182Tungsten Mössbauer spectroscopy of heteropolytungstates†‡
Christian G.
Bochet
ab,
Thérèse
Draper
a,
Bernard
Bocquet
a,
Michael T.
Pope
c and
Alan F.
Williams
*a
aSection of Chemistry and Biochemistry, University of Geneva, 30 quai Ernest Ansermet, CH 1211, Genève 4, Switzerland. E-mail: alan.williams@unige.ch; Fax: +41 22 3796830; Tel: +41 22 3796425
bChemistry Department, University of Fribourg, Ch. du Musee 9, CH-1700, Fribourg, Switzerland. E-mail: christian.bochet@unifr.ch
cDepartment of Chemistry, P.O. Box 571227, Georgetown University, Washington, DC 20057, USA. E-mail: popem@georgetown.edu
First published on 7th May 2009
Abstract
The tungsten-182 Mössbauer spectra of a series of Keggin structure heteropolytungstates, [EW12O40]n− are reported. There is a very considerable variation in quadrupole coupling at the tungsten nucleus indicating considerable asymmetry in the electron distribution for the more electronegative elements E. The quadrupole coupling correlates well with the structural data, in particular with the distance between the tungsten and the oxygen atom of the EO4group. These compounds may be regarded as rigid W12O36 cages interacting more or less strongly with an EO4n−host. The spectra of salts of metatungstate [H2W12O40]6− and [W6O19]2− are also given.
Introduction
The heteropolytungstates are one of the oldest families of self-assembled polynuclear species. Marignac1 reported the preparation of silicotungstates in 1862 although the polyoxomolybdates were prepared even earlier. Despite their venerable age, polyoxotungstates have continued to be of interest to chemists, notably by virtue of their structural diversity, their rich redox chemistry, and their potential application in catalysis.2–6Some years ago, in a preliminary investigation of the application of 182W Mössbauer spectroscopy to inorganic chemistry, we reported the spectrum of a sample of silicotungstic acid, which was remarkable in showing a large quadrupole coupling.7 Since the tungsten atom, formally, has a 5d0 electron configuration and lies in an octahedral environment, this was a surprising observation, and prompted us to investigate a series of these compounds. We report herein the somewhat surprising results of this study.
Mössbauer spectroscopy of tungsten8 has a number of practical disadvantages, including the high γ-ray energy (100 keV), which requires source and absorber to be cooled to liquid helium temperature, and a very weak sensitivity of the isomer shift to changes in the chemical environment. However, the 0 → 2 nuclear spin transition allows complete electric field gradient information (quadrupole coupling, sign and asymmetry parameter, η) to be obtained.
We chose to concentrate our studies on heteropolytungstates of formula [EW12O40]n− with the Keggin structure9 (Fig. 1) although a few other compounds are included for comparison. The Keggin structure has high (432) symmetry such that all tungsten atoms are equivalent, and show a coordination number of six, formed by one terminal oxygen, four co-planar oxygen atoms bridging two tungsten atoms and finally a sixth oxygen, which is formally part of a [EO4]n− anion at the centre of the anion and which bridges three tungsten atoms. Thus, the coordination, although formally octahedral, is quite irregular.
