Synthesis and characterization of new divalent lanthanide complexes supported by amine bis(phenolate) ligands and their applications in the ring opening polymerization of cyclic esters†
Divine T.
Dugah
a,
Brian W.
Skelton
b and
Ewan E.
Delbridge
*c
aChemistry Department, University of North Dakota, Grand Forks, ND 58202, USA. Tel: 701 777 0380/3222
bChemistry M313, School of Biomedical, Biomolecular and Chemical Sciences, The University of Western Australia, Crawley, WA 6009, Australia
cThe Lubrizol Corporation, 29400 Lakeland Blvd, Wickliffe, OH 44092, USA. Tel: 440 943 4200E-mail: eadl@lubrizol.com
First published on 15th January 2009
Abstract
Treatment of an array of bis(phenol)s H2Lx {Lx = [(−OC6H2(2,4-R)(6-CH2))2 NCH2CH2X], where X = CH2NMe2, NMe2, NEt2, OMe and R = But, Pet where Pet = C(CH3)2Et} with [Ln(N(SiMe3)2)2(THF)2] (Ln = Yb, Sm) in a 1:1 molar ratio in hexanes afforded a multitude of new divalent lanthanide bis(phenolate) complexes: [L2cYb]2 (X = NEt2, R = But) 1, [L2bYb]2 (X = CH2NMe2, R = But) 2, [L3aYb]2 (X = NMe2, R = Pet) 3, [L3bYb]2 (X = OMe, R = Pet) 4, [L2cSm] (X = NEt2, R = But) 5, [L3cYb] (X = NEt2, R = Pet) 6, [L3cSm] (X = NEt2, R = Pet) 7. X-Ray crystallographic analyses of compounds 1 and 3 reveal dimeric, centrosymmetric structures with 5-coordinate ytterbium centers arising from bridging and terminal phenolate groups. A selection of divalent compounds (1, 3, 4, 5, 6 and 7) were tested as catalyst precursors in the polymerization of ε-caprolactone and/or L-lactide which resulted in high molecular weight polymers with PDIs of 1.11–2.81 and 1.13–1.56 for ε-caprolactone and L-lactide respectively. All polymerization studies were performed in either toluene or THF and at room temperature (ε-caprolactone) or 70 °C (L-lactide) at a variety of catalyst:monomer ratios (1:100–1:300). Kinetics analyses of the polymerization of L-lactide by compounds 1 and 5 indicated pseudo-first order response with respect to L-lactide.
Introduction
The synthetic challenges, novel chemical properties and availability of the lanthanide elements continue to drive significant research efforts towards further understanding this often misunderstood group of elements. One area to receive increasing research efforts is the synthesis and stabilization of the ephemeral (and often elusive) divalent lanthanide compounds.1–4 More recently, ground-breaking research accomplishments have shattered the commonly held belief that only samarium, ytterbium and europium possess accessible divalent elements under standard laboratory conditions (i.e. solution chemistry)‡.5–8 Key to the stabilization of these divalent lanthanide compounds9–11 is the use of versatile, bulky, high-coordination number ligands (e.g. the cyclopentadienyl family) despite their hydrolytic sensitivity. In addition to the intrinsic interest posed by divalent lanthanide compounds, many researchers have capitalized on their one-electron reducing capabilities12–16 with continued focus on small molecule activation (e.g. CO2 and COS by [(C5Me5)2Sm(THF)2]17) and polymerization reactions {[(C5Me5)Ln(CH(SiMe3)2)(C5Me5)K(THF)2]n (Ln = Eu, Yb) in the polymerization of styrene and ethylene18}.Recently, interest in divalent lanthanides has progressed to include ligands with less organometallic Ln–C character19,20 and more Ln–X (X = N, O, S etc.)21,22 heteroatom bonding character (e.g. amides,4,23 bis(phenolate)s,14,24β-diketiminates25,26). The motivation for this progression away from organometallic ligand systems lies primarily in the ability of these ligands to impart greater stability to the catalytic systems without loss of ligand versatility. This becomes apparent where these divalent lanthanide systems are used as catalyst precursors in polymerization reactions.27–29 Although divalent lanthanide compounds are highly reactive and air- and moisture-sensitive, typically a pre-polymerization oxidation of the lanthanide(II) compound occurs (with concomitant reduction of an equivalent of the monomer)27–30 to afford the catalytically active trivalent lanthanide compound which then is available for the propagating polymerization reactions. The use of multidentate O, N donor ligands (rather than Cp ligands) imparts greater stability to the catalytically active trivalent systems which have significantly reduced propensity towards hydrolysis—inevitably arising from trace moisture/air contamination typically found in monomer feedstocks.
Among the recently reported non-cyclopentadienyl lanthanide compounds ligands, those containing bis(phenolate) ligands31–33 (Chart 1) have received interest due to their ease of preparation and chemical versatility (both electronic and steric). This versatility has led to the generation of a number of lanthanide bis(phenolate) compounds which have displayed impressive catalytic activities towards polar monomers such as lactide,33–37ε-caprolactone14,15,36,38–40 and methylmethacrylate.41 Only a handful of these examples14,15 contained divalent lanthanide precursors, despite their conceptually similar synthesis to their trivalent counterparts (Scheme 1). In addition to their catalytic properties, these divalent lanthanide bis(phenolate) compounds also demonstrate superior pathways to heteroleptic trivalent lanthanide complexes viaoxidation reactions;14,15 compared to the traditional salt metathesis reactions.39,42
 | ||
| Scheme 1 Synthesis of divalent samarium and ytterbium bis(phenolate) compounds. | ||

Previously the authors reported the first solvent-free Yb(II) bis(phenolate) dimer, {Yb[OC6H2(2,4-But)(6-CH2)]2NCH2CH2NMe2}2 which displayed promising catalytic activity towards ε-caprolactone in addition to oxidative reactivity towards a variety of anhydrous alcohols to afford a multitude of ytterbium(III) bis(phenolate) alkoxide heteroleptics.14 Subsequently, three new divalent Yb and Eu bis(phenolate) compounds, similar to that presented above, have been reported by Shen and Yao et al.24 Reported herein is a sizable expansion of the authors’ previous work, where syntheses involving several bis(phenolate) ligands coordinated to ytterbium and samarium, along with structural investigations (solution and solid-state), are presented. In addition, catalytic properties of these lanthanide compound towards ring opening polymerization (ROP) studies of cyclic esters (ε-caprolactone and L-lactide) have also been evaluated.
Results and discussion
Synthesis of bis(phenolate) ligands
The ligands (Chart 1) were prepared following literature procedures31 as analytically pure white powders via a single-step Mannich type condensation reaction of the commercially available formaldehyde, the appropriate substituted phenols and primary amines. Optimal balance between steric and electronic properties in stabilizing these lanthanide complexes were considered in ligand selection. Using similar protocols, ligands H2L3b and H2L3c, which have not been previously reported, were also synthesized with their identities being confirmed by typical spectroscopic techniques including elemental analysis, melting point, high-resolution mass spectrometry, and IR- and NMR-spectroscopy.Synthesis of divalent lanthanide bis(phenolate) complexes
Access to divalent ytterbium and samarium bis(phenolate) compounds was readily achieved by reaction of the appropriate [Ln(N(SiMe3)2)2(THF)2]4amide with one of the various bis(phenol) ligands displayed in Chart 1. A number of trends have been observed which indicated that steric encumbrance of the phenolate ligand along with metal selection play an important role in influencing overall stability and ultimately the solid-state and solution properties of the lanthanide bis(phenolate) compounds.Compounds 1–4. Substituting the methyl groups for the bulkier tertiary butyl (L2b,c) or tertiary pentyl (L3a,b) groups in the bis(phenol) ligands provided a convenient mechanism to accessing the analytically pure divalent ytterbium phenolate compounds 1–4 (Scheme 1) in good yields (65–83%). The proposed formulations (discussed below) are supported by typical analytical techniques (see Experimental) including IR- and NMR-spectroscopy, elemental analysis (which in some cases returned %C values which were low, but within the commonly held acceptable range for extremely air-sensitive divalent Ln compounds) and for compounds 1 and 3, X-ray crystallography.
