chem. A, 2002, 106, 6750 CrossRef CAS.
- J. Kim, D. W. Hwang, H. G. Kim, J. S. Lee, W. Li and S. H. Oh, Top. Catal., 2005, 35(3–4), 295 CrossRef CAS.
- K. Domen, J. N. Kondo, M. Hara and T. Takata, Bull. Chem. Soc. Jpn., 2002, 73, 1307.
- W. Chyn, A. Ihiskawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1098.
- D. Lu, G. Hitoki, E. Kato, M. Hara and K. Domen, Chem. Mater., 2004, 16, 1603 CrossRef CAS.
- A. Ishikawa, Y. Yamada, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Mater., 2003, 15, 4442 CrossRef CAS.
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inue and K. Domen, Nature, 2006, 440(7082), 295 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CrossRef CAS.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029 CrossRef CAS.
- D. W. Hwang, H. G. Kim, J. S. Lee, J. Kim, W. Li and S. H. Oh, J. Phys. Chem. B, 2005, 109, 2093 CrossRef CAS.
- W. Y. Choi, A. Termin and M. R. Hoffman, J. Phys. Chem., 1994, 84, 13669 CrossRef.
- M. Anpo, Pure Appl. Chem., 2000, 72, 1787 CrossRef CAS.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505 CrossRef CAS.
- M. Kitano, H. Kikuchi, T. Hosoda, M. Takeuchi, M. Matsuoka, T. Eura and M. Anpo, Key Eng. Mater., 2006, 317–318, 823 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- T. Ohno, Z. Miyamoto, K. Nishijima, H. Kanemitsu and X. Y. Feng, Appl. Catal., A, 2006, 302, 62 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- A. Kudo, R. Niishiro, A. Iwase and H. Kato, Chem. Phys., 2007, 339, 104 CrossRef CAS.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441 CrossRef CAS.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inue and K. Domen, J. Phys. Chem. B, 2006, 110, 132107.
- G. Hitoki, T. Takata, J. N. Kondo and K. Domen, Chem. Commun., 2003, 24, 3000 Search PubMed.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547 CrossRef CAS.
- A. Ishikawa, T. Takata, T. Matsumura, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2004, 108, 2637 CrossRef CAS.
- T. Inue, T. Watanabe, A. Fujishima, K. Honda and K. Kohayakawa, J. Electrochem. Soc., 1977, 124, 719 CrossRef.
- J. R. Darwent and G. Porter, J. Chem. Soc., Chem. Commun., 1981, 4, 145 Search PubMed.
- M. Matsumura, Y. Sato and H. Tsubomura, J. Phys. Chem., 1983, 87, 3807 CrossRef CAS.
- L. Borrell, S. Cervera-March, J. Gimenez and R. A. Simarro, Sol. Energy Mater. Sol. Cells, 1992, 25, 25 CrossRef CAS.
- J. F. Rebel and K. Meier, J. Phys. Chem., 1986, 90, 824 CrossRef CAS.
- A. S. K. Sinha, S. Namita, M. K. Arora and S. N. Upadhayay, Catal. Today, 2001, 69, 297 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- M. Sathish, B. Viswanathan and R. P. Viwanath, Int. J. Hydrogen Energy, 2006, 31, 891 CrossRef CAS.
- D. Meissner, R. Memming and B. Kastening, J. Phys. Chem., 1998, 92, 3476 Search PubMed.
- L. Spanhel, H. Weller and A. Henglein, J. Am. Chem. Soc., 1987, 109, 6632 CrossRef CAS.
- J. S. Jang, D. W. Huang and J. S. Lee, Catal. Today, 2007, 120, 174 CrossRef CAS.
- F. Ferro, J. A. Rodriguez, O. Viji, A. Morales-Acevedo and G. F. Contreras-Puente, Phys. Status Solidi B, 2000, 177, 477 CrossRef.
- N. Bühler, K. Meier and J. F. Reber, J. Phys. Chem., 1984, 88, 3261 CrossRef.
- K. Kalyanasundaram, D. E.BorgarelloDuonghong and M. Gratzel, Angew. Chem., Int. Ed. Engl., 1981, 20(11), 987 CrossRef.
- R. M. Navarro, F. del Valle and J. L. G. Fierro, Int. J. Hydrogen Energy, 2008, 33, 4265–4273 CrossRef CAS.
- A. Van Dijken, A. H. Janssen, M. H. P. Smitsmans, D. Vanmaekelbergh and A. Meijerink, Chem. Mater., 1998, 10, 3513 CrossRef CAS.
- Y. Fujishiro, S. Uchida and T. Sato, J. Inorg. Mater., 1999, 1, 67 Search PubMed.
- V. A. Fedorov, V. A. Ganshing and Y. U. N. Norkeshko, Mater. Res. Bull., 1993, 28, 50.
- T. L. Chu, S. S. Chu, J. Britt, C. Ferekides and C. Q. Wu, J. Appl. Phys., 1991, 70, 2688 CrossRef CAS.
- A. Nayeem, K. Yadaiah, G. Vajralingam, P. Mahesh and M. Nagabhooshanam, Int. J. Mod. Phys. B, 2001, 15(7), 2387 CrossRef CAS.
- F. del Valle, R. M. Navarro, A. IshikawaK. Domen, J. L. G. Fierro, International Symposium on Catalysis for Clean Energy and Sustainable Chemistry, 2008, Madrid, Spain Search PubMed.
- B. Pal, T. Torimoto, K. Iwasaki, T. Shibayama, H. Takahashi and B. Ohtani, J. Phys. Chem. B, 2004, 108, 18670 CrossRef CAS.
- F. del Valle, R. M. Navarro, J. L. G. Fierro, 1st Iberian Symposium on Hydrogen, Fuel Cells and Advanced Batteries, 2008, Bilbao, Spain Search PubMed.
- N. F. Zheng, X. H. Bu and P. Y. Feng, J. Am. Chem. Soc., 2005, 127(15), 5286 CrossRef CAS.
- M. Granovskii, I. Dincer and M. A. Rosen, J. Power Sources, 2007, 167, 461 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |