Infrared spectra and density functional theory calculations of coinage metal disulfide molecules and complexes
Xuefeng
Wang
,
Binyong
Liang
and
Lester
Andrews
*
Department of Chemistry, University of Virginia, P.O. Box 400319, Charlottesville, Virginia 22904-4319, USA. E-mail: lsa@virginia.edu
First published on 16th April 2009
Abstract
Sulfur diatomic molecules were reacted with laser ablated Cu, Ag, and Au atoms in excess argon and condensed at 7 K. Several reaction products were identified for each metal from matrix infrared spectra through sulfur-34 isotopic shifts, spectra of sulfur isotopic mixtures, and frequencies calculated by density functional theory. The linear centrosymmetric 63CuS2, 65CuS2, and AuS2 metal disulfide molecules were observed at 508.7, 504.8, and 455.9 cm−1, respectively. The bent CuSS, AgSS, and AuSS isomers were identified from 608.6, 580.7, and 601.1 cm−1 S–S stretching fundamentals, respectively. These group 11 metals also formed cyclic tetrasulfur molecules, MS4, in contrast to group 10 metal atoms, which formed side-bonded disulfur complexes M(S2)2.
Introduction
Transition metal sulfides have attracted considerable attention because of their occurrence in minerals and significant applications in many biological and industrial processes.1 All three coinage metals form dimetal sulfide molecules, and the common tarnish on silver flatware is Ag2S.2,3Copper disulfide in the pryrite form requires high pressure for synthesis.4Copper pyrites are also of interest for superconductor, anion conductor, and electronic structure characteristics.5–7It is interesting to compare oxygen and sulfur in the molecular complexes and the oxides or sulfides that can be formed. There has been extensive investigation of M(O2)n complexes by matrix infrared and ESR spectroscopy, and the question of symmetric side-bonded or asymmetric end-on bonded structures debated.8,9Sulfur analogs of these complexes have not been reported. Very recently AuS2− anion photodetachment has been investigated, and both AuSS− and SAuS− anion and neutral structures reported.10 The analogous neutral OAuO molecule has been prepared from laser-ablated gold atoms and dioxygen and characterized from its matrix infrared spectrum.11,12 Similar OAuO− anion photodetachment has also identified this linear OAuO molecule.10 The linear OCuO molecule has also been prepared in solid argon,13 and the photoelectron spectra of CuO2− anions observed,14 but the sulfur analogs have not been investigated.
Coinage metal carbonyl complexes have been prepared through the reaction of laser-ablated metal atoms and CO in excess noble gas and characterized by matrix infrared spectroscopy and density functional calculations.15 We apply these methods to the investigation of the coinage metal molecular sulfides and complexes using microwave discharge of elemental sulfur and sulfur-34 vapor16 as a source disulfur reagent molecules. Similar investigations with group 4, 5, and 6 metals gave molecular sulfides, and in addition group 10 metal reactions produced disulfur complexes as well.17–20 Metal–sulfur bonding is important for comparison of the analogous sulfide and oxide molecules and in self-assembled monolayers.21
Experimental and computational methods
The technique for conducting reactions of laser-ablated metal atoms has been described in detail previously.22 The Nd:YAG laser fundamental (1064 nm, 10 Hz repetition rate with 10 ns pulse width) was focused onto a rotating high-purity copper, silver (Johnson Matthey), or gold (cut from crucible) metal target. The laser energy was varied from 5 to 10 mJ/pulse. Metal atoms were mixed with a sulfur-doped argon stream and condensed on a 7 K CsI cryogenic window at 2–4 mmol/h for 1 h. Infrared spectra were recorded at 0.5 cm−1 resolution on a Nicolet 550 spectrometer with 0.1 cm−1 accuracy using a mercury cadmium telluride detector. Matrix samples were annealed at different temperatures, and selected samples were subjected to irradiation using a medium-pressure mercury street lamp (> 220 nm) with the globe removed.A microwave discharge in argon entrained with sulfur vapor was used as a source of sulfur atoms and small molecules as reagents. The coaxial quartz discharge tubes used here evolved from the one used in our first experiments.16 Natural isotopic sulfur (Electronic Space Products, Inc.) and enriched sulfur (98% 34S, Cambridge Isotope Laboratories) were used as received: different mixtures of the two isotopic samples were also employed. The vapor pressure of sulfur feeding the discharge was controlled by resistance heating. The microwave discharge was maintained in the argon–sulfur mixture (Opthos Instruments 120 W microwave discharge, 30–50% of the maximum power level) with an Evenson-Broida cavity extended from 5 cm downstream of the sulfur reservoir to the end of the discharge tube.16
