Synthesis of a bulky bis(imino)pyridine compound: a methodology for systematic variation of steric bulk and energetic implications for metalation†
Janelle E.
Steves
,
Margaret D.
Kennedy
,
Karen P.
Chiang
,
William S.
Kassel
,
William G.
Dougherty
,
Timothy J.
Dudley
and
Deanna L.
Zubris
*
Villanova University, Department of Chemistry, Mendel Science Center, Villanova, PA 19085, USA. E-mail: deanna.zubris@villanova.edu; Fax: +1 610 519 7167; Tel: +1 610 519 4874
First published on 18th December 2008
Abstract
A new bis(imino)pyridine compound, 2,6-{(2,6-Me2-C6H3)NC(t-Bu)}2C5H3N (6), has been synthesized with t-butyl substituents on the imino carbon atoms. The stepwise synthetic method for assembly of this compound is novel. Compound 6 and its synthetic precursor, mono(imino)pyridine 3, have been characterized using single-crystal X-ray diffraction. Metalation attempts of 6 using iron(II) chloride under forcing conditions does not yield the desired iron(II) chloride complex; the use of refluxing acetic acid solvent provides a minimal amount of a paramagnetic species that has been characterized by NMR spectroscopy and magnetic susceptibility (NMR method). Computational methods have been used to evaluate the relative energies of three conformations of bis(imino)pyridine ligands with varying alkyl substitution at the imino carbon positions. The relative energies of these closed, open and open-planar conformations of 6 reveal a thermodynamic argument for the difficulty in metalation of 6, as compared to related ligands with less steric hindrance at the imino carbon atoms.
Introduction
Bis(imino)pyridine ligands first came into the spotlight ten years ago when Brookhart1a and Gibson2a described the synthesis of iron and cobalt complexes bearing these ligands and their use as pre-catalysts for polyethylene formation. Follow-up studies by these research groups, as well as DuPont researchers, expanded the scope of these initial reports.1b–c,2b–cIron and cobalt catalysts incorporating the bis(imino)pyridine ligand have been used for ethylene oligomerization, homopolymerization, and copolymerization with α-olefins, as described in a 2006 review;3 some degree of success in α-olefin dimerization, oligomerization and polymerization has been reported by various groups.4The backbone and substitution pattern of the bis(imino)pyridine ligand have been varied extensively, as described in two recent reviews.5,6 The two most common substitution patterns for this ligand (of the general formula: 2,6-{(2-R2,4-R4,6-R3-C6H2)NCR1}2C5H3N) appear in Fig. 1. Ligand modifications have largely focused on substitution of the imino nitrogen atoms (with 2-, 2,6-, and 2,4,6- substituted aryl rings being most common), and on a more limited basis, the use of alkyl-,7amino-, or pyrrolyl-8 substituted imino nitrogen atoms. C1-symmetric bis(imino)pyridine ligands have been synthesized;4b,7a,7b,9 their asymmetry is derived from differentially substituted imino nitrogen atoms. Also, on a more limited basis, the central pyridyl ring has been altered to provide iron(II) pre-catalysts with low activity for ethylene polymerization.10
 | ||
| Fig. 1 Common substitution patterns for the bis(imino)pyridine ligand. | ||
The range of functionality (R1) for the imino carbon atoms is limited, and to date there are no reports of differential substitution of these imino carbon atoms to provide C1-symmetric ligands. Most often, R1 is H or Me, since the corresponding ligands are synthetically derived from commercially available 2,6-diformylpyridine and 2,6-diacetylpyridine, respectively. The use of R1 = Ph has been reported by four research groups; moderate yields are achieved using standard condensation for ligand synthesis,11a and improved yields are achieved using more forcing10,11b or atypical7a reaction conditions. One report describes the installation of heteroatom-substituted imino carbon atoms, specifically, where R1 = OMe, SMe, O(2,6-Me2C6H3) or S(2,6-Me2C6H3).12 The latter three substituents yielded iron(II) pre-catalysts with ethylene polymerization activities comparable to the prototypical (2,4,6-Me3C6H2)-substituted ketimine analogue, [2,6-{(2,4,6-Me3C6H2)NCMe}2C5H3N]FeCl2. Aliphatic R1 substituents with increased steric bulk (relative to R1 = Me) were singularly reported in 2007; pre-assembled bis(imino)pyridine ligands (where R1 = Me) were treated with lithium diisopropylamide, followed by alkyl halide, to yield the following: R1 = CH2CH3, CH(CH3)2, CH2CH2Ph, and CH(CH2Ph)2.13 A notable feature of the corresponding iron(II) pre-catalysts is that as the steric bulk of R1 increases, polyethylene molecular weight increases while maintaining catalyst productivity. The extension of Gibson's synthetic methodology to install a tertiary alkyl as R1 was not reported, though one would anticipate continuation of the favorable trend of increased polyethylene molecular weight with comparable catalyst productivity.
In order to achieve the synthesis of a bis(imino)pyridine compound with R1 = t-Bu, we developed a new synthetic route. In our hands, standard condensation of the appropriate t-butyl disubstituted 2,6-diketopyridine14 with substituted anilines or N-amino-pyrroles was unsuccessful. Furthermore, a range of modified reagents and reaction conditions were employed7a,8c,10,15 but at best the mono(imino)pyridine species was detected. In developing our current synthetic method, we took a cue from the t-butyl substituted β-diketiminate ligand employed by Holland as a component of three coordinate iron(II) complexes.16 We have utilized an imidoyl chloride as a synthetic intermediate, similar to their use in the preparation of sterically hindered β-diketiminate ligands.17,18 Our synthetic route also draws upon the recent literature for selective mono lithium–halogen exchange of 2,6-dibromopyridine.19
Results and discussion
Synthesis of bis(imino)pyridine 6
Preparation of t-butyl substituted amide, 1, was achieved in a high yield by combination of pivaloyl chloride and 2,6-dimethylaniline in the presence of stoichiometric triethylamine (Scheme 1, step i), as described previously.20,21 Compound 1 was evaluated viasingle-crystal X-ray diffraction; suitable crystals of 1 were obtained from an anhydrous dichloromethane solution cooled to −15 °C for 8 d. The crystal structure for 1 was independently reported while our work was in progress; our crystallographic data is consistent with what appears in the literature.22 | ||
| Scheme 1 Reagents and conditions: (i) 2,6-dimethylaniline, NEt3, CH2Cl2, reflux, 1 h; (ii) PCl5, toluene, 22 °C, 15 h. | ||
Using literature methods, a toluene solution of compound 1 was treated with phosphorous pentachloride to generate the t-butyl substituted imidoyl chloride, 2 (Scheme 1, step ii); small-scale synthesis and characterization of 2 was reported previously.23 Ambient pressure distillation was used to remove toluene and the POCl3 by-product from the product mixture, followed by standard vacuum distillation or Kügelrohr distillation to yield 2 as an off-white semi-solid. Alternatively, we have prepared 2 by stepwise treatment of a 0 °C dichloromethane solution of 1 with 2,6-lutidene (1.66 equiv.) and oxalyl chloride (1.00 equiv.), similar to reported conditions for the synthesis of related imidoyl chlorides.24
We utilized Peterson's method for selective mono-lithiation of 2,6-dibromopyridine19 as a model for our synthesis of brominated mono(imino)pyridine, 3 (Scheme 2, step iii). While Peterson demonstrated the successful addition of a variety of electrophiles to in situ generated 2-bromo-6-lithiopyridine, the use of an imidoyl chloride, such as 2, as an electrophile was not described.
 | ||
| Scheme 2 Reagents and conditions: (iii) n-BuLi, CH2Cl2, −78 °C, 15 min; 1.4 equiv. 2, −78–22 °C, 22 h; (iv) n-BuLi, THF, −78 °C, 15 min; 1.0 equiv. 2, −78–22 °C, 17 h. | ||
Mono-lithiation of a dichloromethane solution of 2,6-dibromopyridine at −78 °C, followed by addition of 1.4 equiv. of 2 and slow warming to 22 °C generates 3 (Scheme 2, step iii). Analytically pure, pale yellow microcrystalline 3 can be obtained via flash silica gel column chromatography. Benzene-d6 is an optimal solvent for 1H NMR spectroscopic analysis; chloroform-d gives overlapping pyridyl and phenyl proton resonances. NOE difference experiments were used to assign the three pyridyl proton resonances and homonuclear decoupling experiments were used to detect long range coupling between the methyl (attached to the phenyl rings) and the ortho-phenyl protons. The 13C{1H} NMR spectroscopic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N chemical shift of 174.8 ppm (in benzene-d6) and a CN infrared stretching frequency of 1647 cm−1 provide further support for our structural assignment.
N chemical shift of 174.8 ppm (in benzene-d6) and a CN infrared stretching frequency of 1647 cm−1 provide further support for our structural assignment.
Single crystals of 3 for X-ray diffraction studies were obtained via slow evaporation of a 2.5% ethyl acetate–hexanes solution (v/v) of 3 over the course of 1 d. Most notably, the pyridyl and phenyl rings exhibit a nearly perpendicular arrangement, with a PlnA–PlnB angle of 110.24(5)° (see Fig. 2 and Table 1).
| a Where PlnA contains N(1), C(1), C(2), C(3), C(4) and C(5); PlnB contains C(11), C(12), C(14), C(15), C(16), and C(17). | |||
|---|---|---|---|
| Br–C(1) | 1.903(2) | N(1)–C(1)–Br | 115.9(1) |
| C(5)–C(6) | 1.510(2) | N(1)–C(5)–C(6) | 117.1(1) |
| N(2)–C(6) | 1.272(2) | C(5)–C(6)–C(7) | 117.9(1) |
| N(2)–C(11) | 1.427(2) | C(5)–C(6)–N(2) | 122.8(1) |
| PlnA–PlnBa | 110.24(5) | C(6)–N(2)–C(11) | 121.0(1) |
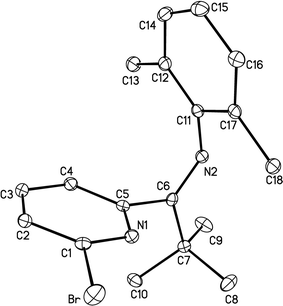 | ||
| Fig. 2 X-Ray crystal structure of 3. Hydrogen atoms are omitted for clarity. | ||
Our initial attempt to prepare the target bis(imino)pyridine compound followed our methodology for preparation of 3; namely, lithium–halogen exchange at −78 °C using dichloromethane solvent, followed by addition of imidoyl chloride, 2, as electrophile. These reaction conditions yielded product 4 (eqn (1)), consistent with alkylation of imidoyl chloride, 2. Presumably, brominated mono(imino)pyridine 3 is not sufficiently soluble in dichloromethane solvent at −78 °C to facilitate lithium–halogen exchange, so instead, imidoyl chloride, 2, is attacked by n-butyl lithium. Upon re-examination of NMR spectra for crude samples of 3, we determined that 4 is also generated as a very minor by-product in formation of 3.
 | (1) |
The use of tetrahydrofuran as solvent, in place of dichloromethane, facilitates our desired lithium–halogen exchange for 3. Treatment of 3 with n-butyl lithium in tetrahydrofuran solvent at −78 °C affords a bright yellow-orange solution immediately upon addition; quenching with excess methanol provides a pale yellow solution. Reaction work-up and purificationvia flash silica gel column chromatography provides a spectroscopically pure sample of compound 5 (eqn (2)), consistent with protonation of a lithiated intermediate.
