DOI:
10.1039/B821030F
(Paper)
Dalton Trans., 2009, 2687-2694
Synthesis and characterization of synthetically useful salts of the weakly-coordinating dianion [B12Cl12]2−†
Received
25th November 2008
, Accepted 4th February 2009
First published on 24th February 2009
Abstract
The closo-dodecahydrododecaborate [NEt3H]2[B12H12] has been prepared on a lab scale by an improved synthesis from cheap and readily available starting materials Na[BH4] and I2 in diglyme (diethylene glycol dimethyl ether). Subsequent chlorination with elemental chlorine in aqueous solution at normal pressure yielded the per-chlorinated weakly coordinating [B12Cl12]2− anion. By simple metathesis reaction a variety of useful salts [cation]2[B12Cl12] (cation = [NEt3H]+, [NBu4]+, Li+, Na+, K+, Cs+) is available. These salts are useful starting materials, which have the potential to open up the chemistry of [B12Cl12]2− as a weakly coordinating dianion. Exemplarily, they were used in further reactions to prepare [NO]2[B12Cl12], [PPN]2[B12Cl12], and [CPh3]2[B12Cl12]. The crystal structures of Cs2[B12Cl12]·SO2, [CPh3]2[B12Cl12]·2C2H4Cl2, and [CPh3]2[B12Cl12]·2SO2 and preliminary crystal structures of [NO]2[B12Cl12]·SO2 and [PPN]2[B12Cl12]·CH2Cl2 were determined. The crystal structure of the SO2 solvate Cs2[B12Cl12]·SO2 is related to the crystal structure of solvent free Cs2[B12Cl12]. [CPh3]2[B12Cl12]·2C2H4Cl2 and [CPh3]2[B12Cl12]·2SO2 have very similar structures in the solid state. In both cases the [CPh3]+ cations form only very weak contacts to the [B12Cl12]2− anion and SO2 or C2H4Cl2solvent molecules respectively. The averaged experimental B–B (178.7 pm) and B–Cl (178.9 pm) bond lengths within [B12Cl12]2− are essentially unchanged in all determined structures and are reproduced well by PBE0/TZVPP quantum chemical calculations (B–B 178.6 pm, B–Cl 179.3 pm). All results indicate that [B12Cl12]2− is a readily accessible weakly-coordinating dianion.
1. Introduction
Weakly-coordinating anions (WCA) have become a major player in fundamental and applied chemical research.1 Among the large variety of WCAs halogenated carborane anions2 have been shown to be the most stable weakly-coordinating anions and allowed the synthesis and characterization of some very reactive cations, which include [Et2Al]+,3a [C60H]+,3b[t-butyl]+,3c [R3Si]+.3d Unfortunately, the synthesis of the carborane anions is time consuming and expensive,4 which prevented their widespread application so far. Halogenated closo-dodecaborates [B12X12]2− (X = F, Cl, Br, I)5 posses a similar stability and they are easier and cheaper to prepare. The higher −2 charge on [B12X12]2− dianions is unusual for a weakly-coordinating anion (a typical WCA is a large (fluorinated) anion with a charge of −1)1 and prevents the often desired “pseudo-gas phase conditions”.1b,c Therefore the closo-dodecaborates [B12X12]2− have been widely neglected so far. The perchlorinated derivative [B12Cl12]2− is the most promising candidate for future applications among the series [B12X12]2− (X = F, Cl, Br, I), because it is more resistant against oxidation than [B12Br12]2− and [B12I12]2−, but more readily accessible than [B12F12]2−, which is prepared by the reaction of [B12H12]2− with elemental fluorine.6 Salts containing the [B12Cl12]2− dianion have already found several applications in the past, some of which are stabilization of cationic Pd and Ni complexes,7 as ionic liquids,8 evaluation as non-aqueous electrolytes in solid cathode lithium batteries9 and in fuel cells.10 Recently the fluorinated [B12F12]2− dianion6 was tested as electrolyte for electrochemical devices.11 Therefore a simple and cheap lab scale synthesis of [B12Cl12]2− and its predecessor [B12H12]2− is key for their widespread investigation. Although [B12H12]2−12 and [B12Cl12]2−5 are well known compounds, full experimental details for a cheap, simple and reproducible lab scale synthesis are not available.
In this paper we present an optimized synthesis of closo-dodecaborate [B12H12]2− from cheap starting materials Na[BH4] and I2 avoiding toxic diborane and expensive decaborane, the complete chlorination of [B12H12]2− to [B12Cl12]2− in aqueous solution with elemental Cl2, and the preparation and characterization of some synthetically useful salts of the [B12Cl12]2− anion, i.e.Li+, Cs+, [NEt3H]+, [NBu4]+, [CPh3]+, and [NO]+. The crystal structures of Cs2[B12Cl12]·SO2 (1), [CPh3]2[B12Cl12]·2C2H4Cl2 (2), and [CPh3]2[B12Cl12]·2SO2 (3) are also reported.
2. Experimental
General remarks
Air and moisture sensitive solid reagents were manipulated using standard vacuum and Schlenk techniques or in a glove box with an atmosphere of dry argon (H2O and O2 <1 ppm). The reactions using liquid sulfur dioxide as a solvent were carried out in H-shaped glass vessels with J. Young Teflon in glass valves and an incorporated fine frit. The solvents liquid sulfur dioxide (Schick), 1,2-dichloroethane (Riedel-de-Haën), and deuterated solvents (Deutero) were dried over CaH2 (Merck) and distilled prior to use. CH3CN, CH2Cl2, and n-pentane were purified using a Grubbssolvent purification system. Na[BH4] (Acros, 98%, powder), I2 (Grüssing, p.a.), diglyme (diethylene glycol dimethyl ether) (Acros), NEt3 (Fluka), LiOH (Merck), NaOH (Grüssing, p.a.), KOH (Roth, p.a.), CsOH (Acros), Cl2 (SBF), [NBu4]Cl (Fluka), [NO][BF4] (Acros, 97%), and CPh3Cl (Alfa Aesar) are commercially available and were used as received. Commercial [CPh3][BF4] (Acros) was purified by washing with n-pentane. [PPN]Cl (bis(triphenylphosphine)iminium chloride) was prepared by a modified literature procedure.13
IR spectra were recorded on a Nicolet Magna-IR 760 equipped with a diamond ATR attachment and FT-Raman spectra were recorded in flame-sealed capillaries on a Bruker Vertex 70 IR spectrometer equipped with a Bruker RAM II Raman module using a highly sensitive Ge detector and a Nd:YAG-Laser (1064 nm). 1H, 11B, 13C and 31P NMR spectra were measured on a Bruker Avance II 400 WB spectrometer in 5 mm NMR tubes at room temperature. Chemical shifts are given with respect to Me4Si (1H, 13C), BF3·OEt2 (11B), H3CNO2 (14N), and 85% H3PO4 (31P). Melting and decomposition points were obtained on a Setaram DSC 131. All actual spectra are included with the ESI.†
[NHEt3]2[B12H12].
A solution of iodine (205.58 g, 0.810 mol) in 350 mL of diglyme was added dropwise over a period of 6 h to a suspension of Na[BH4] (97.91 g, 2.588 mol) in diglyme (400 ml) at 100 °C. During the addition of iodine the amount of insoluble Na[BH4] decreased and at the end a yellow color of the reaction mixture was observed. The reaction mixture was stirred over night at 100 °C to complete the formation of [B3H8]− followed by additional 24 h reflux to completely disproportionate [B3H8]− to [B12H12]2− and [BH4]− (at this stage the yellow color disappeared and a white precipitate started to form). The solvent was distilled off under reduced pressure giving a large amount of white solid. The white solid was dissolved in 600 mL of water and 280 mL of concentrated hydrochloric acid were added carefully to the reaction mixture (caution: hydrogen evolution). The acidified clear solution was stored at +6 °C over night. Colourless crystals of boric acid (ca. 15 g) precipitated and were removed by filtration. The filtrate was treated with 400 mL Et3N (pH = 9–10) and readily a white precipitate formed (ca. 80 g, the white precipitate contained the product [NEt3H]2[B12H12] and remaining boric acid (11B NMR in D2O), which was collected by filtration. The solid was re-suspended in water (ca. 250 ml), stirred for two hours, and then filtrated to remove the more soluble boric acid impurity. The product [NEt3H]2[B12H12] (28.50 g, 0.082 mol, 51%) remained as a white solid. 1H NMR (CD3CN): δ = 0.50–1.90 (mult., br., 12 H) [B12H12]2−, 1.19 (t, 3JHH = 7.3 Hz, 18 H, CH3), 3.11 (q, 3JHH = 7.3 Hz, 12 H, CH2). 11B NMR (CD3CN): δ = −15.3 (d, 1JBH = 125 Hz). A full account of the preparation including all details has been deposited as ESI.†
M2[B12H12] (M = Na, K, Cs).
Solid [NHEt3]2[B12H12] was added to a solution of 2.1 equivalents of MOH (M = Na, K) dissolved in water in a polypropylene beaker. Typically about 100 ml water was used for 10 g [NHEt3]2[B12H12]. The suspension was heated on a water bath until it became a clear solution. Then the solution was evaporated to dryness. Solvent free K2[B12H12] can be prepared by drying the remaining solid in vacuum at 80 °C. The Cs+ salt is much less soluble than the other alkali metal salts and precipitates immediately after CsOH addition.14 It can be re-crystallized from boiling water. 1H NMR (D2O): δ = 0.50–1.90 (mult., br.) [B12H12]2−. 11B NMR (D2O): δ = −15.3 (d, 1JBH = 125 Hz) [B12H12]2−.
Chlorination of Na2[B12H12].
In a typical reaction 10 g Na2[B12H12] were dissolved in 100 ml of water. Chlorine was bubbled through the reaction mixture for five hours at rt and additional 24 hours at 100 °C. Completeness of chlorination was checked by 11B NMR spectra of aliquots every few hours. When the chlorination was complete the excess of chlorine was removed by purging the reaction mixture with argon. The resulting very acidic clear colourless solution of Na2[B12Cl12] was directly used to prepare [NEt3H]2[B12Cl12] and Cs2[B12Cl12] (see below).
[NHEt3]2[B12Cl12].
An excess of NEt3 was added to the acidic aqueous solution from the chlorination reaction (see above) and immediately white [NEt3H]2[B12Cl12] precipitated. The [NEt3H]2[B12Cl12] precipitate was separated by filtration and washed with water (the filtrate was checked for chloride with Ag[NO3]) to give the product as a white solid. 1H NMR (CD3CN): δ = 1.19 (t, 3JHH = 7.3 Hz, 18 H, CH3), 3.11 (q, 3JHH = 7.3 Hz, 12 H, CH2). 11B NMR (CD3CN): δ = −12.7 (s) [B12Cl12]2−. IR (cm−1): ν = 3181 (m), 2988 (w), 1471 (m), 1458 (m), 1397 (m), 1304 (w), 1289 (w), 1172 (w), 1155 (w), 1027 (vs) [B12Cl12]2−, 835 (w), 793 (w), 677 (w), 533 (vs) [B12Cl12]2−, 506 (w), 458 (m), 407 (m). Raman (cm−1): ν = 3187 (5), 2988 (50), 2948 (50), 1453 (17), 300 (90) [B12Cl12]2−, 128 (100) [B12Cl12]2−.
M2[B12Cl12] (M = Li, Na, K).
Solid [NHEt3]2[B12Cl12] was added to a solution of 2.1 equivalents of MOH (M = Li, Na, K) dissolved in water in a polypropylene beaker. Typically about 100 ml water was used for 10 g [NHEt3]2[B12H12]. The suspension was heated on a water bath until it became a clear solution. The solution was evaporated to dryness yielding M2[B12Cl12] as a white solid in quantitative yield. Solvent free M2[B12Cl12] can be prepared by drying in vacuum at 150–200 °C. The product is highly soluble in H2O, and very soluble in liquid SO2 and CH3CN and has low solubility in CH2Cl2. M2[B12Cl12] (M = Li, Na, K, Cs) gives essentially identical spectroscopic data. 11B NMR (D2O): δ = −12.7 (s) [B12Cl12]2−. IR (cm−1): ν = 1029 (vs), 532 (vs), 160 (s), 136 (w), 129 (m). Raman (cm−1): ν = 305 (100), 132 (70). mp > 450 °C.
Cs2[B12Cl12].
2.1 equivalents of CsOH were added to the acidic aqueous solution from the chlorination reaction (see above). The Cs2[B12Cl12] precipitate was separated by filtration and re-crystallized from hot water. Crystals of Cs2[B12Cl12]·SO2 suitable for X-ray diffraction were prepared by dissolving Cs2[B12Cl12] in liquid SO2 and slow removal of the solvent. Cs2[B12Cl12] is soluble in THF and CH3CN, has low solubility in SO2, and is insoluble in toluene, CH2Cl2 and 1,2-C6H4F2.
[NBu4]2[B12Cl12].
An excess of [NBu4]Br was added to the acidic aqueous solution from the chlorination reaction (see above). The [NBu4]2[B12Cl12] precipitate was separated by filtration and washed with water (the filtrate was checked for chloride with Ag[NO3]) to give the product as a white solid. [NBu4]2[B12Cl12] is soluble in CH3CN, CH2Cl2 and SO2. 1H NMR (CD3CN): δ = 0.96 (t, 3JHH = 7.5 Hz, 24H, CH3), 1.34 (sextet, 3JHH = 7.5 Hz, 16H, CH2-CH3), 1.62 (mult., 16H, CH2), 3.09 (mult., 16H, NCH2). 11B NMR (D2O): δ = −12.7 (s) [B12Cl12]2−.13C NMR (CD3CN): δ = 12.7 (CH3), 19.3 (CH2-CH3), 23.2 (CH2), 58.1 (NCH2). IR (cm−1): ν = 2966 (m), 2936 (w), 2878 (w), 1469 (s), 1384 (w), 1169 (w), 1031 (vs) [B12Cl12]2−, 995 (sh), 925 (w), 880 (m), 794 (w), 740 (m), 577 (w), 531 (vs) [B12Cl12]2−. Raman (cm−1): ν = 3145 (2), 2972 (30), 2937 (80), 2875 (30), 2756 (18), 1453 (2), 1355 (1), 1318 (2), 1271 (1), 1111 (6), 1068 (10), 906 (2), 882 (2) 300 (100) [B12Cl12]2−, 127 (80) [B12Cl12]2−. mp 420 °C (dec.).
[PPN]2[B12Cl12].
Li2B12Cl12 (0.172 g, 0.3 mmol) and [PPN]Cl (0.352 g, 0.6 mmol) were dissolved in 50 ml CH2Cl2. The reaction mixture was stirred at rt for 3 h. A white precipitate (LiCl) formed, which was removed by filtration through a fine frit. Removing of the solvent in vacuum yielded [PPN]2[B12Cl12] as a white solid in quantitative yield. Crystals of [PPN]2[B12Cl12] suitable for X-ray diffraction were prepared by dissolving [PPN]2[B12Cl12] in CH2Cl2 followed by slow evaporation of the solvent (see ESI). 1H NMR (CH2Cl2/CDCl3): δ = 7.45–7.55 (mult., 48H, o-/m-C6H5), 7.60–7.70 (mult., 12H, p-C6H5). 11B NMR (CH2Cl2/CDCl3): δ = −12.6 (s, [B12Cl12]2−). 31P NMR (CH2Cl2/CDCl3): δ = 21.1 (s, Ph3N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PNPh3). IR (cm−1): ν = 3059 (w), 2930 (w), 1729 (w), 1587 (w), 1483 (w), 1394 (s), 1303 (m), 1262 (m), 1184 (m), 1115 (m), 1024 (s) [B12Cl12]2−, 997 (s), 854 (w), 799 (w), 750 (m), 721 (s), 691 (2), 617 (w), 527 (s) [B12Cl12]2−, 497 (s), 448 (m). Raman (cm−1): ν = 3062 (61), 1588 (54), 1111 (17), 1027 (31), 1000 (100), 659 (24), 616 (20), 296 (63) [B12Cl12]2−, 235 (29), 129 (89) [B12Cl12]2−. mp 303 °C.
PNPh3). IR (cm−1): ν = 3059 (w), 2930 (w), 1729 (w), 1587 (w), 1483 (w), 1394 (s), 1303 (m), 1262 (m), 1184 (m), 1115 (m), 1024 (s) [B12Cl12]2−, 997 (s), 854 (w), 799 (w), 750 (m), 721 (s), 691 (2), 617 (w), 527 (s) [B12Cl12]2−, 497 (s), 448 (m). Raman (cm−1): ν = 3062 (61), 1588 (54), 1111 (17), 1027 (31), 1000 (100), 659 (24), 616 (20), 296 (63) [B12Cl12]2−, 235 (29), 129 (89) [B12Cl12]2−. mp 303 °C.
[CPh3]2[B12Cl12], method A.
[CPh3][BF4] (0.41 g, 1.23 mmol) and Cs2[B12Cl12] (0.50 g, 0.61 mmol) were dissolved in dry acetonitrile and stirred over night. All volatiles were removed in vacuum and the product was extracted with 1,2-C2H4Cl2 giving [CPh3]2[B12Cl12]·2(1,2-C2H4Cl2) quantitatively as a orange crystalline solid (isolated yield: 0.64 g, 0.52 mmol, 85%). C2H4Cl2 solvate molecules can be removed by dissolving the product in liquid sulfur dioxide followed by drying at 50 °C over night. Crystals suitable for X-ray diffraction of 3 were obtained by dissolving [CPh3]2[B12Cl12] in liquid SO2 followed by slow removal of the solvent and crystals of 2 by slow removal of the solvent from a saturated solution in 1,2-dichloroethane. 1H NMR (CD3CN): δ = 7.73 (d, 3JHH = 7.2 Hz, 12 ortho-H), 7.90 (t, 3JHH = 8.0 Hz, 12 meta-H), 8.30 (t, 3JHH = 7.4 Hz, 6 para-H). 11B NMR (CD3CN): δ = −12.7 (s, [B12Cl12]2−). 13C NMR (CD3CN): δ = 130.6 (meta-C), 139.8 (ipso-C), 142.7 (ortho-C), 143.4 (para-C), 210.9 ([CPh3]+). IR (cm−1): ν = 3064 (w), 1577 (vs), 1480 (s), 1448 (vs), 1352 (vs), 1292 (vs), 1230 (m), 1182 (s), 1166 (sh), 1025 (vs) [B12Cl12]2−, 994 (vs), 983 (sh), 945 (m), 915 (m), 847 (sh), 835 (m), 806 (m), 770 (s), 702 (vs), 687 (sh), 657 (sh), 622 (s), 608 (s), 531 (vs) [B12Cl12]2−, 464 (m), 426 (w), 402 (s). Raman ([CPh3]2[B12Cl12]·2(1,2-C2H4Cl2), cm−1): ν = 3069 (20), 1596 (40), 1580 (100), 1483 (15), 1358 (45), 1296 (5), 1186 (30), 1167 (5), 1027 (10), 998 (40), 916 (10), 849 (5), 772 (5), 708 (5), 623 (10), 469 (15), 405 (40), 298 (5) [B12Cl12]2−, 285 (40), 131 (50) [B12Cl12]2−, 94 (70). We did not succeed in obtaining a Raman spectrum of solvent free [CPh3]2[B12Cl12] presumably due to loss of crystallinity.
[CPh3]2[B12Cl12], method B.
10 ml SO2 were condensed onto a mixture of CPh3Cl (0.80 g, 2.88 mmol) and Li2[B12Cl12] (0.82 g, 1.45 mmol). The reaction mixture turned yellow on warming and was stirred at rt for one hour. The volatiles were removed in vacuum and the product was extracted with 1,2-C2H4Cl2 as described for method A giving [CPh3]2[B12Cl12] as yellow crystalline solid in essentially quantitative yield (isolated yield 1.05 g, 1.01 mmol, 70).
[NO]2[B12Cl12].
10 ml SO2 were condensed onto a mixture of [NO][BF4] (0.21 g, 1.79 mmol) and Li2[B12Cl12] (0.46 g, 0.81 mmol). The reaction mixture was stirred over night giving a red solution and some white precipitate (Li[BF4], IR), which was separated by filtration. Removal of all volatiles in vacuum yielded [NO]2[B12Cl12] as a red solid in quantitative yield (0.40 g, 0.65 mmol, 80%). Red crystals suitable for X-ray diffraction were obtained by slow removal of the solvent (see ESI†). 11B NMR (SO2, unlocked): δ = −12.6 (s, [B12Cl12]2−). 14N NMR (SO2, unlocked): δ = 15 (s, Δω1/2 220 Hz, [NO]+). IR (cm−1): ν = 2214 (m, broad) [NO]+, 1026 (s) [B12Cl12]2−, 530 (s) [B12Cl12]2−. Raman (cm−1): ν = 2220 (100) [NO]+, 1067 (4), 990 (1), 305 (5) [B12Cl12]2−, 144 (6), 126 (4) [B12Cl12]2−.