Following previous work,12,17–20 DFT calculations were performed on anticipated metal sulfide products using the Gaussian 03 program,23 the B3LYP density functional,24 the large Gaussian basis 6-311+G(3df) for sulfur, and the SDD pseudopotential and basis for the metal atoms.25,26
Results and discussion
Infrared spectra
A flowing argon discharge seeded with elemental sulfur vapor gives S2 as evidenced from its blue emission27 and S3 and S4 based on their known infrared absorption spectra.16 Laser-ablated group 11 metal atoms were co-deposited with such an argon stream, and new metal dependent sulfur reaction product spectra will be presented and assigned. Relatively low laser energy was employed, and the concentration of metal atoms in the matrix is low enough to limit the contribution of dimetal species to the observed spectra: resolved natural copper isotopic splittings support this point.Cu + Sx
Several experiments were performed with copper and sulfur vapor to optimize the reagent concentrations. A representative set of spectra is illustrated in Fig. 1, where the S3 absorptions in the 600 cm−1 region (not shown) are as intense as the S4 bands (absorbances 0.14 to 0.16), and weak S3 bands are detected at 584.9, 581.7 cm−1. The major new product absorptions appeared at 608.6, 564.6, 542.1, 520.5, and 508.7 cm−1 as listed in Table 1. Annealing slightly decreased the 608.6 cm−1 band and increased the 542.1 and 520.5 cm−1 bands and ultraviolet irradiation decreased the 564.6 cm−1 band, increased the lower three bands, and brought out a 504.8 cm−1copper isotopic splitting for the 507.8 cm−1 band.28 Higher annealing continued these trends. These new product absorptions exhibited large (16 to 17 cm−1) and small (7 cm−1) shifts on sulfur-34 substitution and 32/34 isotopic frequency ratios from 1.0300 to 1.0151 reflecting the different sulfur and copper participations in the reduced mass of the vibrational mode. Spectra showing the effects of isotopic substitution are also illustrated in Fig. 1. The diagnostic mixed isotopic spectra reveal broad triplets for the two higher new band systems and higher multiplets for the next two. Fortunately, the lowest band is preferentially increased on ultraviolet irradiation, and a sharp triplet absorption emerges at 508.7, 505.2, 501.3 cm−1: although the first two weaker copper isotopic components are covered, the lower one is observed at 497.3 cm−1.| 32S | 34S | 32S + 34S | R(32/34) | identity |
|---|---|---|---|---|
| Copper | ||||
| 608.6 | 591.0 | 608.6, 599.9, 591.1 | 1.0298 | CuSS |
| 564.6 | 549.4 | 564.6, 557.0, 549.3 | 1.0277 | CuSS− |
| 556.6 | 540.4 | 1.0300 | ||
| 542.1 | 526.3 | 542.0, 539.4, 534.8, 531.1, 526.4 | 1.0300 | CuS3 |
| 536.4 | 520.8 | 1.0300 | CuS3 site | |
| 523.6 | 508.2 | 523.6, 518.2, 514.7, 511.3, 508.2 | 1.0303 | CuS4 |
| 522.1 | 506.9 | 522.0, 516.8, 513.8, 509.6, 506.9 | 1.0300 | CuS4 site |
| 520.5 | 505.3 | 1.0301 | CuS4 site | |
| 508.7 | 501.3 | 508.7, 505.2, 501.3 | 1.0148 | linear 63CuS2 |
| 504.8 | 497.3 | —, —, 497.3 | 1.0151 | linear 65CuS2 |
| Silver | ||||
|---|---|---|---|---|
| 584.9 | 567.6 | 585.0, 579.5, 576.2, 572.7, 569.4, 567.6 | 1.0305 | S3, ν1 |
| 582.8 | 565.7 | 582.8, 574.5, 565.6 | 1.0303 | AgSS site |
| 581.5 | 564.4 | 581.6, 578.1, 574.8, 571.3, 566.1, 564.4 | 1.0303 | S3 site, ν1 |
| 580.6 | 563.7 | 580.6, 572.2, 563.7 | 1.0300 | AgSS |
| 550.6 | 534.4 | 1.0303 | ||
| 533.3 | 517.6 | 533.2, 529.3, 525.5, 520.8, 517.9 | 1.0303 | AgS3 site |
| 530.3 | 514.7 | 530.3, 525.8, 522.5, 518.7, 514.8 | 1.0303 | AgS3 |
| 502.3 | 487.6 | 502.1, 498.4, 494.2, 491.4, 487.7 | 1.0301 | AgS4 |
| 476.3 | 462.6 | 1.0296 | (Ag2S4) | |
| 463.2 | 449.6 | 1.0302 | (Ag2S4) | |
| Gold | ||||
|---|---|---|---|---|
| 602.3 | 584.5 | 602.3, 593.4, 584.5 | 1.0305 | AuSS site |
| 601.1 | 583.3 | 601.1, 592.3, 583.3 | 1.0305 | AuSS |
| 520.1 | 506.6 | 1.0266 | AuS3 site | |
| 518.6 | 505.1 | 518.5, 515.7, 513.4, 511.0, 507.4, 505.2 | 1.0267 | AuS3 |
| 509.6 | 496.3 | 1.0268 | AuS4 site | |
| 503.7 | 490.3 | 1.0273 | AuS4 site | |
| 500.2 | 487.2 | 500.2, 497.0, 493.7, 490.4, 487.3 | 1.0267 | AuS4 |
| 494.4 | 479.9 | 1.030 | ?Au(S2) | |
| 455.9 | 445.6 | 456.0, 451.7, 445.6 | 1.0231 | linear AuS2 |
