Investigations using fluorescent ligands to monitor platinum(IV) reduction and platinum(II) reactions in cancer cells
Elizabeth J.
New
,
Ran
Duan
,
Jenny Z.
Zhang
and
Trevor W.
Hambley
*
School of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: t.hambley@chem.usyd.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 3320
First published on 6th March 2009
Abstract
Coordination of the aniline containing fluorophores, coumarin 120 (C120) and coumarin 151 (C151) at the non-leaving group positions of cisplatin analogues (giving cis-[PtCl2(C120)(NH3)] and cis-[PtCl2(C151)(NH3)]) resulted in partial and complete quenching of the fluorescence, respectively. Oxidation of the coumarin 120 complex to the Pt(IV) form (cis,trans,cis-[PtCl2(OH)2(C120)(NH3)]) resulted in further quenching compared to that seen for the Pt(II) complex. The fluorescence profiles of these coumarin complexes were collected to evaluate their suitability for studying the metabolism of cisplatin-based anticancer drugs. C151 has the more suitable profile with a lower energy excitation peak and a better separation between the excitation and emission spectra. The complete damping of fluorescence on coordination to Pt(II) makes it unsuitable for monitoring the reduction process, but does allow it to be used to monitor loss of the aniline type ligand. All of the coumarin complexes revealed moderate cyotoxcities in the range 10–22 μM indicating that they are suitable models of anticancer agents. DNA dampens the fluorescence of both Pt(II) complexes and that of C120 has a much higher DNA binding affinity (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1) than does the complex of C151 (300 M−1). Treatment of A2780 human ovarian carcinoma cells with the Pt-coumarin complexes resulted in fluorescence visible by confocal microscopy, and co-localisation studies with organelle specific dyes suggest they are concentrated in the late endosomes or lysosomes. Cells treated with the Pt(IV) complex of C120 revealed strong fluorescence and a somewhat different distribution to cells treated with the Pt(II) complex indicating reduction following uptake.
000 M−1) than does the complex of C151 (300 M−1). Treatment of A2780 human ovarian carcinoma cells with the Pt-coumarin complexes resulted in fluorescence visible by confocal microscopy, and co-localisation studies with organelle specific dyes suggest they are concentrated in the late endosomes or lysosomes. Cells treated with the Pt(IV) complex of C120 revealed strong fluorescence and a somewhat different distribution to cells treated with the Pt(II) complex indicating reduction following uptake.
Introduction
The earliest investigations of platinum compounds for their antitumour activity by Barnett Rosenberg and his group1–3 identified both Pt(II) and Pt(IV) complexes as active. However, it was the Pt(II) drug cisplatin that was initially studied more extensively. Recent work has seen a return to Pt(IV) complexes in an attempt to circumvent the limitations of cisplatin-based drugs which lie in their toxicity4,5 and tumour resistance.6–10 A number of anticancer active Pt(IV) complexes have been developed, including ormaplatin, or tetraplatin,11iproplatin,12,13 and JM216 (satraplatin).14 In general, Pt(IV) complexes are octahedral, while Pt(II) complexes exhibit a square planar ligand arrangement.15Reduction of the complex from Pt(IV) to the Pt(II) state is therefore accompanied by loss of the axial ligands.†Pt(IV) drugs are believed to be first reduced to Pt(II), whether intra- or extra-cellularly, before binding to proteins and DNA16 and acting in a manner similar to their Pt(II) counterparts. The potential advantages of this class of drugs lie in their activity coupled with a low toxicity while in the Pt(IV) state,17 their activity in the treatment of cisplatin-resistant tumours,14,18,19 and their potential for oral administration due to their kinetic inertness and often higher lipophilicity. In order to harness such properties, a greater understanding of the mode of action of Pt(IV) complexes is required and, in particular, of the mechanisms by which the complexes enter the cell and undergo reduction and metabolism.
Previous studies in our group have utilised X-ray fluorescence techniques such as synchrotron radiation-induced X-ray emission (SRIXE), and X-ray absorption near edge spectroscopy (XANES) to reveal subcellular localisation and reduction.20 While providing some valuable information, such techniques are limited in their resolution, particularly by the limiting probe size. Another technique that has been widely used for the study of platinum complexes is fluorescence microscopy.21–29 While providing good resolution and allowing for clear identification of subcellular organelles, these fluorescence microscopy studies did not give any information about oxidation or coordination state.
Here, we describe work aimed at assessing and utilising coordination and oxidation state dependent fluorescence as a technique for studying drug metabolism. It has been observed that the binding of a number of transition metals to a fluorophore can either increase or decrease its fluorescence properties.30,31 While neither Pt(II) nor Pt(IV) have been reported to induce such fluorescence changes, the results from other d8 and d6 ions are encouraging.32–36 It was hypothesized that changes in fluorescence induced by ligand loss and/or changes in oxidation state could be utilised in microscopy studies to observe the temporal and physical location of these key events.
Results and discussion
Synthesis
The rationale of the present study was to synthesise Pt(II) and Pt(IV) analogues of cisplatin in which a ligand contains a fluorescent moiety. It was anticipated that the fluorescence of this group would be quenched upon coordination or upon oxidation, with the result that the presence or absence of fluorescence would be a marker of metabolic events within the cell such as reduction and/or ligand loss. In the present study, the amine (non-leaving) group of cisplatin was targeted.Cisplatin-based Pt(II) and Pt(IV) complexes were synthesised containing a fluorescent coumarin moiety in place of one of the ammine groups (3–5, Scheme 1). The coumarins used were the aniline group-containing compounds, coumarin 120 (C120) and coumarin 151 (C151). The analogous aniline complexes (1 and 2) were also synthesised for comparison.
 | ||
| Scheme 1 | ||
Pt(II) complexes were synthesised by treatment of the amminetrichloroplatinate(II) anion with aniline or the respective coumarin, to give complexes 1, 3 and 5. Oxidation of these Pt(II) complexes to the Pt(IV) dihydroxo forms was less straight-forward as it was necessary to avoid simultaneous oxidation of the ligand. Heating with peroxide for a short period of time led to successful oxidation of 1 and 3, but was unsuccessful for 5. Oxidation of 5 in other solvents resulted in ligand release and oxidation, suggesting weaker binding of this ligand. This is consistent with the greater electron-withdrawing ability of the trifluoromethyl group, which is expected to result in weaker basicity of the amine group.
Fluorescence properties
The aim of this work was to synthesise complexes suitable for study in cells by fluorescence confocal microscopy. Excitation and emission spectra of C120 and C151 (Fig. 1) show that C151 is a more suitable fluorophore for this purpose, as there is greater excitation at 400 nm, the typical minimum excitation wavelength for single-photon confocal microscopy experiments.37 In addition, the overlap of the excitation and emission spectra of C151 is less than for the spectra of C120. This is an advantage for fluorescence microscopy, where overlap of spectra results in excitation light interfering with the collection of the emission image, decreasing the image contrast.37 | ||
| Fig. 1 Fluorescence excitation (bold lines) and emission (fine lines; excitation at 370 nm) spectra of 0.1 mM solutions of C120 (solid) and C151 (dashed) in DMF. | ||
These differences in excitation and emission energies can be understood by a consideration of the electronics of the systems. The general consensus from theoretical studies of 7-aminocoumarins is that the first three (longest wavelength) excitation peaks result from π–π* transitions38–40 while a fourth, smaller peak at higher energy can be attributed to an n–π* transition.38,41 The π–π* transitions will primarily be considered and various studies have suggested that the excited π* energy levels arise from tautomerisation of the coumarin.42,43 From studying the energy changes that resulted from changing the substituents of 7-aminocoumarins, it was concluded that the longest wavelength singlet–singlet transition (S0–S1) results from an intramolecular charge transfer from a π orbital associated with the benzene ring and nitrogen atom to a π orbital of the pyrone ring and carbonyl group. There is no similarly clear definition of the structures of S2 and S3, but they are believed to differ in charge distribution and dipole moments.44 The electronic structure of the S1 state is of greatest interest as the S0–S1transition gives rise to the highest wavelength excitation absorption, which is the one that will be excited in confocal microscopy and is the primary contributor to the emission.
The longer excitation wavelengths of C151 compared to C120 represent an elevation of the S1 energy compared to the S0 level. This can be explained by the fact that replacement of the methyl group in C120 with the trifluoromethyl in C151 results in decreased electron density in the pyrone ring, thus decreasing the stability of the excited state.
On binding of C120 to Pt(II) (3; Fig. 2a), a 2.5-fold quenching of fluorescence was observed, with a further 7-fold decrease on oxidation of the complex to Pt(IV) (4). This conclusively demonstrates that binding of platinum to a fluorophore results in fluorescence quenching. The effect is considerably more marked for C151 (Fig. 2b), where the Pt(II) complex 5 shows negligible fluorescence. There was no such decrease in fluorescence in solutions containing the Pt(II) or Pt(IV) ions and unbound coumarins, confirming that this quenching is a coordination-mediated event.
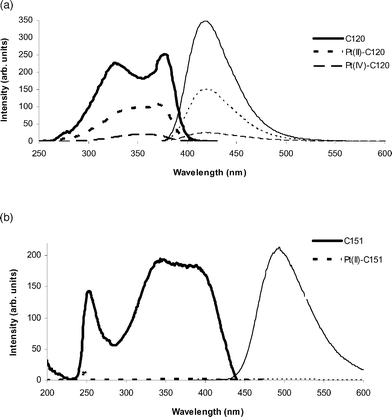 | ||
| Fig. 2 Excitation (heavy lines) and emission (fine lines; excitation at 370 nm) spectra of coumarins and complexes: (a) C120 (solid), 3 (short dashes) and 4 (long dashes); (b) C151 (solid) and 5 (dashes). | ||
The C120 system is likely to be of greater utility in studying the reduction event, as the fluorescence change on oxidation from 3 to 4 is greater than on binding of the coumarin. The Pt(IV) complex 4 exhibits considerable fluorescence, and both complexes should be visible by fluorescence microscopy. However, use of an excitation wavelength in the 380–400 nm range should allow for reduction to be monitored. The shape of the excitation spectrum of the coumarin changes substantially on coordination to Pt(II) and further on oxidation to Pt(IV). Coordination leads to the two primary peaks moving closer together in wavelength and oxidation increases this effect to the point where the peaks are indistinguishable or results in almost complete loss of one of the peaks. As a result negligible excitation of 4 is observed when using excitation wavelengths longer than 380 nm. The C151 system, with the almost complete quenching of fluorescence on binding, would allow for greater differentiation of coordination by microscopy. However, successful oxidation would be likely to yield a similarly non-fluorescent Pt(IV) form, so no information about the reduction event could be obtained.
Given the differences in fluorescence intensities observed for Pt(II) and Pt(IV) complexes, a solution of 4 that had been treated with the cellular reductant ascorbate was monitored over a period of 2 d to observe whether the reduction of the complex to 3 resulted in a concomitant increase in fluorescence. A slight but steady increase in fluorescence was observed over this time (Fig. 3). A solution of 4 alone showed no fluorescence changes over the 2 d period, implicating a reduction event or ligand exchange as being responsible for the fluorescence increase. The observed slow increase in fluorescence suggests slow reduction of the Pt(IV) complex and indicates that it will be resistant to reduction in the medium prior to cellular uptake. This is consistent with the measured negative reduction potential of 4 (−513 mV vs.NHE) and with observations that trans-Pt(IV)-dihydroxo complexes are particularly resistant to reduction in comparison to Pt(IV) complexes with other axial ligands.45,46
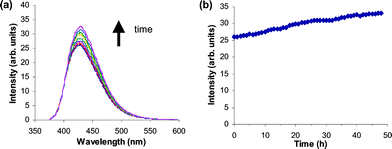 | ||
| Fig. 3 Fluorescence changes following treatment of 4 with ascorbic acid. (a) Emission spectra (with excitation at 370 nm) of 4 collected at various intervals following the addition of ascorbic acid (10-fold excess). (b) Plot of fluorescence intensity at maxima in emission spectra following the addition of ascorbic acid. | ||
Cytotoxicity
The cytotoxicities of the ligands and complexes were determined using the spectrofluorimetric MTT assay. The concentrations of complexes and ligands that inhibited cloned cell survival by 50% (IC50) are shown in Table 1.| Complex/ligand | IC50 (μM) ± S.D.a |
|---|---|
| a Values are means of data from three independent experiments with quadruplicate readings in each experiment. | |
| Cisplatin | 3.2 ± 0.1 |
| cis,trans,cis-[PtCl2(OH)2(NH3)2] | 17.0 ± 2.4 |
| Aniline | 69 ± 26 |
| 1 | 9.3 ± 3.1 |
| 2 | 15.3 ± 4.3 |
| C120 | 250 ± 47 |
| 3 | 21.6 ± 0.6 |
| 4 | 21.2 ± 1.8 |
| C151 | 14.1 ± 0.4 |
| 5 | 12.1 ± 0.6 |
Interestingly, while cisplatin has a 5-fold greater cytotoxicity than its Pt(IV) dihydroxo analogue, cis,trans,cis-[PtCl2(OH)2(NH3)2], there is very little difference between the cytotoxicities of the Pt(II) and Pt(IV) complexes of aniline and C120. This may indicate that the Pt(IV) complexes are being rapidly reduced in vivo to their Pt(II) forms. An increased reduction rate is consistent with the fact that 2 (−568 mV vs.NHE) and 4 (−513 mV vs.NHE) have less negative reduction potentials than cis,trans,cis-[PtCl2(OH)2(NH3)2] (−660 mV vs.NHE). Another possible explanation is that these Pt(IV) complexes are acting via an alternative mechanism, with comparable overall efficacy. Also, a greater cellular uptake of the Pt(IV) complexes 2 and 4 could contribute to the higher than expected cytotoxicity.
While the primary aim of this investigation was to attach a fluorophore for use in microscopy, the Pt-C120 complex was expected to have antitumour activity in its own right, based on the high activity of similar ammine/amine platinum(II) and platinum(IV) complexes.9,47 The fact that these complexes do indeed exhibit cytotoxicity confirms that they are meaningful models of active anticancer complexes. If, for example, a much lower cytotoxicity was observed, any subsequent conclusions about the cellular location of the complex would be less applicable to other complexes with known anticancer activity.
The results indicate that, while they are cytotoxic, 1 and 3 are less cytotoxic than cisplatin, in accord with previous studies, which have demonstrated that substitution of one or both ammine ligands leads to decreased cytotoxicity.48,49 The high cytotoxicity of C151 compared to that of C120 is somewhat surprising, but may be accounted for by the higher reactivity resulting from the presence of the trifluoromethyl group.
Confocal microscopy
Cells that had been treated with ligands and complexes were visualised by confocal fluorescence microscopy, with excitation at 405 nm. As shown in Fig. 4, incubation of A2780 cells with C120, C151 and their complexes resulted in readily-detectable fluorescence. From these images, there appears to be no fluorescence in the nucleus, with fluorescence possibly localised in other organelles in the cytoplasm. When optical sections throughout the cell were taken, this fluorescence could be observed in most layers. As expected, untreated control cells showed no fluorescence. | ||
| Fig. 4 Confocal microscope images of A2780 cells treated for 4 h with 20 μM: (a) C120, (b) 3, (c) 4, (d) C151 and (e) 5. The brightness and contrast of the image have been manually increased. Scale bars represent 10 μm. | ||
The fluorescence observed in cells treated with 3 may be due to this complex or to free C120. However, the C120 is attached to one of the non-leaving group positions of the complex and the nature of the fluorescence is different from that in cells treated with C120, particularly in regard to the speed of photo-bleaching. Therefore, it is most likely to be due predominantly to 3. At longer time points, and following binding to thiol containing biomolecules, it is to be expected that substantial amounts of C120 will be released from 3.
The fluorescence observed in cells treated with 4 (Fig. 4(c)) is most unlikely to be due to the Pt(IV) complex (4) because of its very low fluorescence intensity when excited at 405 nm (Fig. 3). Thus, the observation of substantial fluorescence is consistent with the rapid reduction of various Pt(IV) complexes in vitro, regardless of their reduction potentials, with reduction of cis,trans,cis-[PtCl2(OH)2(NH3)2] being complete within 4 h.50
In comparison to the C120 complex (3), the fluorescence of 5 is negligible (Fig. 2(b)), and the fact that fluorescence is observed in cells treated with 5 (Fig. 4e) is most consistent with metabolism of the complex to yield the free ligand. While cells treated with 3 clearly show organelle localised fluorescence, in cells incubated with 5 the fluorescence is more generally distributed throughout the cytoplasm and is indistinguishable from that produced by C151, consistent with rapid loss of this ligand. These contrasting observations for 3 and 5 are consistent with the lower stability of the latter encountered in the synthetic work.