Single-crystal X-ray structure determinations were carried out on a Rigaku R-AXIS Spider image plate (1 and 3) or a Bruker APEX II CCD diffractometer (2) using Mo Kα (0.71073 Å) radiation. The crystals were mounted onto a kryo loop using fluorinated oil and frozen in the cold nitrogen stream of the goniometer. Details of the crystallographic data collection and refinement parameters are given in Table 1. The structures were solved by direct methods (SHELXS).15 Subsequent least-squares refinement on F2 (SHELXL 97-2) located the positions of the remaining atoms in the electron density maps.15 All atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions using a riding model and were refined isotropically in blocks. The data were corrected for absorption. Graphical representations of the structures were prepared with the program DIAMOND.16
Table 1 Crystallographic parameters
| |
Cs2[B12Cl12]·SO2 (1) |
[CPh3]2[B12Cl12]·2C2H4Cl2 (2) |
[CPh3]2[B12Cl12]·2SO2 (3) |
|
R
1 = ∑‖Fo| − |Fc‖/∑|Fo|, wR2 = (∑[w(Fo2−Fc2)2]/∑[wFo4])1/2.
|
| Formula |
B12Cl12Cs2O2S |
C42H38B12Cl16 |
C38H30B12Cl12O4S2 |
|
M
|
885.00 |
1239.64 |
1169.86 |
| Crystal system |
Trigonal |
Monoclinic |
Monoclinic |
| Space group (no.) |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (148) (148) |
P21/n (14) |
C2/c (15) |
|
a/pm |
1014.32(14) |
993.18(2) |
1620.5(3) |
|
b/pm |
1014.32(14) |
1498.79(4) |
1661.7(3) |
|
c/pm |
2066.8(4) |
1792.43(4) |
1901.0(4) |
|
α/° |
90 |
90 |
90 |
|
β/° |
90 |
91.622(1) |
102.60(3) |
|
γ/° |
120 |
90 |
90 |
|
U
/nm3 |
1.8415(5) |
2667.08(11) |
4.9958(17) |
|
T/K |
110(2) |
114(2) |
173(2) |
|
Z
|
3 |
2 |
4 |
|
μ(Mo Kα)/mm−1 |
4.361 |
0.857 |
0.790 |
|
No. of data collected |
3466 |
62722 |
43480 |
|
No. of unique data |
936 |
6807 |
5725 |
|
R
int
|
0.0418 |
0.0414 |
0.0434 |
|
R
1, wR2 (I >2σ(I))a |
0.0346, 0.0703 |
0.0306, 0.0749 |
0.0355, 0.0889 |
|
R
1, wR2 (all data) |
0.0409, 0.0721 |
0.0393, 0.0802 |
0.0456, 0.0937 |
Quantum chemical calculations
DFT (BP86 and PBE0) calculations were performed with SV(P), TZVPP, and QZVPP basis sets as implemented in the program TURBOMOLE.17 Frequency calculations were performed at the BP86/SV(P) level for all species. All calculated species are true minima on the energy hyper surface as shown by the absence of imaginary frequencies. Calculated coordinates, frequencies and total energies are included with the ESI.†
3. Results and discussion
3.1. Synthesis of [B12H12]2− from [BH4]− and I2
The closo-dodecahydrododecaborate [B12H12]2− is an important starting material in boron chemistry,12 and various syntheses are known.12 Unfortunately, most synthetic routes use either expensive (e.g. [B10H14]) or potentially dangerous (e.g. B2H6) starting materials. A cheap and experimentally simple route is the reaction of M[BH4] (M = Na, K) with I2 in diglyme (diethylene glycol dimethyl ether) giving the anion [B3H8]− as an intermediate in a first step (eqn (1)), which subsequently in a second step can be thermally decomposed to [B12H12]2− (eqn (2)).| | | 3 NaBH4 + I2→ NaB3H8 + 2 NaI + 2 H2↑ | (1) |
| | | 5 Na[B3H8] → Na2[B12H12] + 3 Na[BH4] + 8 H2↑ | (2) |
Both steps are well known independently of each other,18,19 but only limited experimental details are available for the complete two-step synthesis.20–22 We modified and scaled up the published procedures and improved the synthesis of [B12H12]2− from Na[BH4] and I2 to give reproducible yields of 50–60% based on I2 used. The obtained product did not contain smaller boron clusters (e.g. [B6H6]2−, [B10H10]2−) as evident from 11B NMR spectra, which showed a doublet at −15.3 ppm for [B12H12]2− only.23 Our improvements include a modified stoichiometry, longer reaction times, and a modified work up procedure. A discussion of the synthesis including full experimental details is included with the ESI.†
3.2.
Chlorination of [B12H12]2− to [B12Cl12]2−
The chlorination of [B12H12]2− is known since the pioneering work of Muetterties et al.,5 who reacted [B12H12]2− with elemental chlorine in aqueous solution giving partly chlorinated compounds, which were subsequently reacted with an excess of chlorine in an autoclave to give [B12Cl12]2− as the final product (eqn (3)).| | | [B12H12]2− + 12 Cl2→ [B12Cl12]2− + 12 HCl | (3) |
However, incomplete chlorination (i.e. the product consists of a mixture of [B12Cl12]2−, [B12HCl11]2−, and [B12H2Cl10]2−) has been reported.8,10,24 The absence of the B–H vibration in the IR spectrum was suggested as diagnostic tool for checking the completeness of the chlorination process.10 We prepared Na2[B12Cl12] under avoiding a high pressure autoclave5,25 by passing elemental chlorine through an aqueous solution of Na2[B12H12] at room temperature. The sodium salt is preferred over the caesium salt because of its much higher solubility in water.14 The reaction speed decreases with progressing chlorination and therefore after a few hours the reaction mixture was heated to 100 °C. The chlorination progress was checked repeatedly by 11B NMR. In contrast to previous reports10 we found the absence of a B–H vibration in the IR spectrum to be non suitable for checking the chlorination completeness, because the B–H vibration becomes increasingly weak with progressing chlorination and may not be observable even if the chlorination is still incomplete. Fig. 1 shows 11B NMR spectra of [B12H12]2− and [B12Cl12]2− together with several 11B NMR spectra of samples taken from the reaction mixture during chlorination over a time period of 40 h. The spectra of the intermediate steps show two separate regions for H-bonded and Cl-bonded boron atoms, which shift to higher field with progressing chlorination. A preliminary investigation of the composition of the intermediate reaction mixture may be found in the ESI. The existence of a single peak only at about −12.8 ppm indicates complete chlorination.
![11B NMR spectra in D2O at rt of aliquots taken from the reaction of Na2[B12H12] with Cl2 in H2O over a time period of 40 h.](/image/article/2009/DT/b821030f/b821030f-f1.gif) |
| | Fig. 1
11B NMR spectra in D2O at rt of aliquots taken from the reaction of Na2[B12H12] with Cl2 in H2O over a time period of 40 h. | |
The mechanism of the chlorination remains unknown, but electrophilic substitution and electrophile-induced nucleophilic substitution (EINS) have been suggested.4 The stereochemistry of the substitution and the corresponding substitution pattern for the chlorination is controversial. Preetz et al. investigated the first three steps of the chlorination and suggested an ortho substitution pattern to give [1,2-B12H10Cl2]2− and [1,2,3-B12H9Cl3]2− based on 11B NMR and vibrational spectroscopy.26 In contrast a detailed NMR spectroscopic study (based on 11B–11B cosy spectra) together with structure determination of some intermediates for the corresponding fluorination by Kuznetsov et al. showed unambiguously a preferred meta substitution pattern. Correspondingly [1,7-B12F2H10]2− and [1,7,9-B12H9F3]2− were identified.27 Only two partly chlorinated clusters have been structurally characterized, i.e. [B12H11Cl]2−28 and [B12H2Cl10]2−,24 which give no evidence for either of the substitution patterns. Isolation and characterization of the intermediate products of the chlorination will be a part of a future project to experimentally proof the theoretically predicted sequential substitution pattern.29
3.3. Synthesis and characterization of synthetically useful salts of the dianion [B12Cl12]2−
Li2[B12Cl12], Cs2[B12Cl12], and [NBu4][B12Cl12].