X-ray crystallographic analysis of compounds 1 [YbL2c]2 and 3 [YbL3a]2, of which the crystals were obtained from non-donor solutions, revealed dimeric structures with a centrosymmetric YbO2Yb core and bridging and terminal phenolate oxygen donor atoms (vide infra). Detailed 1H NMR spectroscopic studies indicated that the solid-state structure is preserved in non-donor deuterated solvents such as C6D6 or C7D8 but the bis(phenolate) ligands become symmetrical and the ytterbium complex monomeric, upon addition of donor solvents such as C4D8O (Scheme 1). Moreover, this dynamic solution behavior from dimer to monomer is totally reversible and has been noted by the authors previously for a related ytterbium bis(phenolate) complex {Yb[OC6H2(2,4-But)(6-CH2)]2NCH2CH2NMe2}2.14 Subsequent research efforts by Shen and co-workers15 reported monomeric THF analogue of this compound which support the suppositions inferred from the aforementioned solution NMR spectroscopic studies.
Though it was not possible to obtain solid state structures for 2 [YbL2b] and 4 [YbL3b], all spectroscopic and analytical data not only supported the dimeric formulation ‘[YbL]2’ but also the dynamic solution behavior in donor/non-donor solvents such that both solid-state structures are considered dimeric i.e.[YbL]2 since each were isolated as solids from toluene/hexanes solutions.
Compounds 6,7. It was postulated that further increasing the steric constraints exerted by the bis(phenolate) ligands may suppress the propensity of these lanthanide bis(phenolate) complexes to exist as aggregates (or dimers) in the presence of non-donor solvent systems (see discussion above). As such, treatment of the appropriate [Ln(N(SiMe3)2)2(THF)2] with H2L3c (Chart 1), (where the side-arm substituents in H2L3a,b have been modified from X = NMe2 or OMe to the bulkier NEt2 group) in 1:1 molar ratio in hexanes afforded compounds 6 [YbL3c] and 7 [SmL3c]. Unlike compounds 1–4, longer reaction times¶ (9 and 24 h for compounds 6 and 7 respectively cf. 4 h for 1–4) were necessary to achieve quantitative protiolytic ligand exchange. Repeated attempts to obtain X-ray crystallographic quality crystals of compounds 6 and 7 were thwarted by formation of microcrystalline powders. However, the solution properties of both compounds were investigated by 1H and 13C NMR spectroscopic studies and revealed only one phenolate environment (as evidenced by only two unique aromatic C–H groups) which is consistent with a monomeric structural motif; akin to those described above and reported previously for the analogous THF adduct reported by Shen and coworkers.15 Moreover, NMR spectra of both 6 and 7 in C4D8O were almost identical to those recorded in C6D6, further supporting the supposition of predominant monomeric divalent lanthanide bis(phenolate) compounds. Not surprisingly, monomeric compound 7 [SmL3c] exhibited greater air-sensitivity than its ytterbium analogue, compound 6 [YbL3c], and both displayed greater air-sensitivity than the aggregated compounds 1–4 as ascertained semi-quantitatively by relative oxidation rates of the solids in air.
Compound 5. Several attempts to synthesize a number of divalent samarium bis(phenolate) compounds analogous to the ytterbium compounds described above were unsuccessful as indicated by typical auto-oxidation of the divalent samarium compound24 upon addition of the bis(phenol) reagent. Specifically, Shen noted that SmL2a could not be synthesized from [Sm(N(SiMe3)2)2(THF)2] and H2L2a. These observations are not surprising given the relative reducing power of the Sm(II)vs.Yb(II)45 {e.g. (C5Me5)2Ln (Ln = Sm, Yb) reacts with PhC
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH to afford the pureSm(III) complex [(C5Me5)2Sm]2(PhC
CH to afford the pureSm(III) complex [(C5Me5)2Sm]2(PhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCCPh)46vs. the mixed valent Yb complex [(C5Me5)2Yb(III)]2(μ-CCPh)4Yb(II)47}. In addition, the larger ionic radius of Sm(II)vs.Yb(II)45 undoubtedly enhances oxidation reactions by providing greater access of the bis(phenol) ligand substrate to the metal center. Similar oxidation reactions of Sm(II) involving less bulky bis(phenolate) ligands have been observed previously.48 Given these constraints, the successful isolation of a samarium(II) bis(phenolate) compound [SmL2c] 5, is a remarkable achievement (Scheme 1, Chart 1) given the L2c ligand is significantly less sterically demanding than that found in compound 7, L3c (described above). In the absence of any X-ray crystallographic analyses,|| it is difficult to explain the stability of this system; however an intimate balance between metal size and ligand bulk likely saturates the coordination environment about the metal center, protecting the metal center from further redox chemistry. The ‘[SmL2c]’ formulation proposed for compound 5, which may be dimeric or monomeric, was supported by elemental analysis,**49IR spectroscopy and NMR spectroscopy (see Experimental). Compound 5 was insoluble in non-donor solvents such as C6D6 or C7D8 precluding probing the degree of possible solution and solid-state aggregation. However, given similarities between compounds 5 and 1 [LnL2c], similar aggregation properties are likely wherein compound 1 was deemed as dimeric in non-donor solvents (see aforementioned discussion). For this reason, compound 5 is grouped with compounds 1–4 in Scheme 1.
CCCPh)46vs. the mixed valent Yb complex [(C5Me5)2Yb(III)]2(μ-CCPh)4Yb(II)47}. In addition, the larger ionic radius of Sm(II)vs.Yb(II)45 undoubtedly enhances oxidation reactions by providing greater access of the bis(phenol) ligand substrate to the metal center. Similar oxidation reactions of Sm(II) involving less bulky bis(phenolate) ligands have been observed previously.48 Given these constraints, the successful isolation of a samarium(II) bis(phenolate) compound [SmL2c] 5, is a remarkable achievement (Scheme 1, Chart 1) given the L2c ligand is significantly less sterically demanding than that found in compound 7, L3c (described above). In the absence of any X-ray crystallographic analyses,|| it is difficult to explain the stability of this system; however an intimate balance between metal size and ligand bulk likely saturates the coordination environment about the metal center, protecting the metal center from further redox chemistry. The ‘[SmL2c]’ formulation proposed for compound 5, which may be dimeric or monomeric, was supported by elemental analysis,**49IR spectroscopy and NMR spectroscopy (see Experimental). Compound 5 was insoluble in non-donor solvents such as C6D6 or C7D8 precluding probing the degree of possible solution and solid-state aggregation. However, given similarities between compounds 5 and 1 [LnL2c], similar aggregation properties are likely wherein compound 1 was deemed as dimeric in non-donor solvents (see aforementioned discussion). For this reason, compound 5 is grouped with compounds 1–4 in Scheme 1.
X-Ray crystallography of compounds 1 and 3
The results from single crystal X-ray structure determinations of compounds 1 and 3 are consistent with the proposed formulations, [YbL]2, in terms of stoichiometry and connectivity i.e. dimers with bridging and terminal asymmetric phenolate groups (Fig. 1 and 2 and Tables 1 and 2 respectively). Compound 1 was solved in the triclinic space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule of toluene, while compound 3 crystallizes in the monoclinic space groupP21/n.
with one molecule of toluene, while compound 3 crystallizes in the monoclinic space groupP21/n.