![The structure of the [PW12O40]3− ion.10](/image/article/2009/DT/b904101j/b904101j-f1.gif) | ||
| Fig. 1 The structure of the [PW12O40]3− ion.10 | ||
Experimental
Solvents and starting materials were purchased from Fluka and used without purification unless otherwise specified. Compounds were prepared according to literature methods and the references are given in Table 1. Caesium salts were obtained from the acids by precipitation with aqueous caesium chloride solution. Tetrabutylammonium salts were obtained by addition of saturated aqueous tetrabutylammonium chloride solution to an aqueous solution of the polytungstate. The resulting precipitate was filtered and recrystallised from acetonitrile. Full details of the characterisation of samples are given in the ESI.‡| Sample | Quadrupole coupling, e2qQ/mm s−1 | Asymmetry parameter, η | Linewidth Γ/mm s−1 | Synthesis Ref. |
|---|---|---|---|---|
| a Prepared by metathesis from salt or acid.11TBA = tetrabutylammonium. Estimated errors: quadrupole coupling: ± 0.5 mm s−1, asymmetry parameter ± 0.1. | ||||
| H3[PW12O40]·9H2O | −16.6 | 0.29 | 2.4 | 12 |
| Cs3[PW12O40]·6H2O | −16.4 | 0.20 | 2.2 | a |
| (TBA)3[PW12O40] | −16.6 | 0.29 | 2.2 | 11 |
| Na3[PW12O40]·9H2O | −16.3 | 0.29 | 2.5 | 12 |
| Cs3[AsW12O40]·10H2O | −13.5 | 0.42 | 3.0 | 11 |
| (TBA)3[AsW12O40] | −13.7 | 0.40 | 2.4 | 11 |
| H4[SiW12O40]·8H2O | −12.7 | 0.41 | 3.0 | 13 |
| K4[SiW12O40]·10H2O | −12.0 | 0.22 | 3.0 | 11 |
| (TBA)4[SiW12O40] | −12.8 | 0.39 | 2.4 | 11 |
| (TBA)4[GeW12O40] | −10.0 | 0.58 | 2.3 | 11 |
| K5[BW12O40]·11H2O | −13.1 | 0.33 | 2.8 | 11 |
| H5[BW12O40]·15H2O | −12.5 | 0.41 | 2.5 | 11 |
| Cs4H [AlW12O40]·6H2O | −6.5 | 0.59 | 3.0 | a |
| (TBA)4H[AlW12O40]·3H2O | −7.5 | 0.90 | 2.5 | a |
| K5[AlW12O40]·15H2O | −7.4 | 0.51 | 2.7 | 14 |
| (TBA)4H[FeW12O40]·4H2O | −5.8 | 0.84 | 3.0 | 15 |
| K5[CoW12O40]·11H2O | −6.2 | 0.80 | 2.4 | 16 |
| (TBA)4H2[CoW12O40]·6H2O | −6.7 | 0.69 | 2.8 | 17 |
| K4H2[CoW12O40]·6H2O | 6.1 | 0.76 | 2.5 | 16 |
| (TBA)4H2[ZnW12O40]·6H2O | −6.6 | 0.89 | 3.3 | 18 |
| K6[H2W12O40]·2H2O | 6.4 | 1.0 | 3.1 | 19 |
| (TBA)2[W6O19] | −9.4 | 0 | 2.3 | 20 |
Physical measurements
The infrared spectra were recorded from KBr pellets with a Perkin-Elmer TR-850 or IR-883 spectrophotometer. Polarograms were recorded on a Metrohm Polarecord 626 in the DP10 mode (pulse amplitude of −10 mV), with a sweep rate of −2 mV s−1 from 0 to −1.5 V, at 20 °C. The half-wave potentials in aqueous solutions are referenced to Ag/AgCl/satd KCl and those in acetonitrile to Ag/AgNO3/HNO3 and are corrected for the pulse amplitude; the potentials are expressed in mV, and the relative peak intensities are indicated in parentheses. For the tetrabutylammonium salts the polarograms were measured in acetonitrile solution (0.2M in LiClO4) against an Ag/AgNO3/HNO3electrode connected by an acetonitrile/LiClO4 bridge). The potentials were calibrated against ruthenium(II)(tris-bipyridyl). The solutions were deoxygenated by bubbling N2 for 15 min immediately before the measures. pH-dependent polarograms were obtained by progressive basification of a 1 M H2SO4 solution with 5 M NaOH. The pH was measured with a Metrohm E500 pH-meter. TLC-plates (Merck 60, F254) were eluted with a 0.3 M ethanolic solution of LiClO4 and the products were detected under a UV-lamp at 254 nm.182W Mössbauer spectra were measured using a constant acceleration spectrometer with source and absorber cooled to liquid helium temperature in an Oxford Instruments cryostat. The source was 182Ta obtained by neutron irradiation of a natural Ta foil. Because of the low recoil-free fractions relatively thick samples containing 110 mg tungsten cm−2 were used. The spectra were fitted using the least square procedure of Stone using five lines of equal width whose positions are determined by the values of the isomer shift δ, the quadrupole coupling e2qQ and the asymmetry parameter η. Isomer shifts were identical within experimental error and are not discussed here.
Results
Preparation and characterisation of compounds
The compounds were prepared using standard methods from the literature, and the references are given in Table 1. Considerable effort was taken in the characterisation of the compounds. The infra-red spectra were used to confirm the presence of the polyanion, and compared with the detailed survey of Rocchiccioli-Deltcheff et al.11Water content, and in the case of tetrabutylammonium (TBA) salts, the cation content were obtained by thermogravimetric analysis. Following Rocchiccioli-Deltcheff et al. we also studied the electrochemical properties of the anions. The DC polarograms obtained corresponded to those in the literature, but in general we found that differential pulse polarography (DPP) gave a better appreciation of the purity of the sample since well resolved peaks were obtained for pure samples. We noted however that the peaks were very sensitive to the pH of the medium. Fig. 2 represents the changes in the peaks, represented as Gaussian curves, as the pH is raised. It will be noticed that the polarogram of [PW12O40]3− disappears as low as pH 3. For this reason we believe the acetic acid/acetate buffer used by Rocchiccioli-Deltcheff et al. is not suitable for the phosphotungstates. The polarograms also allowed us to exclude the presence of β-isomers.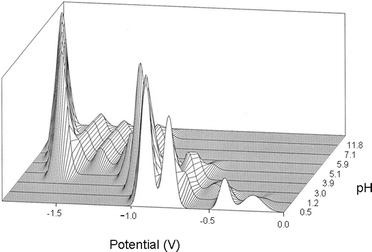 | ||
| Fig. 2 Overlay of the DPP polarograms of phosphotungstic acid as a function of pH. | ||
Finally, we found that TLC on silica may be used to check the purity of the samples. The eluent used was either ethanol with lithium perchlorate 0.3M or acetone–10% hexane–lithium perchlorate 0.3M. The Rf values decreased with increasing charge: typical values in ethanol were 0.62 for 4− ions, 0.58 for 5− ions and 0.38 for 6− ions. Phosphotungstates with a 3− charge showed a spot at an Rf value of 0.66, but other spots were also present suggesting decomposition in the solution. Paratungstate, a frequent impurity in the preparations, does not migrate under these conditions. An unsatisfactory TLC was inevitably confirmed by other measurements such as polarography or infrared spectroscopy.
Mössbauer spectra
The data are summarised in Table 1. Isomer shifts showed no significant variation and we shall concentrate our discussion on the quadrupole coupling and asymmetry parameters. The first observation is the very large range of values. The phosphotungstates (around −16 mm s−1) show the largest couplings, with values among the largest reported for tungsten compounds. There is a steady drop as the electronegativity of the central heteroatom falls to values around 6 mm s−1 for the systems where the heteroatom is a metallic cation. We may note that a value of 6 mm s−1 results in a spectrum which appears as a slightly broadened line, and consequently such couplings are not particularly precise. For couplings of 10 mm s−1 or more the peaks are well separated, and may be fitted with precision (Fig. 3). The value initially reported for silicotungstic acid is anomalously high,7 and we believe this is due to a calibration error; it should be ignored in favour of the value reported here.![182W Mössbauer spectrum of K4[SiW12O40]·10H2O.](/image/article/2009/DT/b904101j/b904101j-f3.gif) | ||
| Fig. 3 182W Mössbauer spectrum of K4[SiW12O40]·10H2O. | ||
The asymmetry parameters increase as the quadrupole coupling falls, to values close to 1 for couplings around 6 mm s−1. In such systems, the sign of the coupling is not well defined, and the apparent change in sign of the gradient is not particularly significant.