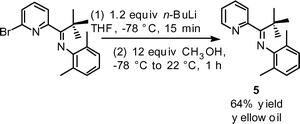 | (2) |
Thus, lithiation of a tetrahydrofuran solution of 3 at −78 °C, followed by addition of 1.0 equiv. of 2 and slow warming to 22 °C generates 6 (Scheme 2, step iv). Compound 5 was detected as the major impurity in this reaction; pale yellow microcrystalline 6 was isolated in 54.2% yield after flash silica gel column chromatography. We postulate that our moderate yield is due to some product loss during silica gel column chromatography, though we found it to be a superior purification method in comparison to recrystallization. The 13C{1H} NMR spectroscopic CN chemical shift of 175.9 ppm (in benzene-d6) and the CN infrared stretching frequency of 1651 cm−1 are consistent with the presence of a bis(imine).
A saturated solution of 6 in anhydrous pentane was cooled to −15 °C for 1 month to produce a single crystal suitable for X-ray diffraction (Table 2 and Fig. 3). As described in the literature for related alkyl or aryl substituted bis(imino)pyridine ligands,25 the pyridyl and phenyl rings are nearly orthogonal, with a PlnA–PlnB angle of 116.22(4)° and a PlnA–PlnC of 105.06(4)° (where PlnA contains the pyridyl ring, PlnB contains the phenyl ring incorporating C(11) through C(16), and PlnC contains the phenyl ring incorporating C(24) through C(29)). Also, the imino CN bonds are short (1.275(2) and 1.270(2) Å, respectively), as observed for related bis(imino)pyridine ligands. The most striking aspect of this structure is that the imino CN bonds are nearly orthogonal to the pyridyl ring, as indicated by the following dihedral angles: N(1)–C(1)–C(6)–N(2), −46.7(2)°; N(1)–C(5)–C(19)–N(3), −87.7(1)°. This conformation differs from the reported structure for the methyl-substituted analogue, 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N25b (vide infra).
| a Where PlnA contains N(1), C(1), C(2), C(3), C(4) and C(5); PlnB contains C(11), C(12), C(13), C(14), C(15), and C(16); PlnC contains C(24), C(25), C(26), C(27), C(28), and C(29). | |||
|---|---|---|---|
| N(1)–C(1) | 1.345(2) | C(19)–N(3)–C(24) | 123.9(1) |
| N(1)–C(5) | 1.342(2) | N(1)–C(1)–C(6) | 116.5(1) |
| C(1)–C(6) | 1.510(2) | N(1)–C(5)–C(19) | 115.5(1) |
| C(5)–C(19) | 1.518(2) | N(1)–C(1)–C(2) | 122.5(1) |
| N(2)–C(6) | 1.275(2) | N(1)–C(5)–C(4) | 122.7(1) |
| N(3)–C(19) | 1.270(2) | C(1)–C(6)–N(2) | 123.3(1) |
| C(6)–C(7) | 1.537(2) | C(5)–C(19)–N(3) | 123.2(1) |
| C(19)–C(20) | 1.533(2) | N(2)–C(6)–C(7) | 117.2(1) |
| N(3)–C(19)–C(20) | 118.8(1) | ||
| C(1)–N(1)–C(5) | 118.3(1) | PlnA–PlnBa | 116.22(4) |
| C(6)–N(2)–C(11) | 127.2(1) | PlnA–PlnCa | 105.06(4) |
 | ||
| Fig. 3 X-Ray crystal structure of 6. Hydrogen atoms are omitted for clarity. | ||
Attempted metalation of 6 with FeCl2 and FeCl2·4H2O
Literature reports have revealed that as the steric bulk at the imino carbon atoms increases (where R1 is Me, Et, i-Pr or Ph, see Fig. 1), longer reaction times or forcing conditions are required for metalation. Gibson and co-workers have described metalation reactions using methyl substituted 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N and R1 substituted 2,6-{(2,4,6-Me3-C6H2)NCR1}2C5H3N (where R1 is Me, Et, i-Pr or Ph), all with the same metalation agent (FeCl2), solvent (n-butanol), and reaction temperature (80 °C).2,10,13 In all cases, a color change is observed immediately upon mixing the ligand and FeCl2; the resulting iron compounds are either blue (where R1 is Me, Et or i-Pr) or green (where R1 is Ph) in color. Where R1 is methyl, the reaction was stopped after only 15 min;2 where R1 is phenyl a 30 min reaction time was used.10 Where R1 is ethyl or isopropyl, a 12 h reaction time was reported.13 Brookhart described milder conditions for metalation of methyl substituted 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N (use of either FeCl2 or FeCl2·4H2O, tetrahydrofuran solvent, ambient temperature, and several hours reaction time).1Attempted metalation of 6 under the following conditions gave no evidence of the desired iron(II) chloride metalated product: FeCl2, n-hexanol solvent, reflux, 48 h; FeCl2, n-butanol solvent, 80 °C, 14 h; FeCl2, tetrahydrofuran solvent, reflux, 24 h; FeCl2·4H2O, tetrahydrofuran solvent, reflux, 20 h. In each case, the anticipated color change (to green, blue or purple) was not detected and unreacted 6 was recovered.