 | ||
| Fig. 1 Infrared spectra in the 620–480 cm−1 region for products formed in the laser-ablated Cu reaction with discharged sulfur vapor during condensation in excess argon at 7 K. (a) Cu and natural isotopic sulfur sample deposited for 60 min, (b) after annealing to 30 K, (c) after > 220 nm irradiation, (d) after annealing to 35 K, and (e) after annealing to 40 K. Scans (f), (g), and (h) are spectra from the Cu reaction using 50/50 mixed S-32 and S-34 after deposition, > 220 nm irradiation, and 40 K annealing, and traces (i), (j), and (k), are analogous spectra from the Cu reaction with S-34 in the discharge. | ||
Ag + Sx
The silver spectra reveal sharp new absorptions at 582.8, 580.6 cm−1 and a stronger band at 530.3 cm−1, which are shown in Fig. 2 and listed in Table 1, along with the results of sulfur-34 substitution. | ||
| Fig. 2 Infrared spectra in the 600–450 cm−1 region for products formed in the laser-ablated Ag reaction with discharged sulfur vapor during condensation in excess argon at 7 K. (a) Ag and natural isotopic sulfur sample deposited for 60 min, (b) after > 220 nm irradiation, (c) after annealing to 35 K, and (d) after annealing to 40 K. Scans (e), (f), (g), and (h) are analogous spectra from the Ag reaction using 50/50 mixed S-32 and S-34. Traces (i), (j), (k), and (l) are analogous spectra from the Au reaction with discharged S-34. | ||
Au + Sx
Reactions of gold and sulfur diatomic molecules produced new bands at 601.1, 518.6, 500.2, and 455.9 cm−1 as shown in Fig. 3. The two middle bands are complicated by satellites. Annealing to 30 K had little effect on the spectrum, but full arc irradiation decreased the upper bands and increased the lowest band slightly. These features exhibited pronounced shifts with sulfur-34 substitution and 32/34 isotopic frequency ratios from 1.0305 to 1.0231 reflecting still different sulfur and gold participations in the reduced masses of the vibrational modes. Fig. 3 also compares mixed and pure sulfur isotopic reaction products. The 601.1 and 455.9 cm−1 bands became sharp triplet absorptions with mixed isotopic sulfur, but the 518.6 cm−1 band developed a sextet and the 500.2 cm−1 band gave most likely a quartet after accounting for satellites. | ||
| Fig. 3 Infrared spectra in the 620–440 cm−1 region for products formed in the laser-ablated Au reaction with discharged sulfur vapor during condensation in excess argon at 7 K. (a) Au and natural isotopic sulfur sample deposited for 60 min, (b) after annealing to 30 K, (c) after > 220 nm irradiation, and (d) after annealing to 40 K. Scans (e), (f), (g), and (h) are analogous spectra from the Au reaction using 50/50 mixed S-32 and S-34. Traces (i), (j), (k), and (l) are analogous spectra from the Au reaction with S-34. | ||
CuSS
The 608.6 cm−1 band decreases on annealing, shifts to 591.0 cm−1 with sulfur-34 defining a 1.0298 sulfur 32/34 isotopic frequency ratio, and develops a triplet absorption at 608.6, 599.9, 591.1 cm−1 with mixed sulfur isotopes. This triplet indicates the participation of two sulfur atoms in the vibrational mode, which is characterized as an almost pure S–S motion from the above ratio. We calculate the global minimum energy CuS2 species to be the 2A″ bent CuSS molecule with strong S–S stretching mode at 587.6 cm−1 whereas the side-bound 2A2 Cu(S2) complex is 4 kcal/mol higher in energy and has a very weak S–S mode at 527.6 cm−1. Our DFT calculations support assignment of the 608.6 cm−1 band to the CuSS molecule (Table 2). Although harmonic B3LYP calculations typically overestimate observed fundamentals, this is not always the case for some transition metal containing molecules and there are examples of underestimates as well since frequency calculation is not an exact science.29,30 The two sulfur atoms are not equivalent, and the calculation finds the Cu32S34S and Cu34S32S modes separated by 0.3 cm−1, which is not resolved in the slightly broader central band component (Fig. 1(f)).| Species | State | Lengths, Å angles, deg | Rel. Ener. (kcal/mol) | Frequencies, cm−1 (symmetry, intensities, km/mol) |
|---|---|---|---|---|
| S2 | 3Σ−g | SS: 1.902 | 0 | 715.9 (0) |
| S3 | 1A1 | SS: 1.922 | 0 | 686.5(b2,134), 594.5(a1,2), 262.3(a1,1) |
| SSS: 118.5 | ||||
| Copper | ||||
|---|---|---|---|---|
| CuS | 2Π | CuS: 2.075 | 0 | Cu32S: 397.5(3); Cu34S: 389.6(3) |
| CuSS | 2A″ | CuS: 2.156 | 0 | Au32S32S: 627.8(a′,67), 313.4(a′,3), 139.3(a′,0) |
| SS: 1.973 | Au32S34S: 618.6(a′,66), 305.1(a′,3), 136.5(a′,0) | |||