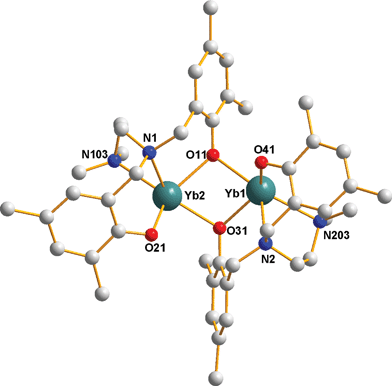 | ||
| Fig. 1 Projection of one molecule of 1, with hydrogen, tert-butyl methyl groups and toluene solvent molecule omitted for clarity. | ||
 | ||
| Fig. 2 Projection of one molecule of 3, with hydrogen and tert-pentyl methyl and ethyl groups omitted for clarity. Only one component of the disordered atoms is shown. | ||
| Bond distances (Å) | |||
| Yb1-O41 | 2.229(4) | Yb2-O21 | 2.245(4) |
| Yb1-O31 | 2.324(4) | Yb2-O11 | 2.328(3) |
| Yb1-O11 | 2.357(3) | Yb2-O31 | 2.371(4) |
| Yb1-N2 | 2.537(4) | Yb2-N1 | 2.521(4) |
| Yb1-N203 | 2.593(4) | Yb2-N103 | 2.582(5) |
| Angles (°) | |||
| O41-Yb1-O31 | 131.66(14) | O21-Yb2-O11 | 133.09(14) |
| O41-Yb1-O11 | 89.56(13) | O21-Yb2-O31 | 90.87(13) |
| O31-Yb1-O11 | 73.20(12) | O11-Yb2-O31 | 72.86(12) |
| O41-Yb1-N2 | 81.07(14) | O21-Yb2-N1 | 82.07(14) |
| O31-Yb1-N2 | 79.17(13) | O11-Yb2-N1 | 79.18(13) |
| O11-Yb1-N2 | 132.83(13) | O31-Yb2-N1 | 133.76(13) |
| O41-Yb1-N203 | 112.11(15) | O21-Yb2-N103 | 112.34(15) |
| O31-Yb1-N203 | 103.08(14) | O11-Yb2-N103 | 102.22(15) |
| O11-Yb1-N203 | 150.65(14) | O31-Yb2-N103 | 148.84(15) |
| N2-Yb1-N203 | 72.37(14) | N1-Yb1-N103 | 72.39(15) |
| Bond distances (Å) | |||
| Yb1-O41 | 2.208(5) | Yb2-O21 | 2.250(3) |
| Yb1-O31 | 2.316(5) | Yb2-O11 | 2.298(5) |
| Yb1-O11 | 2.326(4) | Yb2-O31 | 2.333(4) |
| Yb1-N2 | 2.530(4) | Yb2-N103 | 2.532(5) |
| Yb1-N203 | 2.580(5) | Yb2-N1 | 2.531(6) |
| Angles (°) | |||
| O41-Yb1-O31 | 139.35(17) | O21-Yb2-O11 | 130.66(12) |
| O41-Yb1-O11 | 95.26(17) | O21-Yb2-O31 | 89.23(14) |
| O31-Yb1-O11 | 72.52(16) | O11-Yb2-O31 | 72.72(16) |
| O41-Yb1-N2 | 79.74(16) | O21-Yb2-N103 | 110.37(17) |
| O31-Yb1-N2 | 79.76(15) | O11-Yb2-N103 | 104.76(17) |
| O11-Yb1-N2 | 130.20(16) | O31-Yb2-N103 | 153.12(16) |
| O41-Yb1-N203 | 100.88(18) | O21-Yb2-N1 | 78.85(14) |
| O31-Yb1-N203 | 105.73(17) | O11-Yb2-N1 | 79.73(18) |
| O11-Yb1-N203 | 154.74(16) | O31-Yb2-N1 | 130.93(16) |
| N2-Yb1-N203 | 72.33(15) | N103-Yb2-N1 | 72.78(16) |
Each compound can be described as having an η4-bis(phenolate) ligand coordinated to the ytterbium(II) metal center with coordination saturation being fulfilled through additional oxygen coordination (via bridging) from a neighboring η4-bis(phenolate) unit to give an overall 5-coordinate, dimeric ytterbium complex. These 5-coordinate ytterbium atoms gain additional electron density via agostic interactions of the tert-butyl and tert-pentyl hydrogen atoms in compounds 1 and 3 respectively. As expected, these agostic interactions†† are more significant in 3 which is probably due to the closer proximity of the tert-pentyl hydrogens to ytterbium in 3versus those found in the tert-butyl groups of 1. Ignoring these Yb⋯H interactions, the formal 5-coordination environment about ytterbium does not readily fit either symmetrical trigonal bipyramidal or square-based pyramid models, which is probably attributed to the overall conformational rigidity of the bis(phenolate) ligands. The YbO2Yb cores in both compounds have symmetrical bridging modes with almost equivalent Yb–(μ-O) bond lengths which were longer (by ca. 0.10 Å) than terminal Yb–O bond lengths. In addition, all anionic Yb–O bond lengths are shorter than Yb–N coordinate bonds; all of which are comparable to other bridged Yb(II) bis(phenolate)s including the related {Yb[OC6H2(2,4-But)(6-CH2)]2NCH2CH2NMe2}2.14
Polymerization of ε-caprolactone and L-lactide
While there are a numerous examples of trivalent lanthanide catalyst precursors for the ring opening polymerization (ROP) of cyclic esters such as lactide33,34,36,50,51 and lactones,36,38–40,52,53 there are surprisingly few instances where divalent lanthanide compounds have been employed.14,15,30,54 The divalent ytterbium and samarium bis(phenolate) compounds 1–7 (vide supra) represent ideal candidates for preliminary exploration into the propensity of these compounds to function as catalyst precursors in ROP of L-lactide and/or ε-caprolactone to produce polymers which have numerous applications from bio-friendly packaging materials to bio-assimilable drug-delivery agents.55–57 Reported herein are our results from initialROP studies employing a variety of divalent lanthanide bis(phenolate) compounds. A number of trends were observed, including: 1) Divalent lanthanide bis(phenolate)s effectively polymerize both ε-caprolactone and the more challenging L-lactide quantitatively 2) Samarium bearing catalysts are more active than comparable ytterbium analogues 3) Donor solvents such as THF suppress polymerization rates.| Entry | Initiator | Time/min | [M]0/[I]0 | % Conv.b | 10−4Mn(obs.) c | PDIc |
|---|---|---|---|---|---|---|
| a Polymerization conditions: Toluene as solvent (unless otherwise stated), T = RT, Vtol/VCL = 1.86. b Determined by 1H NMR analysis. c Determined by GPC analysis in toluene with calibration to polystyrene standards. d Solvent = THF. | ||||||
| 1 | 1 | 10 | 200 | 99.6 | 1.59 | 1.22 |
| 2 | 4 | 15 | 300 | 99.8 | 3.82 | 1.99 |
| 3 | 5 | 3 | 100 | 99.6 | 3.72 | 2.81 |
| 4 | 5 d | 15 | 100 | 99.7 | 4.67 | 2.62 |
| 5 | 5 | 7 | 277 | 99.1 | 3.26 | 1.11 |
| 6 | 7 | 8 | 100 | 98.9 | 1.92 | 2.14 |
| 7 | 7 | 13 | 200 | 98.4 | 1.96 | 1.85 |
| 8 | 7 d | 45 | 100 | 97.6 | 1.86 | 1.42 |
In general, samarium-based catalysts polymerized ε-caprolactone more rapidly than their ytterbium counterparts which is in accordance with the findings of other researchers51–53,59,60 where larger lanthanide metal containing catalysts (e.g. Nd vs.Yb) are more active ROP catalysts than those containing smaller lanthanide ions.
Replacing toluene for donor solvents such as THF in the ROP reactions invariably suppressed polymerization rates which again is consistent with previous studies50,52 that have attributed this to probable competition between the THF molecules and monomer substrates for coordination to the metal center (the necessary first step in the polymerization reaction). This, in turn, slows down the polymerization process.
While the molecular structure of the catalyst (i.e. monomer vs. dimer) in solution will undoubtedly have a profound effect on ROP rates, it is difficult to understand the subtleties of these effects in the present study, since the influence of the monomer substrate (i.e. the cyclic ester) on catalyst aggregation disruption is unknown. However, the dimeric compound 1 has a significantly lower catalytic activity compared with the analogous monomeric Yb complex {(THF)2Yb[OC6H2(2,4-But)(6-CH2)]2NCH2CH2NMe2}15 but has comparable activity to other dimeric systems, such as [L*Yb(THF)2]2 {L* = 2,2′-methylene-bis(6-butyl-4-methylphenoxo)}.54
| Entry | Initiator | Time/min | [M]0/[I]0 | % Conv.b | 10−4Mn(obs.) c | PDIc |
|---|---|---|---|---|---|---|
| a Polymerization conditions: toluene as solvent (except stated otherwise), T = 70 °C. b Determined by 1H NMR analysis. c Dertemined by GPC analysis in toluene with calibration to polystyrene standards. d Solvent = THF. e T = 25 °C. | ||||||
| 1 | 1 e | 24 h | 100 | 6.34 | — | — |
| 2 | 5 e | 24 h | 100 | 9.65 | — | — |
| 3 | 1 | 60 | 100 | 86.6 | 1.61 | 1.13 |
| 4 | 5 | 30 | 100 | 95.8 | 2.61 | 1.56 |
| 5 | 5 d | 50 | 100 | 93.6 | 1.96 | 1.25 |
| 6 | 7 | 45 | 100 | 94.7 | 3.1 | 1.3 |
Finally, pseudo-first order kinetics in lactide were observed from ln{[LA0]/[LA]t} vs. time plots from systems employing catalysts 1 and 5 at initiators:monomer ratios of 1:50 and 1:100 respectively (kobs(I) = 0.0416 min−1 and kobs(II) = 0.0321 min−1) (Fig. 3). These data suggest a mechanism which requires one molecule of lactide per active site of catalyst (i.e. one molecule LA per monomeric lanthanide catalyst or two molecules per dimeric lanthanide catalyst).