Under more forcing conditions (FeCl2, acetic acid solvent, reflux, 3 h to 90 h), a color change was observed (a purple solution), but only upon concentration in vacuo (after 3 h reflux) or extended reaction times (>24 h reflux). Similar conditions were described by Chirik et al. for metalation of phenyl substituted 2,6-{(2,6-i-Pr2-C6H3)NCMe}2C5H3N (reflux, 4 h).11b For our metalation attempts using acetic acid solvent, small amounts of a purple, paramagnetic species were isolated upon reaction work-up (10–15 mg recovery, where 200–250 mg of 6 was used for metalation). 1H NMR spectroscopic analysis using CD2Cl2solvent reveals two broad downfield signals at 79.04 and 49.38 ppm that we tentatively assign as pyridyl proton resonances. Other resonances appear in the 12.95 to −32.24 ppm range, with a greater number of signals present than we would anticipate for a pure monomeric iron(II) chloride complex of 6. Magnetic susceptibility measurements (Evans NMR method) give evidence of paramagnetism, though also suggest that impurities and/or multiple paramagnetic species are present. Recrystallization attempts using solvents such as acetone-d6 and CD2Cl2 have led to precipitation of an iron(II) acetate species (presumably formed under metalation conditions). While these metalation conditions provide reproducible results, the poor recovery makes this reaction preparatively ineffective. We have considered the possibility that a given molecule of 6 may coordinate to more than one metal center, potentially yielding an oligomeric species. Nonetheless, experiments are under way to obtain X-ray quality single crystals of this purple, paramagnetic species.
Computational studies
Two facts led us to pursue computational studies for ligands of the form 2,6-{(2,6-Me2-C6H3)NCR1}2C5H3N (where R1 is Me, Et, i-Pr or t-Bu): (1) mild conditions are sufficient for metalation of methyl substituted 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N with iron(II) chloride,1 in contrast to t-butyl substituted 6, and (2) methyl substituted 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N adopts a different conformation in its crystalline form25b than t-butyl substituted 6. For each ligand, the energies of three conformers were calculated: closed (Fig. 3 gives the prototype for this conformer), open (the crystal structure of 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N gives the prototype for this conformer), and open-planar (the crystal structure of [2,6-{(2,4,6-Me3-C6H2)NCMe}2C5H3N]FeCl2 gives the prototype for this conformer).2b The closed, open, and open-planar conformers of 6 are illustrated in Fig. 4. | ||
| Fig. 4 Closed, open, and open-planar conformers of 6 (geometries were calculated at the B3LYP/6–31G(d) level). | ||
Based on the ease of metalation of the methyl substituted ligand and the energies presented in Table 3, the open conformer appears to be the reactive conformer for metalation. For comparison, the open conformers of methyl substituted 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N and t-butyl substituted 6 are shown in Fig. 5. For both the methyl- and ethyl-substituted ligands, the open conformer is the more thermodynamically stable one. This agrees well with the previously reported crystal structure for the methyl-substituted ligand.25b When the size of the alkyl chain is increased (i.e., i-propyl and t-butyl), marked differences are observed in relative energies of the reactive conformation. The closed structure becomes more thermodynamically stable for the i-propyl and t-butyl substituted species, with a lower calculated energy for the t-butyl substituted species as compared to the i-propyl substituted species. This agrees with the crystal structure for 6 (Fig. 3) and may offer some insight into the difficulty in metalating this species. The t-butyl-substituted species also shows a significant geometrical difference compared to the other ligands in that the imino C–N bonds are almost perpendicular to the plane formed by the pyridine ring. This is most likely due to the steric strain the t-butyl groups would experience if the imino nitrogen atoms were in a configuration similar to that of the other ligands (i.e., nearly planar with the pyridine ring).
 | ||
| Fig. 5 Open conformers of methyl substituted 2,6-{(2,6-Me2-C6H3)NCMe}2C5H3N and t-butyl substituted 6 (geometries were calculated at the B3LYP/6–31G(d) level). | ||
Budzelaar and Zhu26 recently published theoretical calculations on similar species in order to parameterize binding of these ligands to metal complexes. In their analyses, they broke down differential binding energies into three terms: σ-donation, π-donation, and reorganization energy associated with the open conformation of the ligand adopting the open-planar conformation. Their studies showed that, referenced to a structure almost identical to our methyl-substitued species, reorganization energies were typically lower or negligibly higher for structures with various substitutions on the imino carbons. However, they did not report reorganization energies based on closed conformations. For the i-propyl and t-butyl cases, this will significantly increase the reorganization energy. The t-butyl structure has the highest reorganization energy of the species considered, suggesting that metalation of this species will be the most difficult to accomplish.