To reveal the subcellular location of the complexes, cells were treated with nuclear or lysosomal stains following incubation with C120 and its complexes. Examination of these images reveals that the localisation of the Pt(II) and Pt(IV) complexes is similar in most respects. Images taken from cells treated with Pt(II)-C120 or Pt(IV)-C120 and the syto21 nuclear stain (Fig. 5) reveal an absence of fluorescence arising from the complexes in the nucleus. This does not preclude the possibility that either complex is present in the nucleus, as the fluorophore could interact with DNA, resulting in quenching of its fluorescence or shifting of the fluorescence out of the range being measured. Cells were also incubated with both C120 and the syto21 nuclear stain to determine the localisation of the fluorophore alone. These images suggest that the ligand localises similarly to the complexes, with no observable fluorescence in the nucleus.
 | ||
| Fig. 5 Confocal microscope images of A2780 cells treated with complexes, C120 and nuclear stain. A2780 cells under confocal fluorescence microscope 4 h after treatment with (a) C120, (b) 3 and (c) 4 with 10 min staining with syto21 nuclear stain. Part (i) shows image collected through blue emission filter (C120 and Pt complexes), (ii) image through green emission filter (syto21) and (iii) superposition of blue and green images. The brightness and contrast of the image have been manually increased. Scale bars represent 10 μm. | ||
The green-blue colour observable in the superposition of images obtained from co-staining with the complexes and the LysoTracker Green lysosomal stain (Fig. 6) indicates co-localisation. However, the blue fluorescence of the complexes is not restricted to the regions showing green fluorescence, indicating that the complexes have entered the lysosomes, but are also present elsewhere in the cytoplasm, or in other organelles. In general, the Pt(II) treated cells show more of the fluorescence localised in punctuate spots corresponding to the lysosomes, whereas Pt(IV) treated cells have a more diffuse distribution of the fluorescence. In some cells, particularly noticeable in Fig. 6(c), the green fluorescence does not correspond to distinct “spots” indicating lysosomes, but appears to be relatively evenly spread throughout the cytoplasm. This indicates that the stain was not completely specific for the lysosomes. Since the stain has high specificity for acidic organelles51 there could have been an aberration in the cells due either to the nature of the cell line or to a generalised decrease in pH within the cytoplasm resulting from treatment with the complex. Given that the effect was most noticeable in Pt(IV)-treated cells, it is more likely to be a result of treatment with the complex. If the Pt(IV) complex is able to either avoid lysosomal uptake or contribute to lysosomal destruction more so than its Pt(II) counterpart, this could contribute to its higher than expected relative cytotoxicity.
 | ||
| Fig. 6 Confocal microscope images of A2780 cells treated with complexes and lysosomal stain. (a) Two A2780 cells and (b) multiple cells after 4 h treatment with 3; (c) two cells and (d) multiple cells after four h treatment with 4. All cells were treated for 1.5 h with LysoTracker Green lysosomal stain. Part (i) shows image collected through blue emission filter (Pt complexes), (ii) image through green emission filter (LysoTracker) and (iii) superposition of blue and green images. Blue-green colour indicates co-localisation. The brightness and contrast of the image have been manually increased. Scale bar represents 10 μm. | ||
It is interesting to compare the localisation results of this experiment with the results of studies that determined the cellular localisation by other means, to establish whether attaching a coumarin moiety has affected the subcellular distribution of the complex. X-Ray imaging of cisplatin in bone marrow cells indicated that most drug was present in the cytoplasm.52 Later electron microscopic analysis of cisplatin in A2780 cells, making use of the electron-dense nature of platinum, detected platinum on the plasma membrane and the nuclear envelope as well as in the nucleus and cytoplasm, but there was no platinum within other organelles.53 These results differ considerably from the findings of the current work, in which the platinum complexes have been found to localise in the lysosomes and perhaps in other organelles, or in the cytoplasm. This suggests that the presence of the coumarin does affect the localisation of the complex. This localisation pattern is similar to those of other platinum–fluorophore complexes.24,27 While this limits the usefulness of C120- and C151-based complexes in providing information about the localisation and ligand exchange of cisplatin itself, there is still much to be learnt about the bioaccumulation and metabolism of other classes of platinum complexes.
If the complexes are not reaching the nucleus, they must be killing the cells by an alternative mechanism, since cytotoxicity data (Table 1) confirmed their role in inducing cell death. One possible mechanism is the disruption of lysosomes, which has long been identified as a trigger for cell death,54 and it is worth noting in this context that the great bulk of the platinum that enters the cell does not reach the nucleus.55 More recently it has been found that disruption of lysosomes and the resultant release of their acidic contents into the cytoplasm causes disruption of the plasma membrane, and thus the death of the cell.56 Lysosomal disruption is a putative cause of the nephrotoxicity of cisplatin57 and has also been identified as a necrosis-inducing pathway triggered for other platinum complexes.24 As mentioned, such lysosomally-induced apoptosis could be an explanation for the unusually high cytotoxicity of 4 and would also contribute to the more diffuse fluorescence distribution.
In addition to gaining an understanding of the cellular localisation of the complexes, these results allow a comparison between the Pt(II) and Pt(IV) complexes. While they appear to have similar localisation, the Pt(IV) complex gave rise to more fluorescence than its Pt(II) counterpart. This difference can be approximately quantified by comparing the photomultiplier tube voltage levels required to give comparable images; for Pt(II)-C120 the voltage was set at the highest possible value while for Pt(IV)-C120 it was set at approximately 93–95% of maximum. This could be due to a higher cellular accumulation of the Pt(IV) complex, a hypothesis that would have to be confirmed by performing cellular uptake studies since uptake of Pt(IV) complexes is generally lower than that of their Pt(II) analogues.58 It was also noticed that the fluorescence of cells treated with uncomplexed C120 was weaker still than that of the Pt(II) complex, which could be due to greater fluorescence bleaching, or to decreased cellular uptake.