![Pseudo-first order kinetics plot of L-lactide polymerization catalyzed by compounds 1 and 5 in toluene at 70 °C. (I) [1]:[LA] = 1:50, (II) [5]:[LA] = 1:100.](/image/article/2009/DT/b816916k/b816916k-f3.gif) | ||
| Fig. 3 Pseudo-first order kinetics plot of L-lactide polymerization catalyzed by compounds 1 and 5 in toluene at 70 °C. (I) [1]:[LA] = 1:50, (II) [5]:[LA] = 1:100. | ||
Conclusions
A number of novel divalent ytterbium and samarium bis(phenolate) complexes have been synthesized by straight-forward amine elimination reactions. The less sterically demanding bis(phenolate) ligands were found to produce trivalent lanthanide complexes. However, more bulky ligand systems produced isolable divalent lanthanide species which exist as either dimers (Ln = Yb) or monomers in solution (Ln = Yb, Sm); as determined by the intimate relationship between metal-ion size and steric demands of the ligands. X-Ray crystallographic characterization of compounds 1 and 3 revealed dimeric ytterbium(II) compounds with symmetrically-bridging bis(phenolate) ligands; supporting the proposed formulations ascertained by solution NMR spectroscopic studies. A selection of these divalent lanthanide compounds has been evaluated as catalysts in the ROP of ε-caprolactone and L-lactide cyclic esters. In most cases quantitative polymer conversion was established, making these catalysts the firstgroup of divalent lanthanide bis(phenolate) complexes to display such activity towards L-lactide, the more difficult of the cyclic esters to polymerize. Finally, kinetic studies revealed that these reactions propagate with pseudo-first order kinetics with respect to L-lactide.Experimental
General
Since all lanthanide complexes described herein are air- and moisture-sensitive all manipulations were performed under nitrogen atmospheres using typical Schlenk line and glove-box techniques. Solvents were dried and purified by distillation under nitrogen from sodium or potassium metal mixed with benzophenone while deuterated solvents (C4D8O, C6D6 and C7D8) were dried over sodium metal and purified by vacuum transfer. The lanthanide precursors [Ln(N(SiMe3)2)2(THF)2] (Ln = Sm, Yb) were synthesized by the reported procedure.4 The starting materials used for the synthesis of the ligands H2LR {LR = [−OC6H2(2,4-R)(6-CH2)]2NCH2CH2X} (R = Me, But, Pet)31,32 were purchased from Acros and used without further purification. Melting points were obtained from sealed capillaries on a Mel-Temp apparatus and are uncorrected. IR spectra (4000–450 cm−1) were recorded as KBr Nujol mulls on an ATI Mattson Genesis Series FTIR Spectrometer. The 1H and 13C NMR spectra were recorded on a Bruker AVANCE-500 NMR spectrometer and referenced to C6D5H or C4D7HO or CHCl3 (1H) and C6D6 or C4D8O or CDCl3 (13C). Elemental analyses (sealed ampoules under inert atmosphere) were performed by Midwest Microlab, Indianapolis, IN.![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) max/cm−1 3370 s, 2623 m, 2036w, 1600w, 1299 s, 1261 m, 1211w, 1095 s, 1060 m, 1018 s, 872 m, 802 m, 725 m, 601w (Nujol); MS: m/z 568.5 [M + H]+. δH (500.1 MHz, CDCl3, 298 K) 0.63 (6H, t, J 7.5 Hz, CCH2CH3), 0.65 (6H, t, J 7.6 Hz, CCH2CH3), 1.25 (12H, s, C(CH3)2), 1.33 (12H, s, C(CH3)2), 1.57 (4H, q, J 7.5 Hz, CCH2CH3), 1.92 (4H, q, J 7.6 Hz, CCH2CH3), 2.72 (2H, t, J 5.2 Hz, NCH2CH2O), 3.45 (3H, s, OCH3), 3.51 (2H, t, J 5.2 Hz, NCH2CH2O), 3.78 (4H, s, ArCH2N), 6.83 (2H, s, Ar), 7.13 (2H, s, Ar), 8.54 (2H, s, OH), δC, (125.8 MHz, CDCl3, 298K) 9.6 (CCH2CH3), 10.0 (CCH2CH3), 28.2 (C(CH3)2Et), 29.0 (C(CH3)2Et), 33.2 (C(CH3)2Et), 37.5 (CCH2CH3), 37.6 (CCH2CH3), 38.9 (C(CH3)2Et), 51.7 (NCH2CH2O), 58.5 (NCH2CH2O), 59.2 (OCH3), 71.8 (ArCH2N), 121.8 (arom-CH), 125.9 (arom-CPet), 126.0 (arom-CH), 134.6 (arom-CPet), 139.3 (arom-CCH2N), 153.1 (arom-CO).
max/cm−1 3345 s, 2730w, 2356 m, 1600 m, 1261 m, 1095 s, 1049 m, 1022 s, 872w, 802 s, 725 m, 609 w (Nujol); MS: m/z 609.5 [M + H]+. δH (500.1 MHz, C4D8O, 298 K) 0.65 (6H, t, J 7.0 Hz, CCH2CH3), 0.70 (6H, t, J 8.1 Hz, CCH2CH3), 1.51 (6H, t, J 7.0 Hz, N(CH2CH3)2), 1.29 (12H, s, C(CH3)2), 1.41 (12H, s, C(CH3)2), 1.63 (4H, q, J 7.0 Hz CCH2CH3), 1.95 (4H, q, J 8.1 Hz, CCH2CH3), 2.68-2.63 (8H, overlapping signals of NCH2CH2N and N(CH2CH3)2), 3.75 (4H, s, ArCH2N), 6.88 (2H, s, Ar), 7.12 (2H, s, Ar), 9.7 (2H, s, OH), δC{H} (125.8 MHz, C4D8O, 298K) 9.6 (CCH2CH3), 10.0 (CCH2CH3), 28.14 (C(CH3)2Et), 29.0 (C(CH3)2Et), 33.2 (C(CH3)2Et), 34.0 (C(CH3)2Et), 37.6 (CCH2CH3), 38.9 (CCH2CH3), 45.7 (NCH2CH3), 48.1 (NCH2CH3), 49.2 (NCH2CH2N), 50.4 (NCH2CH2N), 57.1 (ArCH2N), 121.6 (arom-CH), 125.9 (arom-CPet), 127.2 (arom-CH), 124.4 (arom-CPet), 138.6 (arom-CCH2N), 155.5 (arom-CO).
max/cm−1 1738 w, 1655 m, 1260 s, 1623 m, 1260 s, 1091 s, 1022 s, 866 w, 800 s, 697 w, 570 w, 546 m (Nujol); δH (500.1 MHz, C4D8O, 298K) 0.66 (6H, t, J 7 Hz, N(CH2CH3)2), 1.24 (18H, s, But), 1.47 (18H, s, But), 2.05 (2H, t, J 6.9 Hz, NCH2CH2N), 2.24 (4H, q, J 7 Hz, N(CH2CH3)2), 2.25 (2H, t, J 6.9 Hz, NCH2CH2N), 2.89 (2H, d, J 12.3 Hz, ArCH2N), 4.55 (2H, d, J 12.3 Hz, ArCH2N), 6.87 (2H, s, Ar), 7.09 (2H, s, Ar), δH (500.1 MHz, C6D6) 0.67 (6H, t, J 7.0 Hz, N(CH2CH3)2), 1.34 (6H, t, J 6.7 Hz, N(CH2CH3)2), 1.36–1.80 (80H, overlapping signals of But and NCH2CH2N), 1.95 (4H, q, J 7.0 Hz, N(CH2CH3)2), 2.97 (4H, q, J 6.7 Hz, N(CH2CH3)2), 3.25 (2H, d, J 12.2 Hz, ArCH2N), 3.47 (2H, d, J 12.2 Hz, ArCH2N), 4.88 (2H, d, J 12.2 Hz, ArCH2N), 5.68 (2H, d, J 12.2 Hz, ArCH2N), 7.03 (2H, s, Ar), 7.14 (2H, s, Ar), 7.35 (2H, s, Ar), 7.54 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298 K) 5.3 (N(CH2CH3)2), 27.7 (CMe3), 29.8 (CMe3), 33.2 (CMe3), 37.7 (CMe3), 41.0 ((N(CH2CH3)2), 44.7 (NCH2CH2N), 49.2 (NCH2CH2N), 62.3 (ArCH2), 120.9 (arom-CH), 122.1 (arom-CBut), 124.4 (arom-CH), 129.0 (arom-CCH2N), 132.6 (arom-CBut), 164.2 (arom-CO).