Alkali metal and ammonium salts are fundamental starting materials for exploring the chemistry of [B12Cl12]2−. Cs[B12Cl12] is the least soluble alkali metal salt and can be prepared directly from the chlorination reaction mixture by precipitating with CsOH (eqn (4). Similarly, neutralization of the acidic chlorination reaction mixture with NEt3 affords only sparingly soluble [NEt3H]2[B12Cl12], which can be reacted with MOH (M = Li, K) in hot water in a second step to obtain the more soluble alkali metal salts (eqn (5)).| | 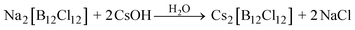 | (4) |
| |  | (5) |
M = Li, Na, K, Cs.
The lithium, caesium and tetrabutylammonium salts of [B12Cl12]2− are especially useful, because they show complementary properties (e.g. solubility in different solvents)30 and have been turned out to be ideal starting materials. For instant Li2[B12Cl12] has some solubility in CH2Cl2 and undergoes easily metathesis reactions with chlorides to form the desired [B12Cl12]2− salts and LiCl. This methodology may be useful to prepare low melting salts or ionic liquids containing the [B12Cl12]2− anion. Exemplarily, we prepared [PPN]2[B12Cl12] from Li2[B12Cl12] and [PPN]Cl in CH2Cl2 (eqn (6)).| |  | (6) |
A preliminary crystal structure of [PPN]2[B12Cl12] has been determined and shows well defined [B12Cl12]2− anions and heavily disordered [PPN]+ cations, which prevent a detailed discussion of the structure (see ESI†).
[CPh3][B12Cl12].
Trityl (= [CPh3]+) salts have been recognized as important reagents for hydride and alkyl abstraction.1b Therefore, [CPh3]2[B12Cl12] is a worthwhile synthetic target to extend the chemistry of [B12Cl12]2−. We developed two independent synthetic routes to [CPh3]2[B12Cl12]. Cs2[B12Cl12] reacts with [CPh3][BF4] in CH3CN in a metathesis reaction to [CPh3]2[B12Cl12] and Cs[BF4] (eqn (7)).| |  | (7) |
However, this reaction uses expensive [CPh3][BF4] as a starting material. Thus we investigated routes using the cheaper trityl chloride as a starting point. Li2[B12Cl12] reacts with CPh3Cl in liquid SO2 to [CPh3]2[B12Cl12] and LiCl (eqn (8)).
| | 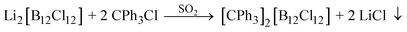 | (8) |
The cleavage of the C–Cl bond is overcompensated by the high lattice enthalpy of LiCl. The solid state reaction is favourable by about 100 kJ mol−1 as evident from the Born–Fajans–Haber cycle in Fig. 2. However, in acetonitrile, which dissolves the starting materials and the products very well, the reaction is not complete. Instead, in acetonitrile solution there is a reversible balance between the starting materials and the products. In contrast, in SO2LiCl is insoluble, which forces the reaction to the product side.
![Born–Fajans–Haber cycle for the formation of [CPh3]2[B12Cl12] from CPh3Cl and Li2[B12Cl12]. Details on the calculation of the gas phase and the lattice enthalpies are included with the ESI.](/image/article/2009/DT/b821030f/b821030f-f2.gif) |
| | Fig. 2 Born–Fajans–Haber cycle for the formation of [CPh3]2[B12Cl12] from CPh3Cl and Li2[B12Cl12]. Details on the calculation of the gas phase and the lattice enthalpies are included with the ESI.† | |
Independent of the synthetic route used [CPh3]2[B12Cl12] has to be separated from the insoluble side-products Cs[BF4] or LiCl respectively. It turned out that 1,2-dichloroethane seems to be the only solvent, in which [CPh3]2[B12Cl12] has significant solubility and the side-products are insoluble. Unfortunately, the solvent gets incorporated in the crystal lattice and thus solid [CPh3]2[B12Cl12] contains two equivalents of 1,2-dichloroethane (1H NMR and crystal structure determination), which cannot be removed in vacuum. The solvated 1,2-dichloroethane can be replaced by other solvent molecules (e.g. SO2, CH3CN, CH2Cl2) by dissolving the compound in an excess of the respective solvent and subsequent removal of all volatiles. The solid state structures of the 1,2-dichloroethane and the sulfur dioxide solvates have been determined by single crystal X-ray diffraction (see below). Solvate free [CPh3]2[B12Cl12] is available by dissolving the compound in liquid sulfur dioxide followed by drying in vacuum at 50 °C over night.
[NO]2[B12Cl12].
[NO]+ salts are strong oxidizers and important reagents in organic chemistry.31 Recently [NO]+ salts of some weakly-coordinating anions have been reported.32,33a We decided to prepare [NO]2[B12Cl12], because it has the potential to oxidize a substrate and at the same time gives direct access to desired salts [substrate][B12Cl12]. It has been shown before that the metathesis of lithium salts of weakly coordinating anions with [cation][MF6] (M = As, Sb) in liquid sulfur dioxide represents a simple straight forward route to the respective salts of the weakly-coordinating anion.33 Following this methodology the metathesis reaction of Li2[B12Cl12] with [NO][BF4] in liquid sulfur dioxide yielded [NO]2[B12Cl12] as a red solid in quantitative yield (eqn (9)).| |  | (9) |
The 14N NMR spectrum in liquid SO2 shows a signal at 15 ppm for [NO]+, which is shifted downfield by about 20 ppm compared to previously published data.33a,34 The NO stretching frequency at 2214 cm−1 (IR) and 2220 cm−1 (Raman) fits well into the wide range of nitrosonium ion stretching frequencies ranging from 2150 to 2400 cm−1 and is similar to [NO]+ stretching frequencies in other salts containing chlorinated counter anions, cf. [NO][SbCl6] 2189 cm−1, [NO]2[SnCl6] 2191 cm−1, [NO][AlCl4] 2242 cm−1.35 In general, the [NO]+ stretching frequency in a salt with a chlorinated counter anion is smaller than in a salt with a similar fluorinated counter anion (e.g.[NO][SbCl6] 2189 cm−1vs. [NO][SbF6] 2385 cm−1), which has been explained in terms of polarization.35
A preliminary crystal structure of [NO]2[B12Cl12]·SO2 has been determined. The [NO]+ cations are disordered, which prohibits a detailed discussion of the structure (see ESI† for X-ray data).