DNA-binding studies
The effect of DNA-binding on coumarin fluorescence was evaluated as an explanation for the observed lack of fluorescence in the nucleus. Titration of DNA into a solution of coumarin or complex resulted in a decrease in fluorescence intensity across the spectrum as shown in Fig. 7.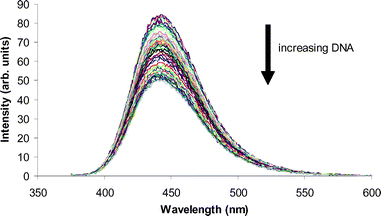 | ||
| Fig. 7 Changes in the fluorescence emission spectrum of 3 on the addition of DNA (λex = 370 nm). | ||
The addition of DNA resulted in a linear decrease in the fluorescence of all solutions tested, demonstrating that DNA quenches the fluorescence of these systems. While quenching was incomplete under the conditions studied, it is feasible that extrapolation to the higher DNA concentrations found in the nucleus would result in complete quenching of fluorescence. Therefore, the absence of fluorescence in the nucleus might be explained by association with DNA.
In addition, there was a dramatic difference between the DNA binding affinities of the coumarins, which were calculated to be 300 for C151 and 10000 for C120. These values suggest that the addition of the trifluoromethyl group interferes with the DNA binding ability of the coumarin. The fact that no shift in the position of the fluorescence maximum of C151 was observed on addition of DNA is consistent with non-intercalation. The fact that C151 is more cytotoxic than C120 (Table 1), despite this apparent lack of interaction with DNA suggests that it may be acting by an alternative mechanism of toxicity.
Conclusions
Our studies confirm that coordination-sensitive fluorescence is a valuable technique for the study of the cellular metabolism of platinum complexes. It was hoped that a Pt(II) complex with a fluorophore in the non-leaving group position would show appreciable fluorescence that would be quenched on oxidation to Pt(IV). In this way, the appearance of fluorescence would indicate that the reduction event had occurred. While this was the case for the C120 complexes, the use of C151 in an attempt to optimise the fluorescence properties of the fluorophore resulted in almost complete quenching of fluorescence on coordination to Pt(II). Even if the Pt(II) complex could be successfully oxidised, it is unlikely that this would result in further useful fluorescence changes. Rather than providing information about the reduction event, therefore, the C151 complex can be used to study loss of the amine ligand and this is seen to be substantial. The C120 complexes provided valuable insights into differences between the uptake of the Pt(II) and Pt(IV) complexes and suggested that the Pt(IV) complex interacts differently with the lysosomes. However, a coumarin with fluorescence properties intermediate between those of C120 and C151 may allow an even clearer distinction between the Pt(II) and Pt(IV) states, enabling the reduction process to be followed in live cells.Methods and instrumentation
Reagents
All solvents used were laboratory grade and were used without further purification unless otherwise stated. Silver nitrate was obtained from Merck. Hydrogen peroxide (30% v/v) was obtained from Ajax Chemicals and stored in the dark at 5 °C. The Advanced DMEM for cellular studies was sourced from Gibco, and Leibovitz's L-15 Medium from Invitrogen. Tetrapropylammonium perchlorate was obtained from the Eastman Kodak Company. Cisplatin, (SP-4-2)-diamminedichloroplatinum(II), was synthesised from K2PtCl4 obtained from Aithaca Chemical Corporation. All other reagents were acquired from Sigma Aldrich. For cytotoxicity and confocal studies, the cell line used was the human ovarian carcinoma cell line A2780. Cells were maintained in exponential growth as monolayers at 37 °C in 5% CO2 in Advanced DMEM supplemented with 2.5 mM glutamine and 2% fetal calf serum.Physical measurements
Infrared spectra were collected as diffuse reflectance infrared transform spectra (DRIFTS) using a Bio-Rad FTS-40 spectrophotometer equipped with Win-IR Windows 3.1 based software. KBr was used as both the background and the matrix over the range 500–4000 cm−1. 195Pt NMR spectra were collected by Dr I. Luck using a Bruker AMX 400 MHz spectrometer. Spectra were collected at 298 K and chemical shifts were referenced to Na2[PtCl6]. 1H and 13C NMR spectra were collected at 300 K on a Bruker 300 MHz spectrometer using commercially available deuterated solvents. For spectra recorded in D2O, TSP was used as an internal standard, while for other solvents, isotopic impurities were used as internal reference signals. Microanalysis (C, H, N) was performed by Chemical and Micro Analytical Services, Belmont, Victoria and the Microanalytical Unit, Australian National University, Australian Capital Territory. Mass spectra were collected using a Finnigan LCQ MS detector. Microfiltration of aqueous solutions was achieved using a Minisart filter unit (0.2 μm pore size) with a disposable 1 mL syringe (Becton-Dickinson).Absorption spectra were collected using a Varian Cary Eclipse 1E UV-vis spectrophotometer, with a 1 cm × 1 cm silica cuvette (Starna). Scans were run on 0.03 mM DMF solutions at a scan speed of 120 nm min−1 and absorbances collected between 250–600 nm. Fluorescence measurements were performed using a Varian Cary Eclipse fluorescence spectrophotometer, with a 1 cm × 1 cm silica cuvette (Starna). Scans were run at a scan speed of 120 nm min−1 with excitation and emission slit widths of 5 nm. Excitation spectra were obtained using a 430–1100 nm emission filter. For in situreduction experiments, emission spectra were recorded at regular time intervals after the addition of ascorbic acid (ten molar equivalents, 29 mM) or cysteine (ten molar equivalents, 45 mM). Parameters were the same as described previously, and emission spectra were collected over the range 375–600 nm with excitation at 370 nm.
Electrochemical measurements were performed using a BAS 100B/W Electrochemical Analyser. Cyclic voltammagrams were collected at room temperature at a scan rate of 100 mV sec−1, using a glassy carbon working electrode, a platinum auxiliary electrode and a silver/silver chloride reference electrode. Complexes were dissolved in acetone containing 0.1 M tetrapropylammonium perchlorate to a final concentration of 1 mM. Solutions were degassed with argon for ten min prior to measurement. Ferrocene was added to each sample as an internal reference.