max/cm−1 2370w, 2342w, 1603 s, 1314w, 1261 s, 1093 s, 1024 s, 871 m, 801 s, 680w, 580 m (Nujol); δH (500.1 MHz, C4D8O, 298 K) 1.28 (18H, s, But), 1.41 (18H, s, But), 1.50 (2H, m, NCH2CH2CH2N), 1.79 (2H, t, J 7.1 Hz, NCH2CH2CH2N), 1.84 (6H, s, NMe2), 2.32 (2H, t, J 7.1 Hz, NCH2CH2CH2N), 2.75 (2H, d, J 13.0 Hz, ArCH2N), 3.93 (2H, d, J 13.0 Hz, ArCH2N), 6.19 (2H, s, Ar), 7.08 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 20.1 (NCH2CH2CH2N), 29.1 (CMe3), 31.2 (CMe3), 32.9 (CMe3), 34.6 (CMe3), 44.6 (NMe2), 46.2 (NCH2CH2CH2N), 58.7 (NCH2CH2CH2N), 59.8 (ArCH2N), 121.9 (arom-CH), 125.6 (arom-CBut), 127.5 (arom-CH), 129.9 (arom-CBut), 133.9 (arom-CCH2N), 165.5 (arom-CO).
max/cm−1 1739 w, 1600 w, 1299 w, 1261 s, 1214 w, 1161 w, 1095 s, 1022 s, 933 w, 872 m, 802 s, 725 m, 602 m (Nujol); δH (500.1 MHz, C4D8O, 298 K) 0.61–0.64 (12H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 1.20 (12H, s, C(CH3)2CH2CH3), 1.41 (12H, s, C(CH3)2CH2CH3), 1.55 (6H, s, NMe2), 1.72 (4H, q, J 2 Hz, C(CH3)2CH2CH3), 1.81 (4H, q, J 2 Hz, C(CH3)2CH2CH3), 2.34 (2H, t, J 5.5 Hz, NCH2CH2N), 2.87 (2H, t, J 5.5 Hz, NCH2CH2N), 2.89 (2H, d, J 12.1 Hz, ArCH2N), 4.14 (2H, d, J 12.1 Hz, ArCH2N), 6.74 (2H, s, Ar), 6.99 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 6.9 (C(CH3)2CH2CH3), 7.5 (C(CH3)2CH2CH3), 25.4 (C(CH3)2CH2CH3), 25.9 (C(CH3)2CH2CH3), 26.9 (C(CH3)2CH2CH3), 30.5 (C(CH3)2CH2CH3), 34.8 (C(CH3)2CH2CH3), 36.5 (C(CH3)2CH2CH3), 42.9 (NMe2), 44.9 (NCH2CH2N), 57.6 (NCH2CH2N), 62.7 (ArCH2N), 123.3 (arom-CH), 125.3 (arom-CPet), 126.2 (arom-CH), 126.6 (arom-CPet), 130.8 (arom-CCH2N), 164.2 (arom-CO).
max/cm−1 3370 s, 2623 m, 2036w, 1600w, 1299 s, 1261 m, 1211w, 1095 s, 1060 m, 1018 s, 872 m, 802 m, 725 m, 601 w (Nujol); δH (500.1 MHz, C4D8O, 298 K) 0.62–0.65 (12H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 1.22 (12H, s, C(CH3)2CH2CH3), 1.36 (12H, s, C(CH3)2CH2CH3), 1.56 (4H, q, J 5.1 Hz, C(CH3)2CH2CH3), 1.78 (4H, q, J 5.5 Hz, C(CH3)2CH2CH3), 2.59 (2H, t, J 4.1 Hz, NCH2CH2O), 2.81 (3H, s, OMe), 2.93 (2H, d, J 11.1 Hz, ArCH2N), 2.98 (2H, t, J 4.1 Hz, NCH2CH2O), 6.7 (2H, s, Ar), 7.13 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 7.2 (C(CH3)2CH2CH3), 7.7 (C(CH3)2CH2CH3), 25.6 (C(CH3)2CH2CH3), 25.8 (C(CH3)2CH2CH3), 27.1 (C(CH3)2CH2CH3), 27.2 (C(CH3)2CH2CH3), 30.6 (C(CH3)2CH2CH3), 34.9 (C(CH3)2CH2CH3), 35.7 (NCH2CH2O), 38.8 (NCH2CH2O), 55.6 (OCH3), 62.3 (ArCH2N), 122.0 (arom-CPet), 123.3 (arom-CH), 125.4 (arom-CH), 126.9 (arom-CPet), 131.2 (arom-CCH2N), 164.1 (arom-CO).
max/cm−1 1775w, 1607 m, 1442 m, 1350w, 1367w, 1323w, 1268 s, 1163w, 1098 s, 1027 s, 931w, 876 s, 803 s, 740 s, 645w (Nujol); δH (500.1 MHz, C4D8O, 298K) 0.19 (18H, s, But), 0.33 (18H, s, But), 0.87 (6H, t, J 10 Hz, N(CH2CH3)2), 1.21 (2H, d, J 11.5 Hz, ArCH2), 1.37 (4H, q, J 10.2 Hz, N(CH2CH3)2), 3.53 (4H, m, NCH2CH2N), 4.23 (2H, d, J 11.5 Hz, ArCH2), 6.3 (2H, s, Ar), 7.2 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 11.6 (N(CH2CH3)2), 30.4 (CMe3), 30.9 (CMe3), 32.4 (CMe3), 32.9 (CMe3), 35.8 (NCH2CH2N), 36.1 (NCH2CH2N), 40.5 ((N(CH2CH3)2), 56.3 (ArCH2N), 121.5 (arom-CH), 122.2 (arom-CBut), 122.4 (arom-CH), 123.6 (arom-CCH2N), 125.0 (arom-CBut), 134.2 (arom-CO).
max/cm−11741w, 1612w, 1301 m, 1261 s, 1220w, 1165w, 1100 s, 1024 s, 933w, 875 m, 812 s, 725 m, 603 m (Nujol); δH (500.1 MHz, C4D8O, 298 K) 0.62–0.66 (12H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 1.10 (4H, q, J 7.1 Hz, C(CH3)2CH2CH3), 1.21 (12H, s, C(CH3)2CH2CH3), 1.23 (6H, t, J 6.2 Hz, N(CH2CH3)2), 1.34 (12H, s, C(CH3)2CH2CH3), 1.72 (4H, q, J 7.1 Hz, C(CH3)2CH2CH3), 1.91 (4H, q, J 6.2 Hz, N(CH2CH3)2), 2.26 (2H, t, J 7.0 Hz, NCH2CH2N), 2.88 (2H, t, J 7.0 Hz, NCH2CH2N), 3.08 (2H, d, J 12.3 Hz, ArCH2N), 4.21 (2H, d, J 12.3 Hz, ArCH2N), 6.66 (2H, s, Ar), 7.07 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 5.1 (C(CH3)2CH2CH3), 6.8 (C(CH3)2CH2CH3), 7.0 (C(CH3)2CH2CH3), 7.3 (C(CH3)2CH2CH3), 7.4 (C(CH3)2CH2CH3), 7.5 (C(CH3)2CH2CH3), 7.7 (C(CH3)2CH2CH3), 8.0 (C(CH3)2CH2CH3), 25.7 (N(CH2CH3)2), 43.4 (N(CH2CH3)2), 48.1 (NCH2CH2N), 49.4 (NCH2CH2N), 62.5 (ArCH2N), 119.9 (arom-CH), 123.4 (arom-CPet), 125.3 (arom-CH), 130.8 (arom-CPet), 151.4 (arom-CCH2N), 164.3 (arom-CO).
max/cm−1 1604 m, 1413 w, 1380 m, 1362w, 1324 w, 1262 s, 1203 m, 1165 w, 1094 m, 1023 m, 933 w, 915 w, 880 s, 813 s, 7420 s, 653 w, 521 s (Nujol); δH (500.1 MHz, C4D8O, 298 K) −6.21 (6H, t, J 5.3 Hz C(CH3)2CH2CH3), −0.83 (6H, t, J 5.1 Hz, C(CH3)2CH2CH3), −0.03 (12H, s, C(CH3)2CH2CH3), 0.16 (12H, s, C(CH3)2CH2CH3), 0.21 (6H, t, J 7.1 Hz, N(CH2CH3)2), 0.48 (8H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 0.51 (4H, q, J 7.1 Hz, N(CH2CH3)2), 1.26 (4H, m, NCH2CH2N), 1.62 (2H, d, J 11.1 Hz, ArCH2N), 1.72 (2H, d, J 11.1 Hz, ArCH2N), 5.22 (2H, s, Ar), 5.46 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 2.2 (C(CH3)2CH2CH3), 5.9 (C(CH3)2CH2CH3), 22.5 (C(CH3)2CH2CH3), 26.6 (C(CH3)2CH2CH3), 26.9 (C(CH3)2CH2CH3), 27.2 (C(CH3)2CH2CH3), 29.8 (C(CH3)2CH2CH3), 32.8 (C(CH3)2CH2CH3), 34.6 (N(CH2Me)2), 35.7, (N(CH2Me)2), 37.1 (NCH2CH2N), 37.7 (NCH2CH2N), 44.7 (ArCH2), 103.6 (arom-CH), 120.6 (arom-CPet), 122.9 (arom-CH), 127.7 (arom-CPet), 133.7 (arom-CCH2N), 167.5 (arom-CO).