3.4. X-Ray single crystal structure determinations
Several single crystal structures containing the [B12Cl12]2− anion have been reported in recent years, i.e. Cs2[B12Cl12] (three polymorphs are known),24,25,36 Cs2[B12Cl12]·nH2O (n = 1, 2),24 Cs2[B12Cl12]·2CH3CN,24 Ag2[B12Cl12],37 and [C2mim]2[B12Cl12].8
Cs2[B12Cl12]·SO2 (1).
Cs2[B12Cl12]·SO2 (1) crystallizes in the trigonal space groupR in a structure type similar to solvate-free Cs2[B12Cl12] and the hydrates Cs2[B12Cl12]·nH2O (n = 1, 2).25Fig. 3 shows the unit cell of 1. The SO2 molecule inserts between two Cs+ cations along the c-axis and increases the Cs+⋯Cs+ distance between these two cations from 763 pm in solvate-free Cs2[B12Cl12]25 to 924 pm in 1. The Cs–O distance of 338 pm is in good agreement with the sum of the ionic radius of Cs+ and the van der Waals radius of oxygen (345 pm),38 which indicates that the interaction is weak and the compound may be described as a solvate rather than a Cs–OSO complex. This is in agreement with almost unchanged O–S bond lengths (137(9)–140(8)) compared to free SO2 (142.97(4)).39 The Cs+ cation is surrounded by four [B12Cl12]2−cluster and one molecule SO2 giving a total coordination number of ten (Fig. 4).
![Unit cell of Cs2[B12Cl12]·SO2. The SO2 molecules are rotationally disordered over six positions. Selected averaged bond length [pm]: B–B 178.5, B–Cl 178.8, O1–S1 138.5.](/image/article/2009/DT/b821030f/b821030f-f3.gif) |
| | Fig. 3 Unit cell of Cs2[B12Cl12]·SO2. The SO2 molecules are rotationally disordered over six positions. Selected averaged bond length [pm]: B–B 178.5, B–Cl 178.8, O1–S1 138.5. | |
![Coordination sphere around Cs+ in Cs2[B12Cl12]·SO2. Selected bond length [pm]: O1–Cs1 337.9(9), Cl1–Cs1 365.4(2) (3×), Cl2–Cs1 349.5(2) (3×), Cl2′–Cs1 373.7(2) (3×).](/image/article/2009/DT/b821030f/b821030f-f4.gif) |
| | Fig. 4 Coordination sphere around Cs+ in Cs2[B12Cl12]·SO2. Selected bond length [pm]: O1–Cs1 337.9(9), Cl1–Cs1 365.4(2) (3×), Cl2–Cs1 349.5(2) (3×), Cl2′–Cs1 373.7(2) (3×). | |
[CPh3]2[B12Cl12]·2C2H4Cl2 (2) and [CPh3]2[B12Cl12]·2SO2 (3).
Fig. 5 shows the crystal structures of 2 and 3. Unit cell representations are deposited as ESI.† Both structures are very similar. The dianion [B12Cl12]2− forms two weak contacts via the chlorine atoms in 1 and 12 position to the carbenium centers of the planar trityl cations (sum of the angles around C1 is 360°). These contacts (Cl5⋯C1 346.1(2) (2), 350.4(2) (3) are longer than the sum of the van der Waals radii (328 pm)38 indicating a very weak interaction. In addition, on the opposite side of the trityl cation solvate molecules 1,2-C2H4Cl2 (2) or SO2 (3), respectively, coordinate very weakly to the carbenium center (C1⋯Cl8 347.9(2) (2), C1⋯O1 309.5(3) (3)). The C1–O1 distance is a little bit shorter than the sum of the van der Waals radii (325 pm),38 however, the SO2 molecule shows no distortion and bond length (142.0(2) and 142.3(2) pm) in agreement with free SO2 (142.97(4)).39 A similar structure without incorporated solvate molecules has been found for [CPh3]2[B12F12],6 with C⋯F distance of 308.7(2) pm in agreement with the sum of the van der Waals radii (300 pm).38 These results indicate that [B12Cl12]2−, despite being a dianion, is only weakly coordinating and comparable to its fluorinated relative [B12F12]2−.
![Crystal structures of [CPh3]2[B12Cl12]·2SO2 (top) and [CPh3]2[B12Cl12]·2C2H4Cl2 (bottom). Thermal ellipsoids are shown at the 50% probability level. Selected bond length [pm] and angles [°]: 2: av. B–B 178.2, av. B–Cl 178.8, Cl8–C21 176.5(3), C21–C20 147.3(5), C20–Cl7 182.3(4), Cl5–C1–Cl7 167.82(5), Cl5⋯C1 346.1(2), C1⋯Cl8 347.9(2); 3: av B–B 178.5, av. B–Cl 179.6, O1–S1 142.0(2), S1–O2 142.3(2), Cl5–C1–O1 166.67(7), Cl5⋯C1 350.4(2), C1⋯O1 309.5(3).](/image/article/2009/DT/b821030f/b821030f-f5.gif) |
| | Fig. 5 Crystal structures of [CPh3]2[B12Cl12]·2SO2 (top) and [CPh3]2[B12Cl12]·2C2H4Cl2 (bottom). Thermal ellipsoids are shown at the 50% probability level. Selected bond length [pm] and angles [°]: 2: av. B–B 178.2, av. B–Cl 178.8, Cl8–C21 176.5(3), C21–C20 147.3(5), C20–Cl7 182.3(4), Cl5–C1–Cl7 167.82(5), Cl5⋯C1 346.1(2), C1⋯Cl8 347.9(2); 3: av B–B 178.5, av. B–Cl 179.6, O1–S1 142.0(2), S1–O2 142.3(2), Cl5–C1–O1 166.67(7), Cl5⋯C1 350.4(2), C1⋯O1 309.5(3). | |
[NO]2[B12Cl12]·SO2 and [PPN]2[B12Cl12]·CH2Cl2.
Preliminary crystal structures of [NO]2[B12Cl12]·SO2 and [PPN]2[B12Cl12]·CH2Cl2 have been determined. The [B12Cl12]2− anions are well defined in both structures. Unfortunately, in both cases the cations are highly disordered, which prevents a detailed discussion. Crystal structure details and figures are included with the ESI.†
3.5. Structural and spectroscopic properties of [B12Cl12]2−
In all determined solid state structures the [B12Cl12]2− anion has almost Ih symmetry. A comparison of all known [B12Cl12]2− data (Table 2) gives an averaged B–B bond length of 178.7 and an averaged B–Cl bond length of 178.9 pm. These values are reproduced well by PBE0/TZVPP quantum chemical calculations which give 178.6 and 179.3 pm respectively. In accordance with its high Ih symmetry the vibrational spectra of [B12Cl12]2− show only few signals. The IR spectra has two intensive signals at 533 (νB–Cl) and 1028 cm−1 (νB–Cl) (plus three signals in the FIR region at 160 (δBBCl), 136, and 129 cm−1) and the Raman spectra two intense signals at 306 (νB–B) and 133 cm−1. Actual vibrational spectra are included with the ESI.† A detailed assignment of the vibrational spectra can be found in the literature.40 A slight distortion from ideal Ih symmetry of the [B12Cl12]2− anion in the solid state does not result in observable splitting of the vibrational frequencies. The limited number of distinct signals due to [B12Cl12]2− makes vibrational spectroscopy a valuable tool for the investigation of the nature of the counter cations. In the 11B NMR spectrum pure [B12Cl12]2− shows only one sharp resonance at −12.8 ppm.