Conclusions
A sterically hindered bis(imino)pyridine compound, 2,6-{(2,6-Me2-C6H3)NC(t-Bu)}2C5H3N (6), has been synthesized and characterized using single-crystal X-ray diffraction. Relative energies of three conformations of this species (closed, open, and open-planar) and the thermodynamic preference for the closed form give a potential explanation for the resistance of 6 towards metalation with iron(II) chloride. Similar calculations are under way in order to identify new, promising synthetic targets in the bis(imino)pyridine ligand family.Experimental
General considerations
All chemical reactions were carried out under an atmosphere of argon using standard Schlenk techniques27 unless otherwise noted. Argon gas was purified by passage over Drierite™. All chemicals were purchased from Aldrich and used as received unless otherwise noted. Chloroform-d, methylene chloride-d2, benzene-d6, and toluene-d8 were purchased from Cambridge Isotope Laboratories and dried over molecular sieves (8–12 mesh, 4 Å, activated) before use. A MBraun Manual Solvent Purification System (MB-SPS) was used to obtain the following anhydrous solvents: toluene, tetrahydrofuran, diethyl ether, pentane and dichloromethane; solvents were submitted to three freeze–pump–thaw cycles before use when rigorously deoxygenated solvent was required.28 Anhydrous n-hexanol and n-butanol were purchased from Aldrich (Sure/Seal bottle™) and used as received. Molecular sieves (8–12 Mesh, 4 Å) were purchased from J. T. Baker and activated before use. Silica gel used for flash chromatography29 was purchased from Aldrich (200–425 mesh). n-BuLi was purchased from Aldrich as a 1.6 M solution in hexanes (Sure/Seal bottle™) and titrated before use.30 Before use, 2,6-dibromopyridine was recrystallized using 200 proof absolute ethanol.31NMR spectra were recorded on a Varian Mercury spectrometer at 300 MHz (1H) and 75 MHz (13C) at 298 K; all chemical shifts are referenced relative to the NMR solvent (either residual protio or 13C signals for the solvent peak(s)). The following abbreviations are used for NMR splitting patterns: pd (pseudo doublet), pt (pseudo triplet), and br s (broad singlet). MS data were obtained using an Applied Biosystems API2000 triple quadrupole mass spectrometer with electrospray ionization. MS samples, containing approximately 1 μg mL−1 of analyte in methanol with 0.1% formic acid, were analyzed by direct infusion. Mass scans were performed from 20–300 m/z, and the signals were time-averaged over 2 min. Infrared spectra were recorded using a Perkin-Elmer Spectrum One FTIR System; samples were prepared by placing the compounds on a diamond attenuated total reflectance (ATR) plate in either solid or liquid form. Elemental analyses were performed at Atlantic Microlab, Inc. in Norcross, Georgia.
Multi-step synthesis of 6
O). 1H NMR (300 MHz, CDCl3, 22 °C) δ/ppm: 1.37 (9H, s, C(CH3)3), 2.21 (6H, s, CH3), 6.87 (1H, br s, N–H), 7.07 (3H, br s, C6H3). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.09 (9H, s, C(CH3)3), 2.05 (6H, s, CH3), 6.03 (1H, br s, N–H), 6.95 (3H, m, C6H3). 13C NMR (75 MHz, CDCl3, 22 °C) δ/ppm: 18.10 (CH3), 27.56 (C(CH3)3), 39.01 (C(CH3)3), 126.55 (C6H3), 127.63 (C6H3), 134.26 (C6H3), 135.38 (C6H3), 176.52 (CO). Single crystals of 1 suitable for X-ray diffraction were obtained from an anhydrous dichloromethane solution cooled to −15 °C for 8 d.
N). 1H NMR (300 MHz, CDCl3, 22 °C) δ/ppm: 1.43 (9H, s, C(CH3)3), 2.06 (6H, s, CH3), 6.96 (1H, m, C6H3), 7.04 (2H, pd, J = 7.4 Hz, C6H3). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.25 (9H, s, C(CH3)3), 2.03 (6H, s, CH3), 6.96 (3H, m, C6H3). 13C NMR (75 MHz, CDCl3, 22 °C) δ/ppm: 17.67 (CH3), 28.53 (C(CH3)3), 43.85 (C(CH3)3), 123.73 (C6H3), 125.89 (C6H3), 127.60 (C6H3), 145.36 (C6H3), 155.45 (CN). Alternatively, Kügelrohr distillation (95 °C, 1 Torr) may be used to collect the desired product. 31P NMR was used to qualitatively monitor the absence of the POCl3 by-product. Purificationvia flash silica gel chromatography is not viable because it hydrolyzes 2, thus reverting to 1.
N). 1H NMR (300 MHz, CDCl3, 22 °C) δ/ppm: 1.38 (9H, s, C(CH3)3), 2.07 (6H, s, CH3), 6.70 (2H, m, Ar), 6.81 (2H, m, Ar), 6.25 (2H, m, Ar). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.37 (9H, s, C(CH3)3), 2.11 (6H, s, CH3), 6.25 (1H, pt, J = 7.7 Hz, Hd), 6.32 (1H, dd, J = 0.8 Hz, 7.7 Hz, He), 6.55 (1H, dd, J = 0.8 Hz, 7.7 Hz, Hc), 6.70 (1H, m, Hb), 6.79 (2H, pd, J = 7.4 Hz, Ha). 1H NMR (300 MHz, toluene-d8; 22 °C) δ/ppm: 1.35 (9H, s, C(CH3)3), 2.07 (6H, s, CH3), 6.32 (2H, two overlapping m, pyr), 6.54 (1H, pd, J = 7.5 Hz, pyr), 6.65 (1H, m, Hb), 6.74 (2H, pd, J = 7.4 Hz, Ha). 1H NMR (300 MHz, CD2Cl2, 22 °C) δ/ppm: 1.35 (9H, s, C(CH3)3), 2.03 (6H, s, CH3), 6.68 (1H, m, Hb), 6.75 (1H, m, Hc), 6.79 (2H, pd, J = 7.4 Hz, Ha), 7.25 (1H, m, He), 7.30 (1H, m, Hd). 13C NMR (75 MHz, CDCl3, 22 °C) δ/ppm: 18.35 (CH3), 28.87 (C(CH3)3), 40.49 (C(CH3)3), 120.04, 122.54, 125.48, 126.96, 127.38, 137.27, 140.78, 147.67, 156.74, 175.21 (CN). 13C NMR (75 MHz, benzene-d6, 22 °C) δ/ppm: 18.74 (CH3), 29.11 (C(CH3)3), 30.35 (C(CH3)3), 120.23, 123.08, 125.64, 127.03, one aromatic resonance not detected, 137.31, 141.20, 148.60, 157.22, 174.81 (CN). NOE difference experiments were used to identify the three pyridyl proton signals. Homonuclear decoupling experiments were used to detect long range coupling between the phenyl-methyl protons and the ortho-phenyl 1H NMR resonances. Single crystals of 3 suitable for X-ray diffraction were obtained via slow evaporation of a 2.5% ethyl acetate–hexanes (v/v) solution over the course of 1 d.