The examination of the results leads to two questions. Firstly, why is the quadrupole coupling so large in the phosphotungstates, and why does it fall so sharply as the heteroatom changes? This change is remarkable in that the change of the central atom, from phosphorus to aluminium, for example, drops the quadrupole coupling by a factor of 2 for all twelve tungsten atoms. Secondly, can we explain the variation in the asymmetry parameter? We will discuss the two non-Keggin systems, K6[H2W12O40]·2H2O and (TBA)2[W6O19] later.
As mentioned in the introduction, the tungsten coordination, although it is usually described as octahedral, is in fact highly distorted, as is often the case for the earlier transition metals.21Fig. 4 shows the coordination sphere for two examples, [PW12O40]3− and [FeW12O40]5−22 which correspond to the two extremes of the quadrupole coupling range.
![The coordination sphere of tungsten in: (a) [PW12O40]3−10 and (b) [FeW12O40]5−.](/image/article/2009/DT/b904101j/b904101j-f4.gif) | ||
| Fig. 4 The coordination sphere of tungsten in: (a) [PW12O40]3−10 and (b) [FeW12O40]5−. | ||
The coordination sphere consists of one oxygen atom at a distance of approximately 1.7 Å, the distance corresponding to a tungsten-oxygen double bond,21 and four at roughly 1.9 Å, corresponding to a single bond distance. The sixth site of the octahedron, trans to the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond, is occupied by an oxygen of the EO4 tetrahedron, and the bond distance is much greater, 2.437 Å for the phosphotungstate and 2.229 Å for the ferrotungtstate. This corresponds to a very low bond valence, perhaps not altogether surprisingly given that the bond valence sum for the first five bonds is close to 6 and the sixth oxygen is shared between three tungsten atoms. We may therefore consider the structure to be a W12O36 cage which interacts more or less strongly with the central EO4 unit. Each tungsten is part of a square pyramidal (SP) WO5 unit, which interacts weakly with an oxygen bound to the heteroatom. This structure would be expected to give rise to a large negative quadrupole coupling as observed since the square pyramid may be considered as an octahedron from which electron density has been removed along the z-axis, giving a positive electric field gradient, q, and the quadrupole moment, Q, of the excited state is negative.23
O double bond, is occupied by an oxygen of the EO4 tetrahedron, and the bond distance is much greater, 2.437 Å for the phosphotungstate and 2.229 Å for the ferrotungtstate. This corresponds to a very low bond valence, perhaps not altogether surprisingly given that the bond valence sum for the first five bonds is close to 6 and the sixth oxygen is shared between three tungsten atoms. We may therefore consider the structure to be a W12O36 cage which interacts more or less strongly with the central EO4 unit. Each tungsten is part of a square pyramidal (SP) WO5 unit, which interacts weakly with an oxygen bound to the heteroatom. This structure would be expected to give rise to a large negative quadrupole coupling as observed since the square pyramid may be considered as an octahedron from which electron density has been removed along the z-axis, giving a positive electric field gradient, q, and the quadrupole moment, Q, of the excited state is negative.23
The quadrupole coupling will decrease as the interaction of the tungsten atom with the EO4oxygen increases and charge is donated. The interaction with the EO4 unit can increase for two reasons: (1) an increased basicity of the EO4n− ion; and (2) an increase in the size of E, which will lengthen the E–O bonds, and for simple geometric reasons approach the oxygen to the tungsten.