Table 2 Comparison of averaged experimental (X-ray diffraction) and calculated (PBE0/TZVPP) B–B and B–Cl bond lengths
| |
B–B distance/pm |
B–Cl distance/pm |
Reference |
|
C2mim = 1-etyl-3-methylimidazolium
Preliminary crystal structure data has been deposited as ESI.1
|
| α-Cs2[B12Cl12] |
178.9 |
179.3 |
25
|
| β-Cs2[B12Cl12] |
178.9 |
178.8 |
24
|
| γ-Cs2[B12Cl12] |
179.4 |
177.6 |
24
|
| Cs2[B12Cl12]·2CH3CN |
178.7 |
179.4 |
24
|
| Ag2[B12Cl12] |
178.4 |
178.6 |
37
|
| [C2mim]2[B12Cl12]a |
178.6 |
179.2 |
8
|
| Cs2[B12Cl12]·SO2 (1) |
178.5 |
178.8 |
This work |
| [CPh3]2[B12Cl12]·2C2H4Cl2 (2) |
178.2 |
178.8 |
This work |
| [CPh3]2[B12Cl12]·2SO2 (3) |
178.5 |
179.6 |
This work |
| [NO]2[B12Cl12]·SO2 |
178.8 |
179.1 |
This workb |
| [PPN]2[B12Cl12]·CH2Cl2 |
178.5 |
178.5 |
This workb |
| calc. (PBE0/TZVPP) |
178.6 |
179.3 |
This work |
Acknowledgements
The authors are grateful to Professor Dr Ingo Krossing for support and helpful discussions, Boumahdi Benkmil for help with the X-ray diffraction measurements, and all the students who got involved in this work while doing their advanced inorganic chemistry labs. This work was supported by the Albert-Ludwigs Universität Freiburg and the Deutsche Forschungsgemeinschaft (DFG).
References
-
(a) See for reviews: S. H. Strauss, Chem. Rev., 1993, 93, 927–942 Search PubMed;
(b) I. Krossing and I. Raabe, Angew. Chem., 2004, 116, 2116–2142 CrossRef; I. Krossing and I. Raabe, Angew. Chem. Int. Ed., 2004, 43, 2066–2090 CrossRef CAS;
(c) I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS.
-
(a) Selected references: C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 Search PubMed;
(b) C. A. Reed, Chem. Commun., 2005, 1669–1677 RSC;
(c) S. V. Ivanov, J. J. Rockwell, O. G. Polyakov, C. M. Gaudinski, O. P. Anderson, K. A. Solntsev and S. H. Strauss, J. Am. Chem. Soc., 1998, 120, 4224–4225 CrossRef CAS;
(d) T. Küppers, E. Bernhardt, R. Eujen, H. Willner and C. W. Lehmann, Angew. Chem., 2007, 119, 6462–6465 CrossRef; T. Küppers, E. Bernhardt, R. Eujen, H. Willner and C. W. Lehmann, Angew. Chem. Int. Ed., 2007, 46, 6346–6349 CrossRef;
(e) A. Herzog, R. P. Callahan, C. L. B. Macdonald, V. M. Lynch, M. F. Hawthorne and R. J. Lagow, Angew. Chem., 2001, 113, 2179–2181 CrossRef; A. Herzog, R. P. Callahan, C. L. B. Macdonald, V. M. Lynch, M. F. Hawthorne and R. J. Lagow, Angew. Chem. Int. Ed., 2001, 40, 2121–2123 CrossRef CAS.
-
(a) K. C. Kim, C. A. Reed, G. S. Long and A. Sen, J. Am. Chem. Soc., 2002, 124, 7662–7663 CrossRef CAS;
(b) C. A. Reed, K. C. Kim, R. D. Bolskar and L. J. Mueller, Science, 2000, 289, 101–104 CrossRef CAS;
(c) T. Kato and C. A. Reed, Angew. Chem., 2004, 115, 2968–2971 CrossRef; T. Kato and C. A. Reed, Angew. Chem. Int. Ed., 2004, 43, 2907–2911;
(d) C. A. Reed, Acc. Chem. Res., 1998, 31, 325–332 CrossRef CAS.
- S. Körbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef ; and references therein.
- W. H. Knoth, H. C. Miller, J. C. Sauer, J. H. Balthis, Y. T. Chia and E. L. Muetterties, Inorg. Chem., 1964, 3, 159–167 CrossRef CAS.
- S. V. Ivanov, S. M. Miller, O. P. Anderson, K. A. Solntsev and S. H. Strauss, J. Am. Chem. Soc., 2003, 125, 4694–4695 CrossRef CAS.
-
(a) I. A. Zakharova, N. T. Kuznetsov and Y. L. Gaft, Inorg. Chim. Acta, 1978, 28, 271–274 CrossRef CAS;
(b) Y. L. Gaft, I. A. Zakharova and N. T. Kuznetsov, Russ. J. Inorg. Chem., 1980, 25, 726–729; Y. L. Gaft, I. A. Zakharova and N. T. Kuznetsov, Zh. Neorg. Khim., 1980, 25, 1308–1313 CAS;
(c) M. G. Voronkov, V. B. Pukhnarevich, I. I. Tsykhanskaya, N. I. Uhakova, Y. L. Gaft and I. A. Zakharova, Inorg. Chim. Acta, 1983, 68, 103–105 CrossRef CAS;
(d) Y. L. Gaft, N. T. Kuznetsov and L. M. Sukova, Russ. J. Inorg. Chem, 1983, 28, 89–92; Y. L. Gaft, N. T. Kuznetsov and L. M. Sukova, Zh. Neorg. Khim, 1983, 28, 162–167 CAS.
- M. Nieuwenhuyzen, K. R. Seddon, F. Teixidor, A. V. Puga and C. Viñas, Inorg. Chem., 2009, 48, 889–901 CrossRef CAS.
-
(a) A. N. Dey and J. Miller, J. Electrochem. Soc., 1979, 126, 1445–1451 CrossRef CAS;
(b) J. W. Johnson and M. S. Whittingham, J. Electrochem. Soc., 1980, 127, 1653–1654 CrossRef CAS;
(c) J. W. Johnson and A. H. Thompson, J. Electrochem. Soc., 1981, 128, 932–933 CrossRef CAS;
(d) J. W. Johnson and J. F. Brody, J. Electrochem. Soc., 1982, 129, 2213–2219 CrossRef CAS.
- M. W. Rupich, J. S. Foos and S. B. Brummer, J. Electrochem. Soc., 1985, 132, 119–122 CrossRef CAS.
-
(a)
S. V. Ivanov, W. J. Casteel Jr, G. P. Pez and M. Ulman, EP 1513215 A2, 2005;
(b)
G. P. Pez, S. V. Ivanov, G. Dantsin, W. J. Casteel and J. F. Lehmann, EP 1763099 A2, 2007.
- See for review: I. B. Sivaev, V. I. Bregadze and S. Sjöberg, Collect. Czech. Chem. Commun., 2002, 67, 679–727 Search PubMed.