 N). m/z (ESI) 246.4 ([M + H]+).
N). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.47 (9H, s, C(CH3)3), 2.19 (6H, s, CH3), 6.30 (1H, ddd, J = 7.5 Hz, 4.9 Hz, 1.2 Hz, He), 6.56 (1H, dt, J = 7.7 Hz, 1.0 Hz, Hc), 6.66 (1H, td, J = 7.5 Hz, 1.7 Hz, Hd), 6.72 (1H, pt, J = 7.4 Hz, Hb), 6.82 (2H, pd, J = 7.4 Hz, Ha), 8.21 (1H, ddd, J = 4.9 Hz, 1.7 Hz, 1.0 Hz, Hf). 13C NMR (75 MHz, benzene-d6, 22 °C) δ/ppm: 18.92 (CH3), 29.33 (C(CH3)3), 40.87 (C(CH3)3), 121.45, 122.46, 122.77, 125.77, 127.86, one aromatic resonance not detected, 134.78, 148.60, 156.97, 176.46 (CN). m/z (ESI) 267.5 ([M + H]+).
N). m/z (ESI) 246.4 ([M + H]+).
N). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.47 (9H, s, C(CH3)3), 2.19 (6H, s, CH3), 6.30 (1H, ddd, J = 7.5 Hz, 4.9 Hz, 1.2 Hz, He), 6.56 (1H, dt, J = 7.7 Hz, 1.0 Hz, Hc), 6.66 (1H, td, J = 7.5 Hz, 1.7 Hz, Hd), 6.72 (1H, pt, J = 7.4 Hz, Hb), 6.82 (2H, pd, J = 7.4 Hz, Ha), 8.21 (1H, ddd, J = 4.9 Hz, 1.7 Hz, 1.0 Hz, Hf). 13C NMR (75 MHz, benzene-d6, 22 °C) δ/ppm: 18.92 (CH3), 29.33 (C(CH3)3), 40.87 (C(CH3)3), 121.45, 122.46, 122.77, 125.77, 127.86, one aromatic resonance not detected, 134.78, 148.60, 156.97, 176.46 (CN). m/z (ESI) 267.5 ([M + H]+).
 N). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.29 (18H, s, C(CH3)3), 2.09 (12H, s, CH3), 6.36 (2H, pd, J = 7.5 Hz, Hc), 6.49 (1H, m, Hd), 6.70 (2H, m, Hb), 6.78 (4H, pd, J = 7.5 Hz, Ha). 13C NMR (75 MHz, benzene-d6, 22 °C) δ/ppm: 19.08 (CH3), 29.22 (C(CH3)3), 40.85 (C(CH3)3), 120.05, 122.65, 125.35, 127.90, 134.42, 148.82, 156.13, 175.87 (CN). A saturated solution of 6 in anhydrous pentane was cooled to −15 °C for 1 month to produce a single crystal suitable for X-ray diffraction.
N). 1H NMR (300 MHz, benzene-d6, 22 °C) δ/ppm: 1.29 (18H, s, C(CH3)3), 2.09 (12H, s, CH3), 6.36 (2H, pd, J = 7.5 Hz, Hc), 6.49 (1H, m, Hd), 6.70 (2H, m, Hb), 6.78 (4H, pd, J = 7.5 Hz, Ha). 13C NMR (75 MHz, benzene-d6, 22 °C) δ/ppm: 19.08 (CH3), 29.22 (C(CH3)3), 40.85 (C(CH3)3), 120.05, 122.65, 125.35, 127.90, 134.42, 148.82, 156.13, 175.87 (CN). A saturated solution of 6 in anhydrous pentane was cooled to −15 °C for 1 month to produce a single crystal suitable for X-ray diffraction.