Reduction in situ
Emission spectra of a 0.1 mM solution of Pt(IV)-C120 in DMF were recorded at regular time intervals after the addition of ascorbic acid (ten molar equivalents, 19 mM). Parameters were the same as described previously, and emission spectra were collected over the range 375–600 nm with excitation at 370 nm.DNA fluorescence quenching and binding studies
DNA binding studies and the determination of binding constants were carried out as described previously.59 The complexes were made up in 1% DMF (C120) or 5% (C151) solutions at 20 μM concentrations and aliquots of 1.75 mM (C120) or 3.36 mM (C151) DNA were added.Cytotoxicity assays
Solutions of cisplatin, transplatin, cis-Pt(II)-azaindole, trans-Pt(II)-azaindole and 7-azaindole were prepared as 0.5 mM solutions (in 100 mM aqueous KCl) immediately prior to the assay. For the platinum complexes 1% DMF was added to ensure solubility. IC50 values were determined using the MTT assay, as described by Carmichael et al.,60 which makes use of the conversion of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to a purple formazan product by the mitochondrial dehydrogenase of viable cells. This insoluble formazan was quantified spectrophotometrically upon dissolution in DMSO. Approximately 2 × 104 A2780 cells in 100 μL DMEM were seeded into each well of flat-bottomed 96-well plates (Corning) and allowed to attach overnight. Complexes were diluted in medium to seven concentrations spanning a 4-log range, and 10 μL of each drug solution was added to quadruplicate wells to produce the final desired concentrations (final DMF concentrations were limited to 0.2%). Following 72 h incubation, MTT (1.0 mM) was added to each well, and the plates incubated for a further 4 h. The culture medium was removed, and DMSO (200 μL) was added. The plates were shaken for 5 seconds and the absorbance measured immediately at 600 nm in a Victor3V microplate reader (Perkin Elmer). IC50 values were determined as the drug concentration required to reduce the absorbance to 50% of that in the untreated, control wells.Confocal microscopy
A2780 cells were seeded (4 mL of approximately 5 × 105cells mL−1) in 6-well plates (Iwaki) on glass cover slips and allowed to grow to 40–60% confluence, at 37 °C in 5% CO2. At this stage, cells were treated with 12 μM drug for 24 or 72 h. The cover slips were then washed with phosphate-buffered saline (pH 7.4), mounted on slides and sealed with nail varnish. Confocal images were acquired using a Leica TCS SPII multiphoton microscope. For all images an x63/1.20 water objective was used. Two-photon excitation at 767 nm was provided by a Ti-Sapphire laser and excitation at 633 nm for transmittance images, by a He/Ne laser. Images were collected and processed using Leica acquisition and analysis software.Syntheses
IR (KBr): 3069 (NH3), 1684, 1596 (NH2, NH3) cm−1. 1H NMR (DMF-d7): δ 7.451 (d, 2H, J = 7.96 Hz, aromatic H), 7.389 (broad, 2H, NH2) 7.333 (t, 2H, J = 7.75 Hz, aromatic H) 7.197 (t, 1H, J = 6.80 Hz, aromatic H), 4.184 (broad, 3H, NH3). 195Pt NMR (DMF-d7): δ−2068. Elemental analysis, found: C 17.61, H 3.01, N 6.83. Calculated for C6H10N2Cl2Pt: C 17.57, H 2.95, N 6.83.
IR (KBr): 3457 ((Pt-)O–H), 3102 (NH3), 1700, 1634, 1559 (NH2, NH3) cm−1. Elemental analysis, found: C 19.18, H 2.65, N 7.46. Calculated for C6H12N2O2Cl2Pt: C 19.16, H 2.68, N 7.45.
IR (KBr): 3239 (NH3), 1700 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1623, 1558 (NH2, NH3) cm−1. 1H NMR (DMF-d7): δ 7.745 (d, 1H, J = 8.50 Hz, aromatic H), 7.432 (dd, 1H, J = 8.53, 2.03 Hz, aromatic H), 7.354 (d, 1H, J = 2.00 Hz, aromatic H), 6.334 (d, 1H, J = 1.20 Hz, CH), 4.266 (broad, 3H, NH3), 3.917 (broad, 2H, NH2), 2.484 (d, 3H, J = 1.10, CH3).195Pt NMR (DMF-d7): δ−2054. Elemental analysis, found: C 27.05, H 2.93, N 6.25. Calculated for C10H12N2O2Cl2Pt: C 26.21, H 2.64, N 6.11.
O) 1623, 1558 (NH2, NH3) cm−1. 1H NMR (DMF-d7): δ 7.745 (d, 1H, J = 8.50 Hz, aromatic H), 7.432 (dd, 1H, J = 8.53, 2.03 Hz, aromatic H), 7.354 (d, 1H, J = 2.00 Hz, aromatic H), 6.334 (d, 1H, J = 1.20 Hz, CH), 4.266 (broad, 3H, NH3), 3.917 (broad, 2H, NH2), 2.484 (d, 3H, J = 1.10, CH3).195Pt NMR (DMF-d7): δ−2054. Elemental analysis, found: C 27.05, H 2.93, N 6.25. Calculated for C10H12N2O2Cl2Pt: C 26.21, H 2.64, N 6.11.
IR (KBr): 3598 ((Pt-)O–H), 3239 (NH3), 1732 (CO) 1617 (NH2, NH3) cm−1. 1H NMR (DMF-d7): δ 7.927 (d, 1H, J = 6.37 Hz, aromatic H), 7.653 (broad, 3H, NH3) 7.561 (dd, 1H, J = 6.36, 1.56 Hz, aromatic H), 7.515 (d, 1H, J = 1.48 Hz, aromatic H), 6.513 (d, 1H, J = 0.95 Hz, CH), 4.117 (broad, 2H, NH2), 2.796 (broad, 2H, OH), 2.678 (d, 3H, J = 2.68, CH3).195Pt NMR (DMF-d7): δ 789.