| CuSS: 109.2 | Au34S32S: 618.5(a′,65), 305.4(a′,3), 138.3(a′,0) | |||
| Au34S34S: 609.1(a′,63), 305.1(a′,3), 135.7(a′,0) | ||||
| CuS2 | 2Π | CuS: 2.028 | 39 | 63Cu32S2: 539.5 (σu,26), 378.9(σg,0), 70(πu,3 × 2) |
| SCuS: 180 | 32S63Cu34S: 535.6 (σ,25), 373.2(σ,0), 69(π,3 × 2) | |||
| 63Cu34S2: 531.5 (σu,25), 367.7(σg,0), 69(πu,3 × 2) | ||||
| 65Cu32S2: 535.2 (σu,25), 378.9(σg,0), 70(πu,3 × 2) | ||||
| CuSS− | 3A′ | CuS: 2.262 | −35 | Cu32S32S: 561.6(a′,23), 253.3(a′,6), 93.5(a′,4) |
| SS: 2.008 | Cu32S34S: 553.4(a′,21); Cu34S32S: 553.2(a′,21) | |||
| CuSS: 113.7 | Cu34S34S: 544.8(a′,21) | |||
| CuSS− | 1A′ | CuS: 2.155 | −40 | Cu32S32S: 503.9(a′,130), 325.6(a′,6), 127.5(a′,2) |
| SS: 2.050 | Cu32S34S: 496.5(a′,125); Cu34S32S: 496.4(a′,127) | |||
| CuSS: 112.7 | Cu34S34S: 488.9(a′,123) | |||
| CuS3 | 2A | CuS: 2.302 | 0 | Cu32S3: 528.1 (76), 512.6(2), 326.1(3), three real |
| SS: 2.027 | Cu34S3: 512.4 (72), 497.4(2), 317.3(4), three real | |||
| SSS: 108.0 | ||||
| SCuS:90.9 | ||||
| CuS4 | 2A | CuS: 2.192 | 0 | Cu32S4: 523.2 (7), 515.3(77), 374.4(2), six real |
| SS: 2.03, 2.14 | Cu34S4: 507.6 (6), 500.0(72), 363.2(2), six real | |||
| SSS: 114, SCuS:119.7 | ||||
| SSCuSS | 2B1 | CuS: 2.161 | 28 | Cu32S4: 614.4 (0), 583.2(380), 380.9(9), six real |
| SS: 1.960 | Cu34S4: 596.1 (0), 565.9(358), 375.3(9), six real | |||
| CuSS: 107.2 | ||||
| SCuS: 180 | ||||
| Silver | ||||
|---|---|---|---|---|
| AgSS | 2A″ | AgS: 2.403 | 0 | Au32S32S: 601.1(a′,67), 270.2(a′,1), 117.1(a′,1) |
| SS: 1.962 | Au32S34S: 592.6(a′,65), 269.9(a′,1), 115.1(a′,1) | |||
| AgSS: 111.0 | Au34S32S: 592.9(a′,65), 263.9(a′,1), 116.4(a′,1) | |||
| Au34S34S: 583.7(a′,63), 263.6(a′,1), 114.4(a′,1) | ||||
| AgS2 | 4Σu+ | AgS: 2.275 | 65 | Ag32S2: 336 (σu,2), 283(σg,0), 24(πu,1 × 2)3 |
| SAgS: 180 | ||||
| AgSS− | 1A′ | AgS: 2.408 | − 41 | Ag32S32S: 520.7(a′,154), 253.4(a′,3), 111.8(a′,2) |
| SS: 2.034 | Ag34S34S: 505.2(a′,145), 247.3(a′,3), 109.3(a′,2) | |||
| AgSS: 114.2 | ||||
| AgS3 | 2A | AgS: 2.583 | 0 | Ag32S3: 537.2 (86), 516.6(2), 306.6(2), three real |
| SS: 2.016 | Ag34S3: 521.2 (81), 501.2(2), 297.7(2), three real | |||
| SSS: 110.2 | ||||
| SAgS:79.9 | ||||
| AgS4 | 2A | AgS: 2.442 | 0 | Ag32S4: 536.3 (10), 525.5(104), 358.4(2), six real |
| SS: 2.006, 2.142 | Ag34S4: 520.3 (10), 509.8(98), 347.7(2), six real | |||
| SSS: 116.3 | ||||
| SAgS:108.0 | ||||
| SSAgSS | 2B1 | AgS: 2.373 | 28 | Ag32S4: 632.7 (0), 597.7(560), 308.1(0), six real |
| SS: 1.950 | Ag34S4: 613.9 (0), 579.9(527), 303.6(0), six real | |||
| AgSS: 109.5 | ||||
| SAgS: 180 | ||||
| Ag2S4 | 2A | AgAg: | 0 | Ag232S4: 472 (18), 469(12), 399(3), eight real |
| AgS: 2.397 | Ag234S4: 458 (17), 455(11), 387(3), eight real | |||
| SS: 2.053, 2.096 | ||||
| Gold | ||||
|---|---|---|---|---|
| a Calculations used B3LYP/6-311+G(3df) for S and SDD for Cu, Ag and Au. | ||||
| AuS | 2Π | AuS: 2.228 | 0 | Au32S: 362.8(0.3); Au34S: 353.5(0.3) |
| AuSS | 2A″ | AuS: 2.310 | 0 | Au32S32S: 627.8(a′,67), 313.4(a′,3), 139.3(a′,0) |
| SS: 1.938 | Au32S34S: 618.6(a′,66), 305.1(a′,3), 136.5(a′,0) | |||
| AuSS: 112.7 | Au34S32S: 618.5(a′,65), 305.4(a′,3), 138.3(a′,0) | |||
| Au34S34S: 609.1(a′,63), 305.1(a′,3), 135.7(a′,0) | ||||
| AuS2 | 2Π | AuS: 2.163 | 33 | Au32S2: 467.8 (σu,9), 424.6(σg,0), 90(πu,0 × 2) |
| SAuS: 180 | 32SAu34S: 463.3 (σ,9), 417.6(σ,0.1), 89(π,0 × 2) | |||
| Au34S2: 457.3 (σu,9), 412.0(σg,0), 88(πu,0 × 2) | ||||
| AuSS− | 1A′ | AuS: 2.351 | − 49 | Au32S32S: 541.7(a′,118), 283.9(a′,5), 121.7(a′,2) |
| SS: 2.015 | ||||
| AuSS: 114.1 | ||||
| AuS3 | 2A | AuS: 2.570 | 0 | Au32S3: 524.6 (61), 513.5(4), 312.3(0), three real |
| SS: 2.015 | Au34S3: 509.0 (57), 498.2(4), 303.1(0), three real | |||
| SSS: 108.5 | ||||
| SAuS:79.5 | ||||
| AuS4 | 2A | AuS: 2.38 | 0 | Au32S4: 517.1 (5), 513.0(77), 327.8(4), six real |
| SS: 2.01, 2.18 | Au34S4: 501.7 (5), 497.7(73), 318.0(4), six real | |||
| SSS: 116, SAuS:112.4 | ||||
| SSAuSS | 2B1 | AuS: 2.305 | 12 | Au32S4: 625.4 (1), 596.4 (427), 328.7(0), six real |
| SS: 1.948 | Au34S4: 606.7 (1), 578.7 (402), 323.9(0), six real | |||
| AgSS: 111.7 | ||||
| SAuS: 176 | ||||
CuS2