Attempted synthesis of iron(II) chloride complex of 6
Crystal structure determinations
Crystallographic data are presented in Table 4. Single crystals of 3 and 6, respectively, were mounted using Paratone® oil onto a glass fiber and cooled to the data collection temperature of 100 K. Data were collected on a Brüker-AXS Platform APEX II CCD diffractometer with 0.71073 Å MoKα radiation for compound 3. Data were collected on a Brüker-AXS Kappa APEX II CCD diffractometer with 1.54178 Å CuKα radiation for compound 6. Unit cell parameters were obtained from 90 data frames, 0.5°Φ, from three different sections of the Ewald sphere. The data-set was treated with SADABS absorption corrections based on redundant multi-scan data.32 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were treated as idealized contribution.| 3 | 6 | |
|---|---|---|
| Empirical formula | C18H21BrN2 | C31H39N3 |
| Formula weight | 345.28 | 453.65 |
| Crystal color | Colorless | Colorless |
| Habit | Block | Block |
| T/K | 100(2) | 100(2) |
| λ/Å | 0.71073 | 1.54178 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| a/Å | 9.5839(5) | 11.5027(4) |
| b/Å | 9.2243(5) | 11.6244(4) |
| c/Å | 18.696(1) | 20.8562(7) |
| α/° | 90 | 90 |
| β/° | 91.430(1) | 105.597(1) |
| γ/° | 90 | 90 |
| V/Å3 | 1652.3(1) | 2686.0(2) |
| Z | 4 | 4 |
| D calcd /Mg m−3 | 1.388 | 1.122 |
| Absorption coefficient/mm−1 | 2.484 | 0.496 |
| F(000) | 712 | 984 |
| Crystal size/mm3 | 0.22 × 0.14 × 0.07 | 0.27 × 0.24 × 0.15 |
| Θ range for data collection/° | 2.18–28.37 | 3.99–68.15 |
| Index ranges | −11 ≤h≤ 12, −12 ≤k≤ 11, −23 ≤l≤ 24 | −11 ≤h≤ 13, −13 ≤k≤ 11, −25 ≤l≤ 24 |
| Reflections collected | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 499 499 |
19189 |
| Independent reflections | 3812 [Rint = 0.0364] | 4836 [Rint = 0.0221] |
| Completeness | 99.2% to Θ = 25.00° | 99.5% to Θ = 67.00° |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmission | 0.8473 and 0.6086 | 0.9297 and 0.8785 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3812/0/195 | 4836/0/317 |
| Goodness-of-fit on F2 | 1.048 | 1.065 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0320, wR2 = 0.0859 | R 1 = 0.0402, wR2 = 0.0990 |
| R indices (all data) | R 1 = 0.0360, wR2 = 0.0886 | R 1 = 0.0418, wR2 = 0.1001 |
| Largest diffraction peak and hole | 1.041 and −0.331 e Å−3 | 0.208 and −0.196 e Å−3 |
Computational methods
HF/6–31G(d) optimizations were performed and analytic harmonic frequencies were calculated to determine the nature of the stationary points observed. All geometries were then optimized at the B3LYP/6–31G(d) level of theory with very little change observed in any of the geometries. Relative energies are reported for the B3LYP optimized structures and do not include zero-point energy corrections. MP2/cc-pVDZ energies were also calculated at the B3LYP/6–31G(d) optimized geometries. GAMESS33 was used to optimize structures with the maximum gradient tolerance set to 0.0006 Hartree/Bohr. Default values were used for all other parameters.Acknowledgements
We are grateful to reviewers for helpful comments. Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund and Villanova University for support of this research. M. D. K. acknowledges funding from the United States Coast Guard. We thank Dr Walter Boyko of Villanova University for his assistance with NMR spectroscopy.References
-
(a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS;
(b) B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143–7144 CrossRef CAS;
(c)
World Pat., 9827124, 1998; Chem. Abstr., 1998, 129, 122973x.
-
(a) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC;
(b) G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS;
(c)
World Pat., 9912981, 1999; Chem. Abstr., 1999, 130, 252793.
- C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS.
- (a) C. Pellecchia, M. Mazzeo and D. Pappalardo, Macromol. Rapid Commun., 1998, 19, 651–655 CrossRef CAS; (b) B. L. Small and M. Brookhart, Macromolecules, 1999, 32, 2120–2130 CrossRef CAS; (c) B. L. Small and A. J. Marcucci, Organometallics, 2001, 20, 5738–5744 CrossRef CAS; (d) B. L. Small, Organometallics, 2003, 22, 3178–3183 CrossRef CAS; (e) S. T. Babik and G. Fink, J. Mol. Catal. A: Chem., 2002, 188, 245–253 CrossRef CAS; (f) S. T. Babik and G. Fink, J. Organomet. Chem., 2003, 683, 209–219 CrossRef CAS; (g) K. P. Tellman, V. C. Gibson, A. J. P. White and D. J. Williams, Organometallics, 2005, 24, 280–286 CrossRef CAS.
- V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS.
- Q. Knijnenburg, S. Gambarotta and P. H. M. Budzelaar, Dalton Trans., 2006, 5442–5448 RSC.
- (a) M. A. Esteruelas, A. M. Lopez, L. Mendez, M. Olivan and E. Oñate, Organometallics, 2003, 22, 395–406 CrossRef CAS; (b) C. Bianchini, G. Mantovani, A. Meli, F. Migliacci, F. Zanobini, F. Laschi and A. Sommazzi, Eur. J. Inorg. Chem., 2003, 1620–1631 CrossRef CAS; (c) K. Lappalainen, K. Yliheikkilä, A. S. Abu-Surrah, M. Polamo, M. Leskelä and T. Z. Repo, Z. Anorg. Allg. Chem., 2005, 631, 763–768 CrossRef CAS; (d) Y. Nakayama, K. Sogo, H. Yasuda and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3368–3375 CrossRef CAS.