IR (KBr): 3277, 3205 (NH2, NH3), 1749, 1622 (NH2, NH3) cm−1. 1H NMR (DMF-d7): δ 7.735 (d, 1H, J = 8.04 Hz, aromatic H), 7.661 (broad, 2H, NH2), 7.482 (d, 1H, J = 10.02 Hz, aromatic H), 7.438 (s, 1H, aromatic H), 6.971 (s, 1H, aromatic H), 3.991 (broad, 3H, NH3). 195Pt NMR (DMF-d7): δ−2087. Elemental analysis, found: C 23.31, H 1.80, N 5.53. Calculated for C10H9N2F3Cl2Pt: C 23.45, H 1.77, N 5.47.
References
- B. Rosenberg, L. van Camp, E. B. Grimley and A. J. Thomson, J. Biol. Chem., 1967, 242, 1347–1352 CAS.
- B. Rosenberg, L. van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- B. Rosenberg and L. van Camp, Cancer Res., 1970, 30, 1799–1802 CAS.
- J. P. Berry, P. Brille, A. F. LeRoy, Y. Gouveia, P. Ribaud, P. Galle and G. Mathé, Cancer Treat. Rep., 1982, 66, 1529–1533 CAS.
- K. Wang, J. Lu and R. Li, Coord. Chem. Rev., 1996, 151, 53–88 CrossRef CAS.
- C. R. Wolf, I. P. Hayward, S. S. Lawrie, K. Buckton, M. A. McIntyre, D. J. Adams, A. D. Lewis, A. R. R. Scott and J. F. Smyth, Int. J. Cancer, 1987, 39, 695–702 CrossRef CAS.
- A. Eastman and N. Schulte, Biochemistry, 1988, 27, 4730–4734 CrossRef CAS.
- S. Sekiya, T. Oosaki, S. Andoh, N. Suzuki, M. Akaboshi and H. Takamizawa, Eur. J. Cancer Clin. Oncol., 1989, 25, 429–437 CrossRef CAS.
- S. Y. Loh, P. Mistry, L. R. Kelland, G. Abel and K. R. Harrap, Brit. J. Cancer, 1992, 66, 1109–1115 CAS.
- A. K. Godwin, A. Meister, P. J. O'Dwyer, C. S. Huang, T. C. Hamilton and M. E. Anderson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3070–3074 CAS.
- R. J. Schilder, F. P. LaCreta, R. P. Perez, S. W. Johnson, J. M. Brennan, A. Rogatko, S. Nash, C. McAleer, T. C. Hamilton, D. Roby, R. C. Young, R. F. Ozols and P. J. O'Dwyer, Cancer Res., 1994, 54, 709–717 CAS.
- H. Gurney, D. Crowther, H. Anderson, D. Murphy, J. Prendiville, M. Ranson, P. Mayor, R. Swindell, C. H. Buckley and V. R. Tindall, Ann. Oncol., 1990, 1, 427–433 CAS.
- C. Trask, A. Silverstone, C. M. Ash, H. Earl, C. Irwin, A. Bakker, J. S. Tobias and R. L. Souhami, J. Clin. Oncol., 1991, 9, 1131–1137 CAS.
- L. R. Kelland, G. Abel, M. J. McKeage, M. Jones, P. M. Goddard, M. Valenti, B. A. Murrer and K. R. Harrap, Cancer Res., 1993, 53, 2581–2586 CAS.
- F. R. Hartley, The Chemistry of Platinum and Palladium, Applied Science Publishers, London, 1973 Search PubMed.
- M. L. Tobe and A. R. Khokhar, J. Clin. Hematol. Oncol., 1977, 7, 114–137 Search PubMed.
- M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- V. H. Bramwell, D. Crowther, S. O'Malley, R. Swindell, R. Johnson, E. H. Cooper, N. Thatcher and A. Howell, Cancer Treat. Rep., 1985, 69, 409–416 CAS.
- K. R. Harrap, L. R. Kelland, M. Jones, P. M. Goddard, R. M. Orr, S. E. Morgan, B. A. Murrer, M. J. Abrams, C. M. Giandomenico and T. Cobbleigh, Adv. Enzyme Regul., 1991, 31, 31–43 CrossRef CAS.
- M. D. Hall, C. T. Dillon, M. Zhang, P. J. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, J. Biol. Inorg. Chem., 2003, 8, 726–732 CrossRef CAS.
- B. A. Teicher, S. A. Holden, J. L. Jacobs, M. J. Abrams and A. G. Jones, Biochem. Pharmacol., 1986, 35, 3365–3369 CrossRef CAS.
- K. E. Sandman, S. S. Marla, G. Zlokarnik and S. J. Lippard, Chem. Biol., 1999, 6, 541–551 CrossRef CAS.
- C. Molenaar, J. Teuben, R. J. Heetebrij, H. J. Tanke and J. Reedijk, J. Biol. Inorg. Chem., 2000, 5, 655–665 CrossRef CAS.
- R. Safaei, K. Katano, B. J. Larson, G. Samimi, A. K. Holzer, W. Naerdemann, M. Tomioka, M. Goodman and S. B. Howell, Clin. Cancer Res., 2005, 11, 756–767 CAS.
- X.-J. Liang, D.-W. Shen, K. G. Shen, S. M. Wincovitch, S. H. Garfield and M. M. Gottesman, J. Cell. Physiol., 2005, 202, 635–641 CrossRef CAS.
- G. V. Kalayda, B. A. J. Jansen, C. Molenaar, P. Wielaard, H. J. Tanke and J. Reedijk, J. Biol. Inorg. Chem., 2004, 9, 414–422 CrossRef CAS.