Examination of the data supports this argument. As the formal positive charge on the central E atom falls, the basicity of the EO4n− increases, and quadrupole coupling drops, as shown in the series P > Si > Al. The size effect is shown by comparing two elements in the same column of the periodic table where we see P > As, Si > Ge, and B > Al. The strength of the interaction between tungsten and EO4oxygen may also be measured by the W–O bond distance. Table 2 gives the average W–OEO3 distance for the heteropolytungstate anions extracted from the data in the Cambridge Structural Database (version 5.29, update of January 2009). Fig. 5 shows that the average distances correlate very well with the observed quadrupole couplings for distances greater than 2.25 Å. For shorter distances the quadrupole coupling is both rather small and the environment is distinctly non-axial.
| Atom E | EO–W/Å | Bond order21 | E–W/Å |
|---|---|---|---|
| P | 2.443 | 0.195 | 3.556 |
| As | 2.362 | 0.249 | 3.567 |
| Si | 2.356 | 0.253 | 3.520 |
| Ge | 2.298 | 0.302 | 3.539 |
| B | 2.359 | 0.251 | 3.482 |
| Al | 2.253 | 0.346 | 3.504 |
| Co | 2.180 | 0.432 | 3.496 |
| Fe | 2.210 | 0.394 | 3.515 |
| Zn | 2.160 | 0.459 | 3.499 |
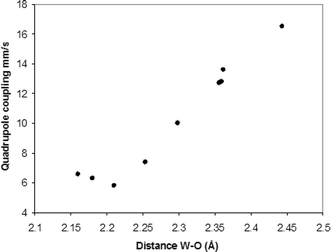 | ||
| Fig. 5 Correlation between quadrupole coupling and W–O bond distance. | ||
The behaviour of the asymmetry parameter is also consistent with this explanation. Fig. 4 shows that the EO4oxygen is displaced from the axis of the WO double bond, giving non-axial symmetry. This is the origin of the small but non-zero asymmetry parameter in the phosphotungstate. As the size of the hetero atom increases the EO4oxygen is moved progressively further away from the axis, and the asymmetry parameter increases. As can be seen from Fig. 4b in the ferrotungstate, the EO4oxygen is much further from the WO axis.
If we consider these compounds as containing W12O36 cages encapsulating [EO4]n− anions, we may ask to what extent the W12O36 cage can shrink or expand to fit in the anion. We may estimate the size of the cage from the mean E–W distance in [EW12O40]n− and the averaged values extracted from the Cambridge Structural Data base are shown in Table 2. Although the values do vary as a function of E, the variation is much less than that of the O–W distances, which would tend to suggest that the cage is rather rigid. A similar conclusion was obtained from recent theoretical calculations on these systems.24
Finally, we may look at the two non-Keggin systems in Table 1. Both are similar to the Keggin systems in that they contain a WO5 square pyramidal unit with a sixth oxygen weakly bound. The metatungstate K6[H2W12O40]·2H2O may be regarded as a Keggin system in which the central heteroatom is missing. The four oxygen atoms that remain of the EO4 tetrahedron are thus formally oxide ions and are quite basic. The two hydrogen atoms are located inside the O4 cavity but cannot be observed by X-ray crystallography. The interaction with the tungsten atoms is thus strong, with a low quadrupole coupling. The average O–W distance is 2.212 Å in agreement with the correlation shown in Fig. 5. As for the Keggin systems a high asymmetry parameter is predicted and observed.
The [W6O19]2− ion in (TBA)2[W6O19] has a central oxygen atom octahedrally coordinated by six WO5 units composed of one terminal oxygen and four oxygen atoms shared with neighbouring tunsgten atoms. The coordination is thus similar to the polytungstate except for the fact that the tungsten atom lies on a four-fold axis. We observe the expected zero asymmetry parameter with a negative coupling of −9.4 mm s−1. The average W–O distance for the central oxygen is 2.323 Å, and this lies close to the correlation in Fig. 5.