The sharp doublet at 508.7, 504.8 cm−1 increases substantially on ultraviolet irradiation and exhibits relative intensities appropriate for copper 63 and 65 isotopes in natural abundance.28 The bands increase slightly on annealing to 35 K as matrix sites coalesce. This doublet shifts to 501.3, 497.3 cm−1 with sulfur-34 and the sulfur 32/34 isotopic frequency ratios for the two copper isotopes, 1.0148 and 1.0151, are substantially lower than the above ratio for the S–S stretching mode, and these low ratios and the copper isotopic splitting both indicate substantial copper participation in the vibrational mode. Although there is some band overlap, ultraviolet irradiation produces a triplet absorption at 508.7, 505.2, 501.3 cm−1 for copper-63 and the lower band at 497.3 cm−1 for copper-65. This triplet indicates the participation of two sulfur atoms, which in this case are equivalent. The central component is just 0.2 cm−1 above the median of pure isotopic values owing to slight interaction with the next lower vibrational mode in the mixed isotopic molecule of lower symmetry. Our B3LYP calculation for CuS2 finds a linear 2Π state, which is 39 kcal/mol higher in energy than the bent CuSS molecule. The analogous linear CuO2 molecule was discovered in the laser-ablated copper atom reaction with dioxygen.13 The antisymmetric S–Cu–S stretching mode is predicted at 539.5 cm−1, which is 6.0% higher than the observed value, but the calculated 32/34 and 63/65 isotopic harmonic frequency ratios, 1.0151 and 1.0080, are only slightly higher than the observed 1.0148 and 1.0077 ratios. This substantiates our identification of the linear CuS2copper disulfide molecule.CuSS−
The broader 564.6 cm−1 band is 80% destroyed by ultraviolet irradiation when the above CuS2 molecule increases more than five-fold. The sulfur-34 counterpart at 549.4 cm−1 behaves similarly and defines a 1.0277 sulfur 32/34 frequency ratio. Such behavior is expected for the CuSS− anion, and its calculated 561.6 cm−1 frequency supports this assignment.CuS3 and CuS4
The 542.1 and 520.5 cm−1 bands increase on uv irradiation and on annealing and although the lower band is complicated by matrix site splittings, each band forms a higher multiplet (five bands observed) using mixed sulfur 32,34. This indicates the involvement of three or four sulfur atoms in the vibrational mode.16Our calculations predict the lowest state of the SSCuSS molecule as 2B1 with a strong S–S stretching mode at 583 cm−1, which is clearly not appropriate for either of the above absorptions. Furthermore, the calculations find cyclic CuS4 to be 28 kcal/mol more stable with a strong fundamental vibration at 515 cm−1, and the mixed isotopic spectrum is predicted to be a broad pentet. Accordingly, the 520.5 cm−1 band is assigned to cyclic CuS4.
Analogous calculations find a stable cyclic CuS3 molecule with a strong antisymmetric S–S stretching mode at 528 cm−1. Although the S3 molecule gave a 1/2/1/1/2/1 mixed isotopic sextet,16 our calculation shows that the two central bands for CuS3 are not resolved and a pentet much like the observed spectrum is predicted. Thus, the 542.1 cm−1 band is assigned to cyclic CuS3. Structures for the coinage metal MSS, SMS, and MS4 molecules are compared in Fig. 4.
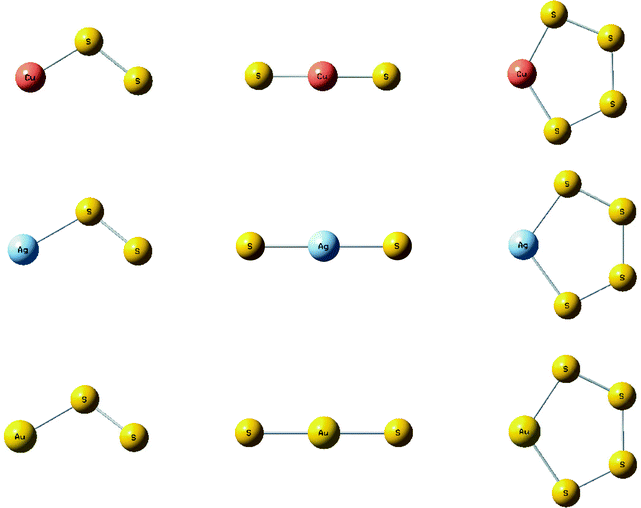 | ||
| Fig. 4 Structures calculated (B3LYP/6-311+G(3df)/SDD) for group 11 metal sulfur species. | ||
AgSS
The first silver reaction product absorptions at 582.8 and 580.6 cm−1 fall near the symmetric stretching modes of S3 split at 584.9 and 581.5 cm−1. The former bands shift to 565.7 and 563.7 cm−1 with sulfur-34 and form two triplets with mixed sulfur-32,34 whereas the S3 bands form sextets as shown in Fig. 1 where the yield is higher. B3LYP calculations find a bent 2A″ ground state for AgSS with strong 601.6 cm−1 S–S stretching fundamental, and the mixed Ag-32,34 and Ag-34,32 isotopic absorptions split by 0.3 cm−1, which is shown by broadening of the central components relative to the pure isotopic outside absorptions in the triplet patterns in Fig. 2(g).AgS3 and AgS4
The most prominent band on sample deposition at 530.3 cm−1 increases on annealing and gives way to a matrix site at 533.3 cm−1. These bands shift to 517.6 and 514.7 cm−1 with sulfur-34 and define the large sulfur 32/34 isotopic frequency ratio (1.0303 in this case), which is characteristic of an almost pure S–S stretching mode. Broad mixed isotopic pentets are observed for each mode, which demonstrates the participation of three or four sulfur atoms in the vibration.B3LYP calculations find cyclic AgS3 in the 2A ground state with a strong 537 cm−1 antisymmetric S–S stretching fundamental. The mixed isotopic spectrum is predicted to be a sextet with 2 cm−1 separation of the middle two components, which is consistent with the broad pentet structure observed here. On the basis of isotopic substitution and calculated spectra, the 530.3 cm−1 band is assigned to cyclic AgS3.