max/cm−1 3370 s, 2623 m, 2036w, 1600w, 1299 s, 1261 m, 1211w, 1095 s, 1060 m, 1018 s, 872 m, 802 m, 725 m, 601w (Nujol); MS: m/z 568.5 [M + H]+. δH (500.1 MHz, CDCl3, 298 K) 0.63 (6H, t, J 7.5 Hz, CCH2CH3), 0.65 (6H, t, J 7.6 Hz, CCH2CH3), 1.25 (12H, s, C(CH3)2), 1.33 (12H, s, C(CH3)2), 1.57 (4H, q, J 7.5 Hz, CCH2CH3), 1.92 (4H, q, J 7.6 Hz, CCH2CH3), 2.72 (2H, t, J 5.2 Hz, NCH2CH2O), 3.45 (3H, s, OCH3), 3.51 (2H, t, J 5.2 Hz, NCH2CH2O), 3.78 (4H, s, ArCH2N), 6.83 (2H, s, Ar), 7.13 (2H, s, Ar), 8.54 (2H, s, OH), δC, (125.8 MHz, CDCl3, 298K) 9.6 (CCH2CH3), 10.0 (CCH2CH3), 28.2 (C(CH3)2Et), 29.0 (C(CH3)2Et), 33.2 (C(CH3)2Et), 37.5 (CCH2CH3), 37.6 (CCH2CH3), 38.9 (C(CH3)2Et), 51.7 (NCH2CH2O), 58.5 (NCH2CH2O), 59.2 (OCH3), 71.8 (ArCH2N), 121.8 (arom-CH), 125.9 (arom-CPet), 126.0 (arom-CH), 134.6 (arom-CPet), 139.3 (arom-CCH2N), 153.1 (arom-CO).
max/cm−1 3345 s, 2730w, 2356 m, 1600 m, 1261 m, 1095 s, 1049 m, 1022 s, 872w, 802 s, 725 m, 609 w (Nujol); MS: m/z 609.5 [M + H]+. δH (500.1 MHz, C4D8O, 298 K) 0.65 (6H, t, J 7.0 Hz, CCH2CH3), 0.70 (6H, t, J 8.1 Hz, CCH2CH3), 1.51 (6H, t, J 7.0 Hz, N(CH2CH3)2), 1.29 (12H, s, C(CH3)2), 1.41 (12H, s, C(CH3)2), 1.63 (4H, q, J 7.0 Hz CCH2CH3), 1.95 (4H, q, J 8.1 Hz, CCH2CH3), 2.68-2.63 (8H, overlapping signals of NCH2CH2N and N(CH2CH3)2), 3.75 (4H, s, ArCH2N), 6.88 (2H, s, Ar), 7.12 (2H, s, Ar), 9.7 (2H, s, OH), δC{H} (125.8 MHz, C4D8O, 298K) 9.6 (CCH2CH3), 10.0 (CCH2CH3), 28.14 (C(CH3)2Et), 29.0 (C(CH3)2Et), 33.2 (C(CH3)2Et), 34.0 (C(CH3)2Et), 37.6 (CCH2CH3), 38.9 (CCH2CH3), 45.7 (NCH2CH3), 48.1 (NCH2CH3), 49.2 (NCH2CH2N), 50.4 (NCH2CH2N), 57.1 (ArCH2N), 121.6 (arom-CH), 125.9 (arom-CPet), 127.2 (arom-CH), 124.4 (arom-CPet), 138.6 (arom-CCH2N), 155.5 (arom-CO).
max/cm−1 1738 w, 1655 m, 1260 s, 1623 m, 1260 s, 1091 s, 1022 s, 866 w, 800 s, 697 w, 570 w, 546 m (Nujol); δH (500.1 MHz, C4D8O, 298K) 0.66 (6H, t, J 7 Hz, N(CH2CH3)2), 1.24 (18H, s, But), 1.47 (18H, s, But), 2.05 (2H, t, J 6.9 Hz, NCH2CH2N), 2.24 (4H, q, J 7 Hz, N(CH2CH3)2), 2.25 (2H, t, J 6.9 Hz, NCH2CH2N), 2.89 (2H, d, J 12.3 Hz, ArCH2N), 4.55 (2H, d, J 12.3 Hz, ArCH2N), 6.87 (2H, s, Ar), 7.09 (2H, s, Ar), δH (500.1 MHz, C6D6) 0.67 (6H, t, J 7.0 Hz, N(CH2CH3)2), 1.34 (6H, t, J 6.7 Hz, N(CH2CH3)2), 1.36–1.80 (80H, overlapping signals of But and NCH2CH2N), 1.95 (4H, q, J 7.0 Hz, N(CH2CH3)2), 2.97 (4H, q, J 6.7 Hz, N(CH2CH3)2), 3.25 (2H, d, J 12.2 Hz, ArCH2N), 3.47 (2H, d, J 12.2 Hz, ArCH2N), 4.88 (2H, d, J 12.2 Hz, ArCH2N), 5.68 (2H, d, J 12.2 Hz, ArCH2N), 7.03 (2H, s, Ar), 7.14 (2H, s, Ar), 7.35 (2H, s, Ar), 7.54 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298 K) 5.3 (N(CH2CH3)2), 27.7 (CMe3), 29.8 (CMe3), 33.2 (CMe3), 37.7 (CMe3), 41.0 ((N(CH2CH3)2), 44.7 (NCH2CH2N), 49.2 (NCH2CH2N), 62.3 (ArCH2), 120.9 (arom-CH), 122.1 (arom-CBut), 124.4 (arom-CH), 129.0 (arom-CCH2N), 132.6 (arom-CBut), 164.2 (arom-CO).
max/cm−1 2370w, 2342w, 1603 s, 1314w, 1261 s, 1093 s, 1024 s, 871 m, 801 s, 680w, 580 m (Nujol); δH (500.1 MHz, C4D8O, 298 K) 1.28 (18H, s, But), 1.41 (18H, s, But), 1.50 (2H, m, NCH2CH2CH2N), 1.79 (2H, t, J 7.1 Hz, NCH2CH2CH2N), 1.84 (6H, s, NMe2), 2.32 (2H, t, J 7.1 Hz, NCH2CH2CH2N), 2.75 (2H, d, J 13.0 Hz, ArCH2N), 3.93 (2H, d, J 13.0 Hz, ArCH2N), 6.19 (2H, s, Ar), 7.08 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 20.1 (NCH2CH2CH2N), 29.1 (CMe3), 31.2 (CMe3), 32.9 (CMe3), 34.6 (CMe3), 44.6 (NMe2), 46.2 (NCH2CH2CH2N), 58.7 (NCH2CH2CH2N), 59.8 (ArCH2N), 121.9 (arom-CH), 125.6 (arom-CBut), 127.5 (arom-CH), 129.9 (arom-CBut), 133.9 (arom-CCH2N), 165.5 (arom-CO).
max/cm−1 1739 w, 1600 w, 1299 w, 1261 s, 1214 w, 1161 w, 1095 s, 1022 s, 933 w, 872 m, 802 s, 725 m, 602 m (Nujol); δH (500.1 MHz, C4D8O, 298 K) 0.61–0.64 (12H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 1.20 (12H, s, C(CH3)2CH2CH3), 1.41 (12H, s, C(CH3)2CH2CH3), 1.55 (6H, s, NMe2), 1.72 (4H, q, J 2 Hz, C(CH3)2CH2CH3), 1.81 (4H, q, J 2 Hz, C(CH3)2CH2CH3), 2.34 (2H, t, J 5.5 Hz, NCH2CH2N), 2.87 (2H, t, J 5.5 Hz, NCH2CH2N), 2.89 (2H, d, J 12.1 Hz, ArCH2N), 4.14 (2H, d, J 12.1 Hz, ArCH2N), 6.74 (2H, s, Ar), 6.99 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 6.9 (C(CH3)2CH2CH3), 7.5 (C(CH3)2CH2CH3), 25.4 (C(CH3)2CH2CH3), 25.9 (C(CH3)2CH2CH3), 26.9 (C(CH3)2CH2CH3), 30.5 (C(CH3)2CH2CH3), 34.8 (C(CH3)2CH2CH3), 36.5 (C(CH3)2CH2CH3), 42.9 (NMe2), 44.9 (NCH2CH2N), 57.6 (NCH2CH2N), 62.7 (ArCH2N), 123.3 (arom-CH), 125.3 (arom-CPet), 126.2 (arom-CH), 126.6 (arom-CPet), 130.8 (arom-CCH2N), 164.2 (arom-CO).