Conclusions
The wide variation in quadrupole coupling observed in these systems was surprising for compounds which are often regarded as isostructural. The very large quadrupole coupling for the phosphotungstates is particularly remarkable, and lies among the highest values reported for tungsten compounds. The spectra are consistent with a very variable interaction of the central EO4 unit with the W12O36 cage, and this is supported by a survey of bond distances in these systems. If the experimental difficulties of Mössbauer spectroscopy of tungsten limit its widespread use, it is significant that these results show a correlation between electron distribution as measured by the quadrupole coupling and bond distance and bond order as measured by X-ray crystallography.Acknowledgements
We gratefully acknowledge the support of this work by the Swiss National Science Foundation.Notes and references
- J.-C. Galissard de Marignac, Ann. Chim., 1864, 3, 5 Search PubMed.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer Verlag, Berlin, Heidelberg, New York, Tokyo, 1983 Search PubMed.
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
- M. T. Pope and A. Müller, Angew. Chem., Int Ed., 1991, 30, 34 CrossRef.
- M. T. Pope and C. L. Hill, in Comprehensive Coordination Chemistry II, ed. J. McCleverty and T. J. Meyer, Pergamon Press, Oxford, 2004, 635, 679 Search PubMed.
- Special issue devoted to poloyoxometallates: Chem. Rev., ed. C. L. Hill, 1998, 98, 1 Search PubMed.
- A. G. Maddock, R. H. Platt, A. F. Williams and R. Gancedo, J. Chem. Soc., Dalton Trans., 1974, 1341 Search PubMed.
- A. F. Williams, in Mössbauer Spectroscopy Applied to Inorganic Chemistry, ed. G. J. Long, Plenum Press, New York, 1987, 429 Search PubMed.
- J. F. Keggin, Proc. R. Soc. A, 1934, 144, 75 CrossRef CAS.
- G. M. Brown, M.-R. Noe-Spirlet, W. R. Busing and H. A. Levy, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1038 CrossRef.
- C. Rocchiccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, Inorg. Chem., 1983, 22, 207 CrossRef CAS.
- P. Souchay, Ions Minéraux Condensés, Masson et Cie, Paris, 1969 Search PubMed.
- E. O. North, Inorg. Synth., 1936, 1, 129.
- J. A. Mair and J. L. T. Waugh, J. Chem. Soc., 1950, 2372 RSC.
- M. T. Pope and G. M. Varga, Jr., Inorg. Chem., 1966, 5, 1249 CrossRef CAS.
- V. E. Simmons, Ph.D. Thesis, Diss. Abs. 1963, 24, 1391, Boston University, 1963.
- K. Nomiya, R. Kobayashi and M. Miwa, Bull. Chem. Soc. Jpn., 1983, 56, 2272 CAS.
- D. H. Brown and J. A. Mair, J. Chem. Soc., 1958, 2597 RSC.
- C. M. Flynn, Jr. and M. T. Pope, Inorg. Chem., 1973, 12, 1626 CrossRef.
- J. Fuchs and K. F. Jahr, Z. Naturforsch., B: Chem. Sci., 1968, 23, 1380 CAS.
- B. Demengès, N. K. McGuire and M. O'Keefe, J. Solid State Chem., 1985, 56, 94 CAS.
- Feng-Xia Ma, Ya-Guang Chen and D.-M. Shi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m672 CrossRef.
- M. G. Clark, J. R. Gancedo, A. G. Maddock and A. F. Williams, J. Chem. Soc., Dalton Trans., 1975, 120 RSC.
- F.-Q. Zhang, X.-M. Zhang, H.-S. Wu and H. Jiai, J. Phys. Chem. A, 2007, 111, 159 CrossRef CAS.
Footnotes |
| † In memory of Alfred Gavin Maddock, 15th August 1917–5th April 2009. |
| ‡ Electronic supplementary information (ESI) available: Sample characterisation data. See DOI: 10.1039/b904101j |
| This journal is © The Royal Society of Chemistry 2009 |