Similar calculations determine the lowest state of the SSAgSS molecule as 2B1 with a strong S–S stretching mode at 598 cm−1, which is clearly not appropriate for any band in the silver spectrum. In addition, the calculations find cyclic AgS4 to be 28 kcal/mol more stable with a strong fundamental vibration at 525 cm−1, and the mixed isotopic spectrum is in accord with a broad pentet. Thus, the 502.3 cm−1 band, which increases on annealing, is assigned to cyclic AgS4.
(Ag2S4)
Two bands appear at relatively lower frequency with silver, and these are at 476.3 and 463.2 cm−1. These bands increase slightly on full arc irradiation and they sharpen on final annealing. Silver is less reactive than Cu so aggregation of Ag might occur during the deposition and reaction process.The cyclic AgS3 anion frequencies (458, 443 cm−1) and the cyclic AgS4 anion frequencies (422, 397 cm−1) are too low, so we considered disilver species. The strongest computed frequencies (Table 2) at 472 and 469 cm−1 are in reasonable agreement with the observed bands, so this tentative assignment is proposed. It is interesting to note that the reaction of Ag and cyclic AgS4 to give cyclic Ag2S4 is 22 kcal/mol exothermic.
AuSS
The sharp 601.1 cm−1 band shifted to 583.3 cm−1 with sulfur-34, and this 32/34 isotopic frequency ratio, 1.0305, is almost that computed for a harmonic S–S stretching frequency, 1.0307, and it so identifies this vibrational mode. The bent AuSS complex in the 2A″ state is the global minimum energy species for this stoichiometry. The highest computed frequency for this product is at 627.8 cm−1 with significant intensity and shift to 609.1 cm−1 giving a 1.0307 ratio. Significantly, the Au32S34S and Au34S32S molecules are predicted to absorb at 618.6 and 618.5 cm−1, which are contained within the triplet band profile in Fig. 3. The average mixed isotopic band is 0.1 cm−1 higher than the median of pure isotopes, and the observed spectrum has the same relationship. This means that there is no nearby interacting mode, as our calculation (Table 2) indicates. Thus, the correlation between calculated and observed frequencies and isotopic characteristics substantiates the preparation and identification of the bent AuSS molecule. This neutral AuSS molecule is the upper state in the photoelectron spectrum of the AuSS− anion where a vibrational spacing of approximately 610 cm−1 was observed for the neutral molecule.10Alternatively, the side-bound 2B2 state is 29 kcal/mol higher in energy, and the weak S–S stretching mode is computed as 484 cm−1, which is not appropriate for the sharp 601.1 cm−1 absorption. However, a weak 494.4 cm−1 band, which decreases on annealing, could be due to such a Au(S2) species trapped in the matrix.
AuS2
The sharp 455.9 cm−1 band shifted to 445.6 cm−1 with sulfur-34, and this 32/34 isotopic frequency ratio, 1.0231, is quite different from the above 32/34 ratio and indicates significant participation of the gold atom in this vibrational mode. The linear 2Π state for AuS2 is 33 kcal/mol higher in energy than the bent AuSS molecule. The computed antisymmetric stretching mode has observable infrared intensity at 467.8 cm−1 and a predicted 32/34 isotopic ratio of 1.0230, both in excellent agreement with the observed values. Of more importance, the mixed isotopic triplet, which indicates the participation of two equivalent sulfur atoms, is asymmetric as the central component is 4.3 cm−1 below the pure sulfur-32 band and 6.1 cm−1 above the pure sulfur-34 absorption, and the computed 32-Au-34 band is spaced 4.5 and 6.0 cm−1 from the pure isotopic components. This arises from interaction with the nearby symmetric stretching mode, which is of the same lower symmetry in the mixed isotopic molecule, but is too weak to be detected. The excellent agreement between the observed and calculated frequencies and isotopic shifts confirms the isotopic assignments to AuS2. Finally, the symmetric stretching mode computed at 424.6 cm−1 is in excellent agreement with the observed approximately 440 cm−1 spacing for the neutral linear AuS2 molecule in the first photoelectron band for the linear AuS2− anion.10Although most of the AuS2 is formed on the initial reaction with laser ablated Au atoms and S2 molecules, full arc irradiation, Fig. 3(c), increases the higher energy AuS2 molecule at the expense of AuSS. The observation of such a stable but higher energy isomer as AuS2 relative to AuSS requires a significant energy barrier to the rearrangement of AuS2 to AuSS, which is a reasonable expectation for these very different structures. The same situation applies to CuS2.
AuS3 and AuS4
Two bands at 518.6 and 500.2 cm−1 increase on early annealing and shift to 505.1 and 487.2 cm−1 with sulfur-34 substitution. Although both bands are complicated by matrix site splittings, mixed isotopic spectra reveal broad pentets, which again shows the involvement of three or four sulfur atoms. In this case the sulfur 32/34 isotopic frequency ratio (1.0267) is not as large as that for a pure S–S stretching mode, and some mode mixing with Au–S stretching character is indicated.B3LYP calculations find cyclic AuS3 in the 2A ground state with a strong 525 cm−1 antisymmetric S–S stretching fundamental. The mixed isotopic spectrum is again predicted to be a sextet with 2 cm−1 separation of the middle two components, which is consistent with the broad pentet structure observed here for the 518.6 cm−1 band. On the basis of isotopic substitution and calculated spectra, this band is assigned to cyclic AuS3. Our calculation, however, predicts the 1.0306 sulfur 32/34 frequency ratio for a pure S–S stretching mode. We believe that the ground state involves another configuration not accounted for in the DFT calculation, which has more Au–S stretching character in the most intense vibrational mode.