max/cm−1 3370 s, 2623 m, 2036w, 1600w, 1299 s, 1261 m, 1211w, 1095 s, 1060 m, 1018 s, 872 m, 802 m, 725 m, 601 w (Nujol); δH (500.1 MHz, C4D8O, 298 K) 0.62–0.65 (12H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 1.22 (12H, s, C(CH3)2CH2CH3), 1.36 (12H, s, C(CH3)2CH2CH3), 1.56 (4H, q, J 5.1 Hz, C(CH3)2CH2CH3), 1.78 (4H, q, J 5.5 Hz, C(CH3)2CH2CH3), 2.59 (2H, t, J 4.1 Hz, NCH2CH2O), 2.81 (3H, s, OMe), 2.93 (2H, d, J 11.1 Hz, ArCH2N), 2.98 (2H, t, J 4.1 Hz, NCH2CH2O), 6.7 (2H, s, Ar), 7.13 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 7.2 (C(CH3)2CH2CH3), 7.7 (C(CH3)2CH2CH3), 25.6 (C(CH3)2CH2CH3), 25.8 (C(CH3)2CH2CH3), 27.1 (C(CH3)2CH2CH3), 27.2 (C(CH3)2CH2CH3), 30.6 (C(CH3)2CH2CH3), 34.9 (C(CH3)2CH2CH3), 35.7 (NCH2CH2O), 38.8 (NCH2CH2O), 55.6 (OCH3), 62.3 (ArCH2N), 122.0 (arom-CPet), 123.3 (arom-CH), 125.4 (arom-CH), 126.9 (arom-CPet), 131.2 (arom-CCH2N), 164.1 (arom-CO).
max/cm−1 1775w, 1607 m, 1442 m, 1350w, 1367w, 1323w, 1268 s, 1163w, 1098 s, 1027 s, 931w, 876 s, 803 s, 740 s, 645w (Nujol); δH (500.1 MHz, C4D8O, 298K) 0.19 (18H, s, But), 0.33 (18H, s, But), 0.87 (6H, t, J 10 Hz, N(CH2CH3)2), 1.21 (2H, d, J 11.5 Hz, ArCH2), 1.37 (4H, q, J 10.2 Hz, N(CH2CH3)2), 3.53 (4H, m, NCH2CH2N), 4.23 (2H, d, J 11.5 Hz, ArCH2), 6.3 (2H, s, Ar), 7.2 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 11.6 (N(CH2CH3)2), 30.4 (CMe3), 30.9 (CMe3), 32.4 (CMe3), 32.9 (CMe3), 35.8 (NCH2CH2N), 36.1 (NCH2CH2N), 40.5 ((N(CH2CH3)2), 56.3 (ArCH2N), 121.5 (arom-CH), 122.2 (arom-CBut), 122.4 (arom-CH), 123.6 (arom-CCH2N), 125.0 (arom-CBut), 134.2 (arom-CO).
max/cm−11741w, 1612w, 1301 m, 1261 s, 1220w, 1165w, 1100 s, 1024 s, 933w, 875 m, 812 s, 725 m, 603 m (Nujol); δH (500.1 MHz, C4D8O, 298 K) 0.62–0.66 (12H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 1.10 (4H, q, J 7.1 Hz, C(CH3)2CH2CH3), 1.21 (12H, s, C(CH3)2CH2CH3), 1.23 (6H, t, J 6.2 Hz, N(CH2CH3)2), 1.34 (12H, s, C(CH3)2CH2CH3), 1.72 (4H, q, J 7.1 Hz, C(CH3)2CH2CH3), 1.91 (4H, q, J 6.2 Hz, N(CH2CH3)2), 2.26 (2H, t, J 7.0 Hz, NCH2CH2N), 2.88 (2H, t, J 7.0 Hz, NCH2CH2N), 3.08 (2H, d, J 12.3 Hz, ArCH2N), 4.21 (2H, d, J 12.3 Hz, ArCH2N), 6.66 (2H, s, Ar), 7.07 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 5.1 (C(CH3)2CH2CH3), 6.8 (C(CH3)2CH2CH3), 7.0 (C(CH3)2CH2CH3), 7.3 (C(CH3)2CH2CH3), 7.4 (C(CH3)2CH2CH3), 7.5 (C(CH3)2CH2CH3), 7.7 (C(CH3)2CH2CH3), 8.0 (C(CH3)2CH2CH3), 25.7 (N(CH2CH3)2), 43.4 (N(CH2CH3)2), 48.1 (NCH2CH2N), 49.4 (NCH2CH2N), 62.5 (ArCH2N), 119.9 (arom-CH), 123.4 (arom-CPet), 125.3 (arom-CH), 130.8 (arom-CPet), 151.4 (arom-CCH2N), 164.3 (arom-CO).
max/cm−1 1604 m, 1413 w, 1380 m, 1362w, 1324 w, 1262 s, 1203 m, 1165 w, 1094 m, 1023 m, 933 w, 915 w, 880 s, 813 s, 7420 s, 653 w, 521 s (Nujol); δH (500.1 MHz, C4D8O, 298 K) −6.21 (6H, t, J 5.3 Hz C(CH3)2CH2CH3), −0.83 (6H, t, J 5.1 Hz, C(CH3)2CH2CH3), −0.03 (12H, s, C(CH3)2CH2CH3), 0.16 (12H, s, C(CH3)2CH2CH3), 0.21 (6H, t, J 7.1 Hz, N(CH2CH3)2), 0.48 (8H, m, overlapping signals of o-C(CH3)2CH2CH3 and p-C(CH3)2CH2CH3), 0.51 (4H, q, J 7.1 Hz, N(CH2CH3)2), 1.26 (4H, m, NCH2CH2N), 1.62 (2H, d, J 11.1 Hz, ArCH2N), 1.72 (2H, d, J 11.1 Hz, ArCH2N), 5.22 (2H, s, Ar), 5.46 (2H, s, Ar), δC{H} (125.8 MHz, C4D8O, 298K) 2.2 (C(CH3)2CH2CH3), 5.9 (C(CH3)2CH2CH3), 22.5 (C(CH3)2CH2CH3), 26.6 (C(CH3)2CH2CH3), 26.9 (C(CH3)2CH2CH3), 27.2 (C(CH3)2CH2CH3), 29.8 (C(CH3)2CH2CH3), 32.8 (C(CH3)2CH2CH3), 34.6 (N(CH2Me)2), 35.7, (N(CH2Me)2), 37.1 (NCH2CH2N), 37.7 (NCH2CH2N), 44.7 (ArCH2), 103.6 (arom-CH), 120.6 (arom-CPet), 122.9 (arom-CH), 127.7 (arom-CPet), 133.7 (arom-CCH2N), 167.5 (arom-CO).
Crystal data †
Data collection was by means of a Bruker Apex I (for 1) or Oxford Diffraction Gemini (3) diffractometer. Following multi-scan absorption corrections the structures were solved by direct methods and refined by full-matrix refinement on F2 using the SHELXL 97 program.62Hydrogen atoms were placed in calculated positions and refined as part of riding models., specimen: 0.45 × 0.36 × 0.25 mm3, μ = 2.432 mm−1, Dc (Z = 2) = 1.312 Mg m−3, λ(Mo Kα) = 0.71073 Å, Tmin/max = 0.77, 2θmax = 56.5°. A total of 44182 reflections was collected, of which 17646 were unique (Rint = 0.023); R1 = 0.046 (I > 2σ(I)), wR2 = 0.125 (all data), S = 1.09; |ρmax| = 2.84 e Å−3. The methyl groups of two tert-butyl methyl groups and the toluene molecule are each disordered over two sites with site occupancy factors for one tert-butyl methyl group (14n) and for that of the toluene set as being identical (0.62(2) and 1–0.62(2)) after trial refinement showed them to be not significantly different. The site occupancy factors for the two components of the other disordered tert-butyl methyl group (44n) refined to 0.76(1) and 1–0.76(1). Atoms of the minor components of tert-butyl methyl (44n) and also those of the solvent toluene were refined with isotropic displacement parameters. The C–C bond lengths of disordered atoms were restrained to ideal values and the carbon atom of one methyl group (C146) was refined with isotropic displacement parameters. Since the remaining atoms showed no unacceptable anisotropy, no further disordering was attempted. The atoms of both components of the solvent toluene were refined as rigid groups.
Typical polymerization procedures
Acknowledgements
The authors wish to express sincere gratitude to Professor Lothar Stahl for numerous useful suggestions regarding this research. In addition, the author gratefully acknowledge: Professor Edward Kolodka and Mr Kirtipal Barse UND for assistance with GPC analyses. Financial support from ND EPScOR program is gratefully acknowledged.References
- A. Recknagel and F. T. Edelmann, Angew. Chem., 1991, 103, 720 CrossRef CAS.
- A. Recknagel, A. Steiner, S. Brooker, D. Stalke and F. T. Edelmann, Chem. Ber., 1991, 124, 1373 CAS.
- W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2119 CrossRef CAS.
- W. J. Evans, D. K. Drummond, H. Zhang and J. L. Atwood, Inorg. Chem., 1988, 27, 575 CrossRef CAS.
- M. N. Bochkarev, Coord. Chem. Rev., 2004, 248, 835 CrossRef CAS.
- M. Wedler, A. Recknagel and F. T. Edelmann, J. Organomet. Chem., 1990, 395, C26 CrossRef CAS.
- W. J. Evans and N. T. Allen, J. Am. Chem. Soc., 2000, 122, 2118 CrossRef CAS.
- W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2000, 122, 11749 CrossRef CAS.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1981, 103, 6507 CrossRef CAS.
- G. W. Watt and E. W. Gillow, J. Am. Chem. Soc., 1969, 91, 775 CrossRef CAS.
- W. J. Evans, L. A. Hughes and T. P. Hanusa, J. Am. Chem. Soc., 1984, 106, 4270 CrossRef CAS.
- F. Yuan, Q. Shen and J. Sun, J. Organomet. Chem., 1997, 538, 241 CrossRef CAS.
- W. J. Evans, C. A. Seibel and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 6745 CrossRef CAS.
- E. E. Delbridge, D. T. Dugah, C. R. Nelson, B. W. Skelton and A. H. White, Dalton Trans., 2007, 143 RSC.
- H. Zhou, H. Guo, Y. Yao, L. Zhou, H. Sun, H. Sheng, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 958 CrossRef CAS.
- Z. Hou, T.-a. Koizumi, M. Nishiura and Y. Wakatsuki, Organometallics, 2001, 20, 3323 CrossRef CAS.
- W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770 CrossRef CAS.
- Z. Hou, Y. Zhang, M. Nishiura and Y. Wakatsuki, Organometallics, 2003, 22, 129 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2006, 250, 2511 CrossRef CAS.
- J. Gottfriedsen and F. T. Edelmann, Coord. Chem. Rev., 2006, 250, 2347 CrossRef CAS.
- Z.-X. Zhang, Y.-R. Li, R.-T. Liu, Z.-X. Chen, L.-H. Weng and X.-G. Zhou, Polyhedron, 2007, 26, 4986 CrossRef CAS.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021 RSC.
- L. Zhou, J. Wang, Y. Zhang, Y. Yao and Q. Shen, Inorg. Chem., 2007, 46, 5763 CrossRef CAS.
- H. Guo, H. Zhou, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2007, 3555 RSC.
- S. Harder, Angew. Chem., Int. Ed., 2004, 43, 2714 CrossRef CAS.
- C. Ruspic, J. Spielmann and S. Harder, Inorg. Chem., 2007, 46, 5320 CrossRef CAS.
- Y. Zhang, Z. Hou and Y. Wakatsuki, Macromolecules, 1999, 32, 939 CrossRef CAS.
- X. Xi, L. Jiang, J. Ling, W. Sun and Z. Shen, J. Appl. Polym. Sci., 2006, 102, 175 CrossRef CAS.
- L. Jiang, L. Lou, W. Sun, L. Xu and Z. Shen, J. Appl. Polym. Sci., 2005, 98, 1558 CrossRef CAS.
- W. J. Evans and H. Katsumata, Macromolecules, 1994, 27, 2330 CrossRef CAS.
- E. Y. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2001, 20, 3017 CrossRef CAS.
- E. Y. Tshuva, S. Groysman, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2002, 21, 662 CrossRef CAS.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2005, 12, 169.
- H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32 RSC.
- H. Ma and J. Okuda, Macromolecules, 2005, 38, 2665 CrossRef CAS.
- F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046 CrossRef CAS.
- C.-X. Cai, A. Amgoune, W. Lehmann Christian and J.-F. Carpentier, Chem. Commun., 2004, 330 RSC.
- F. M. Kerton, A. C. Whitwood and C. E. Willans, Dalton Trans., 2004, 2237 RSC.
- Y. Yao, M. Ma, X. Xu, Y. Zhang, Q. Shen and W.-T. Wong, Organometallics, 2005, 24, 4014 CrossRef CAS.
- Y. Yao, X. Xu, B. Liu, Y. Zhang, Q. Shen and W.-T. Wong, Inorg. Chem., 2005, 44, 5133 CrossRef CAS.
- C.-X. Cai, L. Toupet, C. W. Lehmann and J.-F. Carpentier, J. Organomet. Chem., 2003, 683, 131 CrossRef.
- O. Runte, T. Priermeier and R. Anwander, Chem. Commun., 1996, 1850 Search PubMed.
- T. Yoshimura, Z. Hou and Y. Wakatsuki, Organometallics, 1995, 14, 5382 CrossRef CAS.
- Z. Hou, T. Yoshimura and Y. Wakatsuki, J. Am. Chem. Soc., 1994, 116, 11169 CrossRef CAS.
- S. Cotton, Lanthanide and Actinide Chemistry, Wiley, 2006 Search PubMed.
- W. J. Evans, R. A. Keyer and J. W. Ziller, Organometallics, 1993, 12, 2618 CrossRef CAS.
- J. M. Boncella, T. D. Tilley and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1984, 710 RSC.
- E. E. Delbridge and D. T. Dugah, unpublished results.
- G. B. Deacon, T. Feng, P. C. Junk, B. W. Skelton and A. H. White, Chem. Ber./Recl., 1997, 130, 851 CrossRef CAS.
- H. Ma, T. P. Spaniol and J. Okuda, Dalton Trans., 2003, 4770 RSC.
- P. I. Binda and E. E. Delbridge, Dalton Trans., 2007, 4685 RSC.
- I. Palard, A. Soum and S. M. Guillaume, Macromolecules, 2005, 38, 6888 CrossRef CAS.
- M. Yamashita, Y. Takemoto, E. Ihara and H. Yasuda, Macromolecules, 1996, 29, 1798 CrossRef CAS.
- M. Deng, Y. Yao, Q. Shen, Y. Zhang and J. Sun, Dalton Trans., 2004, 944 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS.
- R. Langer, Science, 2001, 293, 58 CrossRef CAS.
- I. Westmoreland and J. Arnold, Dalton Trans., 2006, 4155 RSC.
- S. Agarwal and M. Puchner, Eur. Polym. J., 2002, 38, 2365 CrossRef CAS.
- Y. Shen, Z. Shen, F. Zhang and Y. Zhang, Polym. J. (Tokyo, Japan), 1995, 27, 59 Search PubMed.
- H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecules, 2005, 38, 5400 CrossRef CAS.
- G. M. Sheldrick, SHELX-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † CCDC reference numbers 704056 and 704057. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b816916k |
| ‡ Indeed Tm, Nd, Hoetc. divalent compounds, derived from novel solution chemistry, are all well established.5–8 |
| § Indeed a number of other systems involving less encumbered bis(phenol) ligand reproducibly yielded oxidized products.14 |
| ¶ Within 4 h only 60% of compound 6 formed as monitored by 1H NMR spectroscopy. |
| || Several attempts to obtain a solid state structure of 5 were thwarted by crystal twinning. |
| ** Albeit 1.7% low in carbon. Given the extreme air-sensitivity of lanthanide(II) toward oxidation, repeated attempts at obtaining “satisfactory” elemental analyses were thwarted presumably by imperfect analyses techniques.49 |
| †† Which were not observed in solution, as identified by 1H NMR spectroscopy. |
| This journal is © The Royal Society of Chemistry 2009 |