- B. A. J. Jansen, P. Wielaard, G. V. Kalayda, M. Ferrari, C. Molenaar, H. J. Tanke, J. Brouwer and J. Reedijk, J. Biol. Inorg. Chem., 2004, 9, 403–413 CrossRef CAS.
- G. V. Kalayda, B. A. J. Jansen, P. Wielaard, H. J. Tanke and J. Reedijk, J. Biol. Inorg. Chem., 2005, 10, 305–315 CrossRef CAS.
- R. A. Alderden, PhD Thesis, The University of Sydney, 2006.
- A. W. Czarnik, in Fluorescent Chemosensors for Ion and Molecule Recognition, ed. A. W. Czarnik, American Chemical Society, Washington, DC, 1993, pp. 1–9 Search PubMed.
- A. W. Czarnik, Acc. Chem. Res., 1994, 27, 302–308 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, M. Licchelli, C. Mangano, P. Pallavicini, L. Parodi and A. Poggi, Coord. Chem. Rev., 1999, 190–192, 649–669 CrossRef CAS.
- K. Zahir, W. Böttcher and A. Haim, Inorg. Chem., 1985, 24, 1966–1968 CrossRef CAS.
- G. W. Aodeng, W. Y. Bai and Z. Q. Duan, Spectrosc. Spec. Anal., 2001, 21, 846–848 Search PubMed.
- C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1974, 96, 4710–4712 CrossRef CAS.
- C.-T. Lin and N. Sutin, J. Am. Chem. Soc., 1975, 97, 3543–3545 CrossRef CAS.
- A. R. Hibbs, Confocal Microscopy for Biologists, Plenum Publishers, New York, 2004 Search PubMed.
- P.-S. Song and W. H. Gordon, J. Phys. Chem., 1970, 74, 4234–4240 CrossRef CAS.
- T. A. Moore, M. L. Harter and P.-S. Song, J. Mol. Spectrosc., 1971, 40, 144–157 CrossRef CAS.
- P. K. McCarthy and G. J. Blanchard, J. Phys. Chem., 1993, 97, 12205–12209 CrossRef CAS.
- A. R. Reddy, D. V. Prasad and M. Darbarwar, J. Photochem., 1986, 32, 69–80 CrossRef CAS.
- B. N. Mattoo, Trans. Faraday Soc., 1956, 52, 1184–1194 RSC.
- R. Sairam and N. K. Ray, Indian J. Chem., 1980, 19B, 989–991 Search PubMed.
- N. A. Nemkovich, W. Baumann, H. Reis and Y. V. Zvinevich, J. Photochem. Photobiol. A, 1997, 109, 287–292 CrossRef CAS.
- L. T. Ellis, H. M. Er and T. W. Hambley, Aust. J. Chem., 1995, 48, 793–806 CAS.
- T. W. Hambley, A. R. Battle, G. B. Deacon, E. T. Lawrenz, G. D. Fallon, B. M. Gatehouse, L. K. Webster and S. Rainone, J. Inorg. Biochem., 1999, 77, 3–12 CrossRef CAS.
- A. R. Khokhar, Y. Deng, S. Al-Baker, M. Yoshida and Z. H. Siddik, J. Inorg. Biochem., 1993, 51, 677–687 CrossRef CAS.
- N. Farrell, L. R. Kelland, J. D. Roberts and M. Van Beusichem, Cancer Res., 1992, 52, 5065–5072 CAS.
- T. A. Connors, M. Jones, W. C. J. Ross, P. D. Braddock, A. R. Khokhar and M. L. Tobe, Chem-Biol Interact., 1972, 5, 415–424 CrossRef CAS.
- M. D. Hall, G. J. Foran, M. Zhang, P. J. Beale and T. W. Hambley, J. Am. Chem. Soc., 2003, 125, 7524–7525 CrossRef CAS.
- Z. Diwu, Y.-Z. Zhang, and R. P. Haugland, XVII Congress of the International Society for Analytical Cytology, New York, 1994.
- R. G. Kirk, M. E. Gates, C. Chang and P. Lee, Exp. Mol. Pathol., 1995, 63, 33–40 CrossRef CAS.
- G. L. Beretta, S. C. Righetti, L. Lombardi, F. Zunino and P. Perego, Ultrastruct. Pathol., 2002, 26, 331–334 CrossRef.
- D. K. Miller, E. Griffiths, J. Lenard and R. A. Firestone, J. Cell. Biol., 1983, 97, 1841–1851 CrossRef CAS.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS.
- O. Asada, K. Umakoshi, K. Tsuge, S. Yabuuchi, Y. Sasaki and M. Onishi, Bull. Chem. Soc. Jpn., 2003, 76, 549–555 CrossRef CAS.
- H. Kobayashi, H. Ohi and T. Terao, Asia-Oc. J. Obst. Gyn., 1991, 17, 277–288 Search PubMed.
- M. D. Hall, S. Amjadi, M. Zhang, P. J. Beale and T. W. Hambley, J. Inorg. Biochem., 2004, 98, 1614–1624 CrossRef CAS.
- R. M. Whan, B. A. Messerle and T. W. Hambley, Dalton Trans., 2009, 932–939 RSC.
- J. Carmichael, W. G. DeGraff, A. F. Gazdar, J. D. Minna and J. B. Mitchell, Cancer Res., 1987, 47, 936–942 CAS.
- L. Cai, K. Lim, S. Ren, R. S. Cadena and W. T. Beck, J. Med. Chem., 2001, 44, 2959–2965 CrossRef CAS.
Footnote |
| † By convention, ligands that lie above and below the plane of the am(m)ine ligands in Pt(IV) complexes are referred to as “axial” ligands, while those that lie in the same plane as the am(m)ine ligands are designated “equatorial” ligands.57,58 |
| This journal is © The Royal Society of Chemistry 2009 |