Similar calculations determine the lowest state of the SSAuSS molecule as 2B1 with a strong S–S stretching mode at 596 cm−1, and no such band is observed in the gold spectrum. In addition, the calculations find cyclic AuS4 to be 12 kcal/mol more stable with a strong fundamental vibration at 513 cm−1, and the mixed isotopic spectrum is in accord with a broad pentet. Thus, the 500.2 cm−1 band, which increases on annealing, is assigned to cyclic AuS4.
Comparison of O2 and S2 reaction products
The first reaction products to be considered, the linear CuS2 and AuS2 disulfides, are analogous to the linear doublet state CuO2 and AuO2 dioxides, which were first observed by matrix isolation spectroscopy11–13 and later in the photoelectron spectra of the corresponding anions.10, 14 It is noteworthy that linear AgO2 and AgS2 are both higher energy species in quartet ground states and are not observed in these experiments.It is interesting to note that CuO2 is only 14 kcal/mol higher in energy than CuOO, based on small basis set B3LYP calculations, and both isomers were observed on deposition of laser ablated Cu and dioxygen.13 However, we find CuS2 to be 39 kcal/mol higher energy than CuSS, and relatively less CuS2 is produced on sample deposition. As a consequence, the CuO2− and CuSS− anion isomers are observed in the initial matrix spectra. Ultraviolet irradiation leads to a pronounced decrease of CuSS− and increase in the CuS2 infrared absorptions in these experiments.
Next the OOCuOO complex was a major product in dioxygen experiments, but the more stable cyclic CuS4 isomer was instead observed in the present sulfur system. The well-known ability of sulfur to form cyclic species is also apparent here.3
The spectra of silver reaction products are similar in that the major new species are the AgOO, AgO3, AgSS, and AgS3 molecules.12 No anion was detected in the silver sulfur system.
Again, the major gold reaction products are AuOO, AuO2, AuSS, and AuS2. The disulfide is calculated to be 33 kcal/mol higher than AuSS whereas the dioxide is 51 kcal/mol above AuOO, and this is manifested in the relative product yields. We observed stronger AuS2 relative to AuSS than the found analogous dioxygen species. Absorptions are also observed for AuS3 and AuS4 again showing the catenation property of sulfur. Wang et al point out that the observation of stronger AuS bonds relative to AuO bonds is due to the less antibonding nature of the HOMO's and that these bonds are strongly covalent with multiple bond characters due to Au 5d orbital bonding as result of relativistic effects.10,31 Some evidence for this is found in the shorter calculated distance in AuS2 (2.163 Å) as compared to the sum of the covalent radii (2.18 Å).32
Recent gas phase photoelectron spectra of gold anion clusters identified all of the above molecules save AuOO because the Au(O2)− anion precursor is not bound, which is in contrast to the Au(S2)− anion where bonding arises from strong Au 6 s and S2π* interactions.10 However, the Au(O2)2− anion appears to be stable based on the neon matrix spectrum. The neutral AuOO molecule exhibited a large neon–argon matrix shift, which suggests a weak, medium dependent Au–O2 interaction.12 Finally, gas phase investigations of copper sulfide anion clusters are expected to find stable CuSS and CuS2 anions and neutral molecules based on the present work. Analogous silver oxide and sulfide anion investigations should give rise to AgOO and AgSS, based on the observation that AgOO− anion and AgOO neutral complex in solid neon12 and the present observation of AgSS and the calculation of a stable AgSS− anion.
We note that the 2A cyclic MS4 molecules are more stable than the open 2B1 SSMSS molecules. Computed Mulliken charges suggest that the Ag and Au species are more ionic than the Cu derivatives (Table 3). The spin densities are spread more evenly on the four sulfur atoms in the cyclic species than in the open isomers, which is a manifestation of the well-known tendency of sulfur to form rings.3 In contrast the group 10 MS4 molecules form coplanar C2v M(S2)2 structures.20
| S″S′CuS′S″ | S″S′AgS′S″ | S″S′AuS′S″ | |
|---|---|---|---|
| q(M, S′,S″) | −0.33, 0.27, −0.10 | 0.88, −0.35, −0.09 | 0.67, −0.27, −0.07 |
| sd(M, S′,S″) | −0.07, 0.10, 0.44 | −0.01, 0.13, 0.37 | −0.05, 0.10, 0.42 |
| CuS2′S2″ | AgS2′S2″ | AuS2′S2″ | |
|---|---|---|---|
| a Calculations used B3LYP/6-311+G(3df) for S and SDD for Cu, Ag and Au. | |||
| q(M, S′, S″) | 0.02, −0.04, 0.03 | 0.70, −0.37, 0.02 | 0.53, −0.29, 0.02 |
| sd(M, S′, S″) | 0.02, 0.22, 0.27 | 0.00, 0.21, 0.29 | 0.01, 0.22, 0.28 |
Conclusions
Reactions of laser ablated coinage metal atoms with disulfur produced the CuSS, CuS2, AgSS, AuSS, and AuS2 molecules, which are analogous to products observed in the dioxygen systems. The higher energy copper and gold disulfide molecules are also formed on photochemical rearrangement of the disulfur complexes, but the still higher energy silver disulfide molecule is not produced. The same linear 2Π AuS2 and bent 2A″ AuSS molecules have been observed in the photoelectron spectrum of AuS2− anions.10The major difference in products between oxygen and sulfur is the observation of MS3 molecules owing to the presence of S3 in the reaction mixture16 and the cyclic MS4 structures derived from the tendency of sulfur to form cyclic species. The cyclic coinage metal MS4 molecules are 12–28 kcal/mol more stable than the open SSMSS isomers.