- J. K. Ruff and W. J. Schlientz, Inorg. Synth., 1974, 15, 84–90 CAS.
- N. A. Zhukova, E. A. Malinina and N. T. Kuznetsov, Russ. J. Inorg. Chem., 1985, 30, 735–736; N. A. Zhukova, E. A. Malinina and N. T. Kuznetsov, Zh. Neorg. Khim., 1985, 30, 1292–1294 CAS.
-
G. M. Sheldrick, SHELX-97 Programs for Crystal Structure Analysis (Release 97-2), Institut für Anorganische Chemie der Universität Göttingen, 1997 Search PubMed.
-
Diamond-Visual Crystal Structure Information System, Crystal Impact, Bonn, Germany Search PubMed.
-
(a)
TURBOMOLE
Version 5.10; COSMOlogic;
(b) R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Let., 1989, 162, 165–169 CrossRef CAS.
-
(a) K. C. Nainan and G. E. Ryschkewitsch, Inorg. Nucl. Chem. Let., 1970, 6, 765–766 Search PubMed;
(b) G. E. Ryschkewitsch and K. C. Nainan, Inorg. Synth., 1974, 15, 113–118 CAS;
(c) K. G. Myakishev, I. I. Gorbacheva and V. V. Volkov, Russ. J. Inorg. Chem., 1984, 29, 526–529; K. G. Myakishev, I. I. Gorbacheva and V. V. Volkov, Zh. Neorg. Khim., 1984, 29, 912–916 CAS.
-
(a) L. V. Titov, E. R. Eremin and V. Y. Rosolovskii, Russ. J. Inorg. Chem., 1982, 27, 500–520; L. V. Titov, E. R. Eremin and V. Y. Rosolovskii, Zh. Neorg. Khim., 1982, 27, 891–895 CAS;
(b) W. Preetz and G. Peters, Eur. J. Inorg. Chem., 1999, 1831–1846 CrossRef CAS.
-
(a)
G. Brauer, Handbuch der Präparativen Anorganischen Chemie, 3rd edn, vol. 2, F. Enke Verlag, Stuttgart, 1979, pp. 794–795 Search PubMed;
(b)
Synthetic Methods of Organometallic and Inorganic Chemistry, ed. W. A. Herrmann, N. Auner and U. Klingebiel, Thieme Verlag, Stuttgart, 1996, p. 67 Search PubMed.
- M. Komura, K. Aono, K. Nagasawa and S. Sumimoto, Chem. Express, 1987, 2, 173–176 Search PubMed.
- F. Wang, Y. Wang and X. Wang, Yingyong Huaxue (Chin. J. Appl. Chem.), 1998, 15, 111–112 Search PubMed , (only available in Chinese).
- W. L. Smith, J. Chem. Ed., 1977, 54, 469–473 CrossRef CAS.
-
I. Tiritiris, Doctoral Thesis, Universität Stuttgart, 2004.
- I. Tiritiris and T. Schleid, Z. Anorg. Allg. Chem., 2004, 630, 1555–1563 CrossRef.
- H. G. Srebny, W. Preetz and H. C. Marsmann, Z. Naturforsch., 1984, 39B, 189–196 Search PubMed.
- K. A. Solntsev, S. V. Ivanov, S. G. Sakharova, S. B. Katser, A. S. Chernyavskii, N. A. Votinova, E. A. Klyuchishche and N. T. Kuznetsov, Russ. J. Coord. Chem., 1997, 23, 369–376 Search PubMed; K. A. Solntsev, S. V. Ivanov, S. G. Sakharova, S. B. Katser, A. S. Chernyavskii, N. A. Votinova, E. A. Klyuchishche and N. T. Kuznetsov, Koord. Khim., 1997, 23, 403–409.
- O. Haeckel and W. Preetz, Z. Anorg. Allg. Chem., 1995, 621, 1454–1458 CrossRef CAS.
- R. Hoffmann and W. N. Lipscomb, J. Chem. Phys., 1962, 37, 520–523 CrossRef CAS.
- N. A. Zhukova, E. A. Malinina and N. T. Kuznetsov, Russ. J. Inorg. Chem., 1985, 30, 735–736; N. A. Zhukova, E. A. Malinina and N. T. Kuznetsov, Zhur. Neorg. Khim., 1985, 30, 1292–1294 Search PubMed.
- G. I. Borodkin and V. G. Shubin, Russ. Chem. Rev, 2001, 70, 211–230 RSC; G. I. Borodkin and V. G. Shubin, Usp. Khim., 2001, 70, 241–261 ; and references therein.
-
(a) E. Bernhardt, M. Finze and H. Willner, Z. Allg. Anorg. Chem., 2006, 632, 248–250 CrossRef CAS;
(b) A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2008, 130, 11876–11877 CrossRef CAS.
-
(a) A. Decken, H. D. B. Jenkins, G. B. Nikiforov and J. Passmore, Dalton Trans., 2004, 2496–2504 RSC;
(b) C. Knapp, A. Mailman, G. B. Nikiforov and J. Passmore, J. Fluorine Chem., 2006, 127, 916–919 CrossRef CAS;
(c) A. Decken, C. Knapp, G. B. Nikiforov, J. Passmore, M. Rautiainen, X. Wang and X. Zeng, Chem. Eur. J., 2009 Search PubMed , submitted.
- J. Mason and K. O. Christe, Inorg. Chem., 1983, 22, 1849–1853 CrossRef CAS.
- D. W. A. Sharp and J. Thorley, J. Chem. Soc., 1963, 3557–3560 RSC.
- I. Tiritiris and T. Schleid, Z. Anorg. Allg. Chem., 2002, 628, 2212.
- I. Tiritiris and T. Schleid, Z. Anorg. Allg. Chem., 2003, 629, 581–583 CrossRef CAS.
-
(a)
J. Huheey, E. Keiter and R. Keiter, Anorganische Chemie, 2nd edn, de Gruyter, Berlin, 1995 Search PubMed;
(b) S. S. Batsanov, Inorg. Mater., 2001, 37, 871–855 CrossRef CAS; S. S. Batsanov, Neorg. Mater., 2001, 37, 1031–1046.
- R. Mews, E. Lork, P. G. Watson and B. Görtler, Coord. Chem. Rev., 2000, 197, 277–320 CrossRef CAS.
-
(a) L. A. Leites, S. S. Bukalov, A. P. Kurbakova, M. M. Kaganski, Y. L. Gaft, N. T. Kuznetsov and I. A. Zakharova, Spectrochim. Acta, 1982, 38A, 1047–1056 CrossRef CAS;
(b) S. J. Cyvin, B. N. Cyvin and T. Mogstad, Spectrochim. Acta, 1986, 42A, 985–989 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and discussion of the synthesis of [NEt3H]2[B12H12] and additional data for the chlorination of [B12H12]2−, all actual NMR, IR, and Raman spectra, details on the thermodynamics and quantum chemical calculations, preliminary crystal structure determinations of [NO]2[B12Cl12]·SO2 and [PPN]2[B12Cl12]·CH2Cl2, figures of the unit cells of [CPh3]2[B12Cl12]·2C2H4Cl2 (2) and [CPh3]2[B12Cl12]·2SO2 (3). CCDC reference numbers 710773–715826. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b821030f |
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.