-
(a)
L. S. Moody,
P. Mackenzie,
C. M. Killian,
G. G. Lavoie,
J. A. Ponasik Jr.,
A. G. Barrett,
T. W. Smith and J. C. Pearson, World Pat., 0050470, 2000; Chem. Abstr., 2000, 133, 208316;
(b) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, S. Mastroianni, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2001, 1639–1644 RSC;
(c) C. Amort, M. Malaun, A. Krajete, H. Kopacka, K. Wurst, M. Christ, D. Lilge, M. O. Kristen and B. Bildstein, Appl. Organomet. Chem., 2002, 16, 506–516 CrossRef CAS.
- (a) Z. Ma, W.-H. Sun, Z.-L. Li, C.-X. Shao, Y.-L. Hu and X.-H. Li, Polym. Int., 2002, 51, 994–997 CrossRef CAS; (b) C. Bianchini, G. Giambastiani, I. R. Guerro, A. Meli, E. Passaglia and T. Gragnoli, Organometallics, 2004, 23, 6087–6089 CrossRef CAS; (c) B. L. Small, M. J. Carney, D. M. Holman, C. E. O'Rourke and J. A. Halfen, Macromolecules, 2004, 37, 4375–4386 CrossRef CAS; (d) A. S. Ionkin, W. J. Marshall, D. J. Adelman, A. L. Shoe, R. E. Spence and T. Xie, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2615–2635 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson, O. D. Hoarau, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Inorg. Chem., 2003, 42, 3454–3465 CrossRef CAS.
- (a) N. Kleigrewe, W. Steffen, T. Blömker, G. Kehr, R. Fröhlich, B. Wibbeling, G. Erker, J.-C. Wasilke, G. Wu and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 13955–13968 CrossRef CAS; (b) A. M. Archer, M. W. Bouwkamp, M.-P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269–4278 CrossRef CAS.
- T. M. Smit, A. K. Tomov, V. C. Gibson, A. J. P. White and D. J. Williams, Inorg. Chem., 2004, 43, 6511–6512 CrossRef CAS.
- S. McTavish, G. J. P. Britovsek, T. M. Smit, V. C. Gibson, A. J. P. White and D. J. Williams, J. Mol. Catal. A: Chem., 2007, 261, 293–300 CrossRef CAS.
- A. S. Ionkin and W. J. Marshall, Heteroat. Chem., 2002, 13, 662–666 CrossRef CAS.
- Y. Chen, C. Qian and J. Sun, Organometallics, 2003, 22, 1231–1236 CrossRef CAS.
- (a) J. M. Smith, R. J. Lachicotte and P. L. Holland, Chem. Commun., 2001, 1542–1543 RSC; (b) A. R. Sadique, E. A. Gregory, W. W. Brennessel and P. L. Holland, J. Am. Chem. Soc., 2007, 129, 8112–8121 CrossRef CAS.
- R. Knorr and A. Weiss, Chem. Ber., 1982, 115, 139–160 CAS.
- P. H. M. Budzelaar, A. B. van Oort and A. G. Orpen, Eur. J. Inorg. Chem., 1998, 1485–1494 CrossRef CAS.
- M. A. Peterson and J. R. Mitchell, J. Org. Chem., 1997, 62, 8237–8239 CrossRef CAS.
- F. El-Zahraa, S. El-Basil, M. El-Sayed, K. M. Ghoneim and M. Khalifa, Pharmazie, 1979, 34, 12–13 CAS.
- B. T. Gowda, K. M. Usha and K. Jyothi, Z. Naturforsch., A: Phys. Sci., 2004, 59, 69–76 CAS.
- B. T. Gowda, I. Svoboda and H. Fuess, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3324 CrossRef.
- (a) E. Alonso, D. J. Ramón and M. Yus, Tetrahedron Lett., 1997, 38, 8903–8906 CrossRef CAS; (b) E. Alonso, D. J. Ramón and M. Yus, Tetrahedron, 1998, 12007–12028 CAS.
- (a) P. J. Manley and M. T. Bilodeau, Org. Lett., 2002, 4, 3127–3129 CrossRef CAS; (b) R. F. Cunico and R. K. Pandey, J. Org. Chem., 2005, 70, 5344–5346 CrossRef CAS.
- (a) Structural data for 2,6-{(C6H5)NCCH3}2C5H3N: A. Mentes, J. Fawcett and R. D. W. Kemmitt, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o424–o425 Search PubMed; (b) Structural data for 2,6-{(2,6-(CH3)2-C6H3)NCCH3}2C5H3N: Y.-B. Huang, X.-L. Ma, S.-N. Zheng, J.-X. Chen and C.-X. Wei, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o3044–o3045 Search PubMed; (c) Structural data for 2,6-{(2,6-(i-Pr)2-C6H3)NC(C6H5)}2C5H3N: see ref. 11a.
- D. Zhu and P. H. M. Budzelaar, Organometallics, 2008, 27, 2699–2705 CrossRef CAS.
- D. F. Shriver and M. A. Drezdon, The Manipulation of Air-Sensitive Compounds, Wiley, New York, 2nd edn, 1986 Search PubMed.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- W. G. Kofron and L. M. Baclawski, J. Org. Chem., 1976, 41, 1879–1880 CrossRef CAS.
- W. L. F. Armarego, and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Woburn, MA, 4th edn, 2000 Search PubMed.
- G. M. Sheldrick, SADABS, Program for area detector adsorption correction, Institute for Inorganic Chemistry, University of Göttingen, Germany, 1996 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
Footnote |
| † CCDC reference numbers 702075 and 702076. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b815987d |
| This journal is © The Royal Society of Chemistry 2009 |