Acknowledgements
We gratefully acknowledge financial support from NSF Grant CHE 00–78836 and an NCSA computing Grant No. CHE07–0004N to L. A.References
- Transition Metal Sulfur Chemistry, Biological and Industrial Significance, ed. E. I. Stiefel and K. Matsumoto, American Chemical Society, Washington, 1997 Search PubMed.
- (a) A. F. Wells, Structural Inorganic Chemistry, 4th edn., Clarendon Press, Oxford, 1975 Search PubMed; (b) C. Sourisseau, R. Cavagnat and M. Fouassier, J. Phys. Chem. Solids, 1991, 52, 537 CrossRef CAS.
- F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic Chemistry, 6th edn. Wiley, New York, 1999 Search PubMed.
- T. A. Bither, R. J. Bouchard, W. H. Cloud, P. C. Donahue and W. J. Siemons, Inorg. Chem., 1968, 47, 2208 CrossRef.
- M. Kontani, T. Tutui, T. Moriwakat and T. Mizukoshi, Physica B, 2000, 284, 675 CrossRef.
- H. Ueda, M. Nohara, K. Kitazawa, H. Takagi, A. Fujimori, T. Mizokawa and T. Yagi, Phys. Rev. B, 2002, 65, 155104 CrossRef.
- Z. F. Hou, A. Y. Li, Z. Z. Zhu and M. C. Huang, J. Mat. Sci. Tech. B, 2004, 20, 429 Search PubMed.
- (a) D. E. Tevault, J. Chem. Phys., 1982, 76, 2859 CrossRef CAS; (b) G. A. Ozin, S. A. Mitchell and J. Garcia-Prieto, J. Am. Chem. Soc., 1983, 105, 6399 CrossRef CAS; G. A. Ozin, S. A. Mitchell and J. Garcia-Prieto, J. Phys. Chem., 1982, 86, 473 CrossRef CAS; (c) D. McIntosh and G. A. Ozin, Inorg. Chem, 1976, 15, 2869 CrossRef CAS; D. McIntosh and G. A. Ozin, Inorg. Chem, 1977, 16, 59 CrossRef CAS , (Ag, Au + O2).
- (a) P. H. Kasai and P. M. Jones, J. Phys. Chem, 1986, 90, 4239 CrossRef CAS; (b) J. A. Howard, R. Sutcliffe and B. Mile, J. Phys. Chem, 1984, 88, 4351 CrossRef CAS.
- H.-J. Zhai, C. Burgel, V. Bonacic-Koutecky and L.-S. Wang, J. Am. Chem. Soc., 2008, 130, 9156 CrossRef CAS.
- A. Citra and L. Andrews, J. Mol. Struct., 1999, 189, 95 and unpublished results (Ag, Au + O2).
- X. Wang and L. Andrews, J. Phys. Chem. A, 2001, 105, 5812 CrossRef CAS (Ag, Au + O2 in neon).
- G. V. Chertihin, L. Andrews and C. W. Bauschlicher, Jr., J. Phys. Chem. A, 1997, 101, 4026 CrossRef CAS (Cu + O2 in argon).
- H. Wu, S. R. Desai and L.-S. Wang, J. Phys. Chem. A, 1997, 101, 2103 CrossRef CAS.
- (a) M. Zhou and L. Andrews, J. Chem. Phys., 1999, 111, 4548 CrossRef CAS (Cu + CO in neon); (b) B. Liang and L. Andrews, J. Phys. Chem. A, 2000, 104, 9156 CrossRef CAS (Ag, Au + CO in neon).
- G. D. Brabson, Z. Mielke and L. Andrews, J. Phys. Chem., 1991, 95, 79 CrossRef CAS.
- B. Liang and L. Andrews, J. Phys. Chem. A, 2002, 106, 3738 CrossRef CAS (V, Nb, Ta + S2).
- B. Liang and L. Andrews, J. Phys. Chem. A, 2002, 106, 6295 CrossRef CAS (Ti, Zr, Hf + S2).
- B. Liang and L. Andrews, J. Phys. Chem. A, 2002, 106, 6945 CrossRef CAS (Cr, Mo, W + S2).
- B. Liang, X. Wang and L. Andrews, J. Phys. Chem. A, 2009, 113, 3336 CrossRef CAS (Ni, Pd, Pt + S2).
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS.
- (a) L. Andrews and A. Citra, Chem. Rev, 2002, 102, 885 CrossRef CAS , and references therein; (b) L. Andrews and H.-G. Cho, Organometallics, 2006, 25, 4040 CrossRef CAS , and references therein (Review article).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, Y. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062 CrossRef.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CAS.
- (a) S. R. Long and G. C. Pimentel, J. Chem. Phys., 1977, 66, 2219 CrossRef CAS; (b) R. R. Smardzewski, J. Chem. Phys., 1978, 68, 2878 CrossRef.
- The natural abundances of 63Cu and 65Cu are 69.17% and 30.83%. CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 1985 Search PubMed.
- L. Andrews and H.-G. Cho, Organometallics, 2006, 25, 4040 CrossRef CAS , and references therein (Review article).
- J. T. Lyon, H.-G. Cho and L. Andrews, Organometallics, 2007, 26, 6373 CrossRef CAS (Cr, Mo, W + CHX3, CX4).
- P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef CAS.
- P. Pyykkö, S. Riedel and M. Patzschke, Chem.–Eur. J., 2005, 12, 3511 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |