DOI:
10.1039/B815056G
(Paper)
Dalton Trans., 2009, 202-209
Stereoelectronic effects in a homologous series of bidentate cyclic phosphines. A clear correlation of hydroformylation catalyst activity with ring size†
Received
29th August 2008
, Accepted 14th October 2008
First published on 25th November 2008
Abstract
The homologous series of diphosphines (CH2)n−1P(CH2)3P(CH2)n−1 where n = 5 (L5), 6 (L6), or 7 (L7) have been synthesized from the corresponding PhP(CH2)n−1. Treatment of [PtCl2(cod)] with L5–7 gave the 6-membered chelates cis-[PtCl2(L5–7)], the crystal structures for which reveal that L5–7 have very similar steric bulk and bite angles. Treatment of [Rh2Cl2(CO)4] with L5–7 gave the binuclear trans-[Rh2Cl2(CO)2(μ-L5–7)2] with syn and anti orientations of the CO and Cl ligands suggested by the 31P NMR spectra and the crystal structures of syn–trans-[Rh2Cl2(CO)2(μ-L5)2] and anti–trans-[Rh2Cl2(CO)2(μ-L7)2]. The ν(CO) values for trans-[Rh2Cl2(CO)2(μ-L5–7)2] indicate that the donor strength increases in the order L5 < L6 < L7. A study of rhodium-catalysed hydroformylation of 1-octene using diphosphines L5–7 is described. The catalyst activity decreases with increasing phosphacycle ring size: L5 > L6 > L7.
Introduction
Monodentate and bidentate phosphacycles of a variety of types and sizes are excellent ligands for hydroformylation catalysis as illustrated by the examples LA–H given in Fig.1.
Phosphacyclic ligands often give more active, selective and stable catalysts than their linear analogues. Delineating factors that influence the efficiency of catalysts for hydroformylation is apparently easy, understanding them remains a challenge.1,2 In the case of phosphacycles, the features that lead to their advantages include: (i) the entropic stabilisation of the P–X bonds in phosphacycles, (ii) the conformational rigidity of the ring and (iii) the constraints on the X–P–X angle imposed by the ring. Thus, the kinetic stabilisation of the P–O bonds in cyclic phosphites,3 such as LA or the P–N bonds in cyclic phosphamides,4 such as LB make them less susceptible to hydrolysis than acyclic analogues and this has greatly contributed to their utility. The rigidity of ligands such as LC,5LD6 and LE,7 make the α-substituents sterically demanding, which may contribute to the high hydroformylation catalytic activity of their rhodium complexes. The ring rigidity in LF results in a highly defined chiral space around the metal centre, which may account for the success of LF and related ligands in asymmetric hydroformylation.8 The constrained C–P–C angle imposed by the phosphacycle influences the frontier orbital energies9 thereby modulating the σ-donor and π-acceptor characteristics of the ligand and this may partly explain the high activity of hydroformylation catalysts derived from phosphacycles such as LG10 and LH.11
As is evident from the examples in Fig. 1, phosphacycles with ring sizes from 5–7 are important for hydroformylation catalysis. We are interested in understanding the effect the size of the ring has on the coordination chemistry and catalytic performance of P-ligands.7,12,13 Recently, we reported a systematic study of the homologous series of cyclic monophosphines 1a–c and their application in rhodium-catalysed hydroformylation.12 From spectroscopic measurements, it was found that the donor order was 1a < 1b < 1c, which is in line with the prediction that the more acute the C–P–C angle is made, the lower the HOMO and LUMO energies on the ligand become.9 The steric properties of the monophosphacycles (particularly 1c) were shown to depend critically on the ring conformation but no clear trend emerged from the hydroformylation catalysis study with 1a–c and related ligands.12 Part of the problem with discerning structure–activity relationships with monodentate ligands is the variable number (between 0 and 3) of ligands that may be coordinated to the metal and the cis/trans/axial/equatorial geometric relationships to each other that they adopt in the catalytic intermediates. We reasoned that the number of variables would be reduced, and therefore clearer trends may emerge, using the cyclic diphosphines L5–7.
Results and discussion
The series of bidentate cyclic phosphines L5–7 were synthesised by the route shown in Scheme 1. The phosphacycles121a–c were quaternized with 1,3-dibromopropane to give the corresponding phosphonium salts 2a–c as air-stable solids. Refluxing these salts in aqueous sodium hydroxide14 yielded the phosphine oxides 3a–c as white waxy solids. The reduction of 3a–c with phenylsilane, followed by distillation directly from the hot reaction mixture, produced L5–7. Diphosphines L5 and L6 are colourless liquids and L7 is a low-melting solid (see Experimental for the characterising data); L5 has been previously reported15 but L6 and L7 are new.
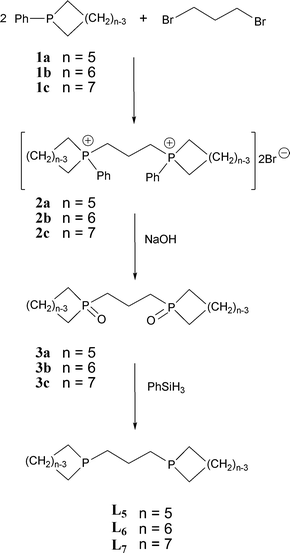 |
| | Scheme 1 | |
Platinum(II) complexes
The addition of one equivalent of diphosphines L5–7 to [PtCl2(cod)] in dichloromethane afforded the corresponding complexes cis-[PtCl2(L5–7)] (4a–c) as air-stable, white solids in quantitative yields (eqn (1), see Experimental for the characterising data).| |  | (1) |
Crystal structures for all three chelates 4a–c were obtained (see Fig. 2–4 and Tables 1–3). The chloroform solvate of 4a crystallises in the space groupP212121 from a saturated CHCl3 solution. The molecule of 4a (see Fig. 2) has approximate mirror symmetry. Selected bond lengths and angles are given in Table 1.
Table 1 Selected bond distances (Å) and angles (°) for 4a·CHCl3
| Bond distances/Å |
| Pt1–P1 |
2.2143(11) |
Pt1–P2 |
2.2089(11) |
| Pt1–Cl1 |
2.3604(10) |
Pt1–Cl2 |
2.3533(10) |
| P1–C1 |
1.835(4) |
P1–C4 |
1.828(4) |
| P1–C5 |
1.807(4) |
P2–C7 |
1.819(4) |
| P2–C8 |
1.822(5) |
P2–C11 |
1.838(4) |
| Bond angles/° |
| P1–Pt1–P2 |
96.02(3) |
C1–P1–C5 |
104.1(2) |
| C1–P1–C4 |
96.26(19) |
C4–P1–C5 |
104.1(2) |
| C7–P2–C8 |
104.5(2) |
C7–P2–C11 |
103.5(2) |
| C8–P2–C11 |
95.81(19) |
|
 |
| | Fig. 2 Thermal ellipsoid plot of the structure of 4a in 4a·CHCl3. Displacement ellipsoids are shown at the 50% probability level. All hydrogen atoms have been removed for clarity. | |
 |
| | Fig. 3 Thermal ellipsoid plot of the structure of 4b. Displacement ellipsoids are shown at the 50% probability level. All hydrogen atoms have been removed for clarity. | |
 |
| | Fig. 4 Thermal ellipsoid plot of the ordered molecule (of three) in the asymmetric unit of the structure of 4c. Displacement ellipsoids are shown at the 50% probability level. All hydrogen atoms have been removed for clarity. | |
The five-membered phospholane rings can adopt a range of conformations of which four idealised forms may usefully be identified (Fig. 5). If the phosphorus atom and the two neighbouring carbon atoms are considered to define a reference plane, the other two carbons in the ring can be: (A) in plane with these atoms (planar), (B) one in plane and one out of the plane (asymmetric envelope), (C) both above or both below the plane (envelope) or (D) one above and one below the plane (twist). In 4a, both rings adopt an asymmetric envelope conformation, with the torsion angle C–C–C–C = ± 47.6°.
 |
| | Fig. 5 Conformations of five-membered phospholane rings. | |
Crystals of 4b crystallised from a saturated MeOH solution in the space groupPbca with one molecule in the asymmetric unit. Selected bond lengths and angles are given in Table 2. The PC5 rings adopt a chair conformation with the C3 chelate backbone and the metal in axial and equatorial sites, respectively (see Fig. 3).
Table 2 Selected bond distances (Å) and angles (°) for 4b
| Bond distances/Å |
| Pt1–P1 |
2.2228(12) |
Pt1–P2 |
2.2289(13) |
| Pt1–Cl1 |
2.3716(13) |
Pt1–Cl2 |
2.3774(13) |
| P1–C1 |
1.825(5) |
P1–C5 |
1.822(5) |
| P1–C6 |
1.823(4) |
P2–C8 |
1.817(5) |
| P2–C9 |
1.827(5) |
P2–C13 |
1.827(5) |
| Bond angles/° |
| P1–Pt1–P2 |
96.48(5) |
C8–P2–C9 |
104.8(2) |
| C8–P2–C13 |
104.0(2) |
C9–P2–C13 |
104.0(2) |
| C1–P1–C5 |
104.5(2) |
C1–P1–C6 |
104.0(2) |
| C5–P1–C6 |
105.6(2) |
|
Crystals of 4c crystallised from a saturated MeCN solution in the space groupPc with three molecules in the asymmetric unit. In two of the molecules, significant disorder is present in the PC6 rings. The molecular geometry (see Table 3) of the only ordered molecule in the crystal structure is shown in Fig. 4. The seven-membered rings in the molecule containing Pt1 adopt twist-chair and boat conformations. In the molecule containing Pt2 they adopt chair conformations with the P atom in a different position in each of the rings and in the molecule containing Pt3 they adopt twist-chair, and chair conformations.12,16
Table 3 Selected bond distances (Å) and angles (°) for 4c.a Parameters for the atoms that were restrained have been omitted
| Bond distances/Å |
|
Equivalent distances for the second and third independent molecules in the asymmetric unit are given in columns 4 and 6. Atoms which are disordered are labelled A or B.
|
| Pt1–P1 |
2.2275(17) |
Pt2–P3 |
2.2258(16) |
Pt3–P5 |
2.2303(18) |
| Pt1–P2 |
2.2330(15) |
Pt2–P4 |
2.2293(15) |
Pt3–P6 |
2.2347(18) |
| Pt1–Cl1 |
2.3719(16) |
Pt2–Cl3 |
2.3740(16) |
Pt3–Cl5 |
2.3636(18) |
| Pt1–Cl2 |
2.3884(16) |
Pt2–Cl4 |
2.3898(15) |
Pt3–Cl6 |
2.3921(18) |
| P1–C1 |
1.837(6) |
P3–C16 |
1.835(6) |
P5–C31 |
1.811(7) |
| P1–C4 |
1.821(6) |
P3–C19 |
1.829(6) |
|
|
| P1–C9 |
1.825(6) |
P3–C24 |
1.821(6) |
| P2–C3 |
1.818(6) |
P4–C18 |
1.810(6) |
P6–C33 |
1.814(6) |
| P2–C10 |
1.827(6) |
P4–C25 |
1.823(6) |
P6–C40 |
1.823(7) |
| P2–C15 |
1.831(6) |
P4–C30 |
1.825(6) |
P6–C45 |
1.827(7) |
| Bond angles/° |
| P1–Pt1–P2 |
97.09(6) |
P3–Pt2–P4 |
97.00(6) |
P5–Pt3–P6 |
95.24(6) |
| C1–P1–C4 |
102.6(3) |
C16–P3–C24 |
102.2(3) |
|
|
| C1–P1–C9 |
103.3(3) |
C16–P3–C19 |
103.5(3) |
| C4–P1–C9 |
109.5(3) |
C19–P3–C24 |
110.0(3) |
| C3–P2–C10 |
104.1(3) |
C18–P4–C25 |
103.4(3) |
C33–P6–C40 |
102.6(3) |
| C3–P2–C15 |
103.6(3) |
C18–P4–C30 |
103.7(3) |
C33–P6–C45 |
102.2(3) |
| C10–P2–C15 |
110.4(3) |
C25–P4–C30 |
110.5(3) |
C40–P6–C45 |
108.6(3) |
From the crystallographic parameters collected in Table 4, it can be deduced that: (1) the intracyclic C–P–C angles increases significantly with increasing ring size, (2) the ligand bite angles increase slightly with increasing ring size and (3) the ligand cone angles (calculated by Tolman's method17) show that the ligands have essentially the same bulk.
Table 4 Selected angles (°) from the crystal structures of 4a–c
| Angles/° |
4a
|
4b
|
4c
|
|
Average of two.
Average of five.
Average of three.
|
| Intracyclic C–P–C |
96.0a |
104.3a |
109.8b |
| P–Pt–P |
96.0 |
96.5 |
97.0a |
| Tolman cone angle |
228 |
229 |
225c |
Rhodium(I) complexes
Addition of two equivalents of L5–7 to [Rh2Cl2(CO)4] in CH2Cl2 gave two species in each case, which are assigned to the binuclear complexes trans-[Rh2Cl2(CO)2(μ-L5–7)2] as a mixture of anti-5a–c and syn-5a–c isomers (eqn (2), see Experimental for characterising data). These structures are assigned on the basis of the 31P NMR spectra, which showed, in each case, two doublets with similar δ(P) and J(RhP) having values typical of a trans-RhCl(CO)(PR3)2 structure.18 A similar ligand-bridged binuclear structure has been reported for trans-[Rh2Cl2(CO)2(μ-Ph2P(CH2)3PPh2)2].19| | 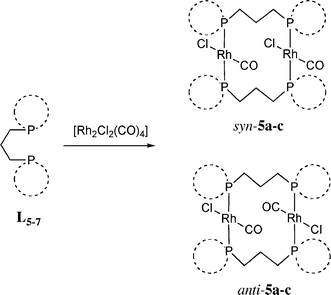 | (2) |
The crystal structures of syn-5a and anti-5c have been determined (see Fig. 6 and 7 and Tables 5 and 6). Crystals of syn-5a were grown by slow diffusion of diethyl ether into a saturated CH2Cl2 solution. The crystals form in the space groupP21/c with one binuclear molecule in the asymmetric unit. The phospholane rings all adopt distorted asymmetric envelope conformations with four out of five atoms in the ring (including phosphorus) coplanar. The angle between the mean planes of Rh1, P1, P4 and Cl1 and Rh2, P2, P3 and Cl2 is 15.1(1)°. Selected geometric parameters are given in Table 5.
Table 5 Selected bond distances (Å) and angles (°) for syn-5a
| Bond distances/Å |
| Rh1–C23 |
1.805(4) |
Rh1–Cl1 |
2.3714(9) |
| Rh1–P1 |
2.3183(9) |
Rh1–P4 |
2.3049(9) |
| Rh2–C24 |
1.811(4) |
Rh2–Cl2 |
2.3746(9) |
| Rh2–P2 |
2.3017(9) |
Rh2–P3 |
2.3077(9) |
| P1–C1 |
1.839(3) |
P1–C4 |
1.852(3) |
| P1–C5 |
1.833(4) |
P2–C7 |
1.831(3) |
| P2–C8 |
1.839(4) |
P2–C11 |
1.836(4) |
| P3–C12 |
1.838(3) |
P3–C15 |
1.847(3) |
| P3–C16 |
1.835(3) |
P4–C18 |
1.838(3) |
| P4–C19 |
1.836(3) |
P4–C22 |
1.844(3) |
| Bond angles/° |
| C1–P1–C4 |
94.56(16) |
C1–P1–C5 |
103.68(17) |
| C4–P1–C5 |
103.21(17) |
C7–P2–C11 |
104.51(17) |
| C7–P2–C8 |
104.57(18) |
C8–P2–C11 |
93.59(18) |
| C12–P3–C15 |
94.05(16) |
C12–P3–C16 |
105.84(16) |
| C15–P3–C16 |
104.71(17) |
C18–P4–C22 |
103.57(16) |
| C18–P4–C19 |
106.55(16) |
C19–P4–C22 |
94.05(16) |
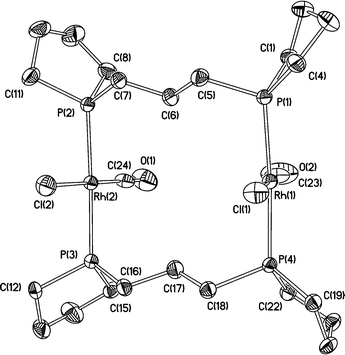
Table 6 Selected bond distances (Å) and angles (°) for anti-5c
| Bond distances/Å |
| Rh1–C31 |
1.927(9) |
Rh1–Cl1 |
2.381(2) |
| Rh1–P1 |
2.3200(17) |
Rh1–P3 |
2.3160(17) |
| Rh2–C32 |
1.807(8) |
Rh2–Cl2 |
2.376(2) |
| Rh2–P2 |
2.3111(18) |
Rh2–P4A |
2.429(10) |
|
Rh2–P4B |
2.242(9) |
|
| P1–C1 |
1.833(6) |
P1–C4 |
1.830(6) |
| P1–C9 |
1.833(6) |
P2–C3 |
1.836(5) |
| P2–C10 |
1.819(6) |
P2–C15 |
1.822(6) |
| P3–C16A |
1.813(7) |
P3–C19 |
1.837(6) |
| P3–C24 |
1.826(6) |
P4A–C25A |
1.812(18) |
| P4A–C30A |
1.81(3) |
|
| Bond angles/° |
| C1–P1–C4 |
104.4(3) |
C1–P1–C9 |
99.7(3) |
| C4–P1–C9 |
101.8(4) |
C3–P2–C10 |
101.8(3) |
| C3–P2–C15 |
102.9(3) |
C10–P2–C15 |
101.7(4) |
| C16A–P3–C19 |
102.8(3) |
C16A–P3-C24 |
101.2(3) |
| C19–P3–C24 |
101.5(3) |
C18A–P4A–C25A |
106.5(10) |
| C18A–P4A–C30A |
101.7(13) |
C25A–P4A–C30A |
101.5(19) |
| C18B–P4B–C25B |
104.2(8) |
C18B–P4B–C30B |
101.9(10) |
| C25B–P4B–C30B |
101.8(14) |
|
 |
| | Fig. 6 Thermal ellipsoid plot of the structure of syn-5a. Displacement ellipsoids are shown at the 50% probability level. All hydrogen atoms have been removed for clarity. | |
 |
| | Fig. 7 Thermal ellipsoid plot of the structure of anti-5c. Displacement ellipsoids are shown at the 50% probability level. All hydrogen atoms have been removed for clarity. | |
Crystals of anti-5c were grown by slow diffusion of hexane into a saturated CH2Cl2 solution, and form in the space groupP212121 with one binuclear molecule in the asymmetric unit (see, Fig. 7). There is disorder in one of the seven membered rings (involving P4) which was modelled as lying over two conformations in the ratio 0.44 : 0.56. The angle between the mean planes of Rh1, P1, P3 and Cl1 and Rh2, P2, P4 and Cl2 is 41.0(1)°. Selected geometric parameters are given in Table 6.
The IR spectra of 5a–c showed single absorptions for ν(CO) at 1968 (5a), 1963 (5b) and 1960 cm−1 (5c), which are consistent with the donor properties20 of the ligands being in the order L5 < L6 < L7. This is the trend expected9 on the basis of the crystallographically determined intracyclic C–P–C angles for the phosphines (see below).
Diphosphines with a propane backbone have previously been used in rhodium-catalysed hydroformylation and it is assumed that mononuclear 6-membered chelates are involved in the catalysis.21 The diphosphines L5–7 were screened for the rhodium-catalysed hydroformylation of 1-octene and the results are given in Table 7. The regioselectivity is similar for the three catalysts which is consistent with the ligands having similar steric bulk.22 The activity of the catalysts is in the order L5 > L6 > L7, which parallels the order of increasing electronic donation of the three ligands. Thus for this series of isosteric diphosphine ligands containing 5-, 6- or 7-membered phosphacycles, the smaller the ring, the higher the hydroformylation catalytic activity of the derived rhodium complex.
| Entry |
Ligand |
% Conversion to nonanals |
n : iso |
|
Reactions conditions: 90 °C, 10 bar CO–H2 (1 : 1) for 4 h in toluene, L–Rh = 5 : 1. See Experimental for details. No decomposition to metallic rhodium was apparent when the autoclaves were opened at the end of a catalytic run. Under more forcing conditions (140 °C, 80 bar), quantitative conversion to aldehyde was observed for all three ligands. No 2-octene was detected in the product of any run and under the same conditions (90 °C, 10 bar, 4 h) the conversions when 2-octene was the substrate were < 1%. The conversions of 1-octene to nonanals are the average of 2 or 3 runs; the differences between individual runs were within 5% for entries 1 and 2 and within 0.5% for entry 3.
|
| 1 |
L
5
|
69 |
2.3 |
| 2 |
L
6
|
48 |
2.2 |
| 3 |
L
7
|
3 |
2.0 |
Diphosphine ligands are widely used for hydroformylation and it has been shown that the ligand bite angle and the bulk of the ligand have a significant effect on the efficiency of the catalyst.1,23 However, the crystal structures of 4a–c described above show that chelated ligands L5–7 are essentially isosteric and have very similar bite angles. Moreover, the ligand giving the most active catalyst (L5), has the smallest bite angle while generally, increasing the ligand bite angle above 90° increases the activity of the derived hydroformylation catalysts.23
Electronegative substituents on the P-donor often increase the hydroformylation catalytic activity24 of the Rh complex although this is not always the case.25 Therefore, we conclude that the phosphacycle ring effect observed here is electronic in origin, confirming the prediction of Orpen and Connelly9 that reducing the C–P–C angle has the effect of reducing the σ-donor and increasing the π-acceptor properties of the tertiary phosphine.
Experimental
General procedures
Unless otherwise stated, all reactions were carried out under a dry nitrogen atmosphere using standard Schlenk line techniques. Dry N2-saturated solvents were collected from a Grubbs solvent system26 in flame and vacuum dried glassware. MeOH was dried over 3 Å molecular sieves and deoxygenated by N2 saturation. Commercial reagents were used as supplied unless otherwise stated. All phosphines were stored under nitrogen at room temperature. Most complexes were stable to air in the solid state and were stored in air at room temperature. Starting materials [PtCl2(cod)]27 and [Rh2Cl2(CO)4]28 were prepared by literature methods. Elemental analyses were carried out by the Microanalytical Laboratory of the School of Chemistry, University of Bristol. Electron Impact and Fast Atom Bombardment mass spectra were recorded by the Mass Spectrometry Service, University of Bristol on a MD800 and an Autospec. Infrared spectroscopy was carried out on a Perkin Elmer 1600 Series FTIR. NMR spectra were measured on a Joel GX 300, Jeol Eclipse 400 or Jeol GX 400. 31P{1H}, 13C{1H} and 1H NMR spectra were recorded at ambient temperature of the probe at 300, 100 and 121 MHz, respectively, using deuterated solvent to provide the field–frequency lock.
Synthesis of 1,3-bis(phenylphospholanium)propane dibromide (2a).
1-Phenylphospholane
1a (2.11 g, 12.9 mmol) and 1,3-dibromopropane (0.65 cm3, 1.30 g, 6.45 mmol) in acetonitrile (20 cm3) were heated at 60 °C for 48 h. The solvent was removed under reduced pressure to give 2a as a white solid (3.40 g, 6.45 mmol, 100%). Elemental analysis, found (calcd for C23H32Br2P2·H2O): C 49.85 (50.38), H 6.28 (6.25). 31P NMR (CD2Cl2) δP/ppm: 49.7 (s). 1H NMR (CD2Cl2) δH/ppm: 8.16–8.03 (m, 4H, ArH), 7.74–7.58 (m, 6H, ArH), 3.48–3.23 (m, 8H), 2.60–2.44 (m, 4H), 2.36–1.94 (m, 10H). 13C NMR (CD2Cl2) δC/ppm: 134.7 (s), 132.6 (m), 130.4 (m), 119.6 (d, ArCipso, 1J(PC) 77.6 Hz), 26.9 (m), 23.5 (m), 23.5 (d, PCH2, 1J(PC) 51.5 Hz), 17.6 (t, PCH2CH2CH2P, 2J(PC) 2.3 Hz).
1,3-Bis(phenylphosphinanium)propane dibromide (2b).
Compound 2b was made in a similar fashion to 2a from 1-phenylphosphinane 1b in 84% yield. Elemental analysis, found (calcd for C25H36Br2P2): C 53.53 (53.78), H 6.74 (6.50). 31P NMR (CD2Cl2) δP/ppm: 21.9 (s). 1H NMR (CD2Cl2) δH/ppm: 8.20–8.10 (m, 4H, ArH), 7.73–7.66 (m, 2H, ArH), 7.66–7.58 (m, 4H, ArH), 3.21–2.94 (m, 12H), 2.24–2.06 (m, 4H), 2.06–1.90 (m, 2H), 1.87–1.72 (m, 2H), 1.70–1.52 (m, 6H). 13C NMR (CD2Cl2) δC/ppm: 134.6 (s), 133.1 (m), 130.4 (m), 117.3 (d, ArCipso, 1J(PC) 79.2 Hz), 24.9 (m), 24.4 (dd, PCH2CH2CH2P, 1J(PC) 48.4 Hz, 3J(PC) 16.1 Hz), 21.4 (m), 18.5 (d, PCH2, cyclo, 1J(PC) 47.7 Hz), 16.1 (t, PCH2CH2CH2P, 2J(PC) 3.1 Hz).
1,3-Bis(phenylphosphepanium)propane dibromide (2c).
Compound 2c was made in a similar fashion to 2a from 1-phenylphosphepane 1c in 90% yield. Elemental analysis, found (calcd for C27H40Br2P2): C 54.71 (55.31), H 7.24 (6.88). 31P NMR (CDCl3) δP/ppm: 33.9 (s). 1H NMR (CDCl3) δH/ppm: 8.02–7.92 (m, 4H, ArH), 7.63–7.51 (m, 6H, ArH), 3.27–3.12 (m, 4H), 3.06–2.95 (m, 4H), 2.93–2.79 (m, 4H), 2.17–1.93 (m, 6H), 1.81–1.62 (m, 8H), 1.56–1.41 (m, 4H). 13C NMR (CDCl3) δC/ppm: 134.1 (s), 131.9 (m), 130.1 (m), 118.7 (d, ArCipso, 1J(PC) 80.0 Hz), 28.7 (s), 25.0 (m), 21.7 (m), 21.4 (d, J(PC) 46.9 Hz), 16.4 (m).
Synthesis of 1,3-diphospholanopropane dioxide (3a).
To 2a (3.40 g, 6.45 mmol) was added NaOH (50 cm3, 20 wt% in distilled water) and the resulting suspension was heated under reflux for 16 h. The product was extracted into CHCl3 (3 × 25 cm3) and the resulting solution dried over MgSO4. The solvents were removed under reduced pressure to give 3a as a white solid (1.43 g, 5.76 mmol, 90%). 31P NMR (CDCl3) δP/ppm: 70.4 (s). 1H NMR (CDCl3) δH/ppm: 2.24–1.66 (m). 13C NMR (CDCl3) δC/ppm: 31.5 (dd, PCH2CH2CH2P, 1J(PC) 60.7 Hz, 3J(PC) 11.5 Hz), 26.9 (d, PCH2, cyclo, 1J(PC) 65.3 Hz), 24.2 (m), 15.7 (t, PCH2CH2CH2P, 2J(PC) 3.8 Hz).
1,3-Diphosphinanopropane dioxide (3b).
Compound 3b was made in a similar fashion to 3a from 2b in 100% yield. 31P NMR (CD2Cl2) δP/ppm: 39.0 (s). 1H NMR (CD2Cl2) δH/ppm: 1.97–1.36 (m). 13C NMR (CD2Cl2) δC/ppm: 28.9 (dd, PCH2CH2CH2P, 1J(PC) 64.6 Hz, 3J(PC) 12.3 Hz), 28.0 (d, PCH2, cyclo, 1J(PC) 62.3 Hz), 26.9 (m), 23.1 (m), 14.2 (t, PCH2CH2CH2P, 2J(PC) 3.9 Hz).
1,3-Diphosphepanopropane dioxide (3c).
Compound 3c was made in a similar fashion to 3a from 2c in 99% yield. 31P NMR (CDCl3) δP/ppm: 52.9 (s). 1H NMR (CDCl3) δH/ppm: 2.10–1.52 (m). 13C NMR (CDCl3) δC/ppm: 31.1 (dd, PCH2CH2CH2P, 1J(PC) 63.68 Hz, 3J(PC) 11.5 Hz), 29.8 (s) 29.8 (d, PCH2, cyclo, 1J(PC) 62.3 Hz), 21.1 (m), 14.6 (t, PCH2CH2CH2P, 2J(PC) 3.9 Hz).
Synthesis of 1,3-diphospholanopropane (L5).
A mixture of 3a (1.43 g, 5.76 mmol) and phenylsilane (0.98 cm3, 0.88 g, 8.1 mmol) was heated at 100 °C for 2 h and the evolution of gas was observed. The resulting opalescent viscous liquid was distilled (0.01 mmHg, fraction collected and boiled over the range 100–140 °C) to give L5 as a clear viscous liquid (0.58 g, 2.69 mmol, 46%). Elemental analysis, found (calcd for C11H22P2): C 60.78 (61.10), H 10.02 (10.25). 31P NMR (CDCl3) δP/ppm: −26.7 (s). 1H NMR (CDCl3) δH/ppm: 1.89–1.61 (m, 12H), 1.59–1.33 (m, 10H). 13C NMR (CDCl3) δC/ppm: 30.4 (dd, PCH2CH2CH2P, 1J(PC) 16.6 Hz, 3J(PC) 10.9 Hz), 27.6 (d, J(PC) 4.2 Hz), 25.7 (d, PCH2, cyclo, 1J(PC) 11.4 Hz), 24.0 (t, PCH2CH2CH2P, 2J(PC) 16.6 Hz).
1,3-Diphosphinanopropane (L6).
L6 was made in a similar fashion to L5 in 85% yield. b.p. = 112–113 °C, 0.2 mmHg. Elemental analysis, found (calcd for C13H26P2) C 64.38 (63.91), H 11.05 (10.73). 31P NMR (CDCl3) δP/ppm: −41.9 (s). 1H NMR (CDCl3) δH/ppm: 1.87–1.73 (m, 14H), 1.73–1.62 (m, 4H), 1.58–1.44 (m, 12H), 1.33–1.17 (m, 6H). 13C NMR (CDCl3) δC/ppm: 28.7 (dd, PCH2CH2CH2P, 1J(PC) 13.1 Hz, 3J(PC) 11.5 Hz), 27.8 (d, J(PC) 2.3 Hz), 24.5 (d, PCH2, cyclo, 1J(PC) 10.8 Hz), 23.2 (d, J(PC) 2.3 Hz), 22.1 (t, PCH2CH2CH2P, 2J(PC) 15.4 Hz).
1,3-Diphosphepanopropane (L7).
L7 was made in a similar fashion to L5 in 78% yield. b.p. = 117–126 °C, 0.2 mmHg. Elemental analysis, found (calcd for C15H30P2): C 66.45 (66.15), H 11.10 (11.27). 31P NMR (CDCl3) δP/ppm: −33.2 (s). 1H NMR (CDCl3) δH/ppm: 1.90–1.65 (m, 8H), 1.63–1.20 (m, 22H). 13C NMR (CDCl3) δC/ppm: 31.0 (t, J(PC) 10.7 Hz), 29.0 (d, J(PC) 13.1 Hz), 28.3 (d, 4.6 Hz), 25.8 (d, J(PC) 7.7 Hz), 22.6 (t, PCH2CH2CH2P, 2J(PC) 14.6 Hz).
Synthesis of [PtCl2(L5)] (4a).
To a solution of L5 (0.090 g, 0.416 mmol) in dichloromethane (2 cm3) was added [PtCl2(cod)] (0.140 g, 0.375 mmol). After stirring the reaction for 12 h, the solvent was removed and the resulting white solid was washed with methanol (10 cm3) to give the desired product (0.142 g, 0.294 mmol, 79% yield). Elemental analysis, found (calcd for C11H22Cl2P2Pt): C 27.82 (27.40), H 4.56 (4.60). 31P NMR (CDCl3) δP/ppm: 2.8 (s, J(PtP) 3323 Hz). 1H NMR (CDCl3) δH/ppm: 2.88–2.57 (m, 4H), 2.40–1.59 (m, 18H). 13C NMR (CDCl3) δC/ppm: 27.7 (m), 27.3 (m), 23.8 (m), 20.8 (s). EI mass spectrum: m/z 482 (M+), 446 (M+− Cl), 409 (M+− 2Cl).
[PtCl2(L6)] (4b).
Compound 4b was made in a similar fashion to 4a from L6 in 64% yield. Elemental analysis, found (calcd for C13H26Cl2P2Pt): C 30.42 (30.78), H 5.07 (5.14). 31P NMR (CDCl3) δP/ppm: −17.1 (s, J(PtP) 3343 Hz). 1H NMR (CDCl3) δH/ppm: 2.99–2.81 (m, 4H), 2.05–1.76 (m, 14H), 1.75–1.65 (m, 2H), 1.60–1.38 (m, 6H). 13C NMR (CDCl3) δC/ppm: 25.7 (m), 23.2 (m), 22.2 (m), 19.3 (s), 17.8 (m). EI mass spectrum: m/z 474 (M+− Cl), 437 (M+− 2Cl).
[PtCl2(L7)] (4c).
Compound 4c was made in a similar fashion to 4a from L7 in 70% yield. Elemental analysis, found (calcd for C15H30Cl2P2Pt): C 33.43 (33.47), H 5.48 (5.62). 31P NMR (CDCl3) δP/ppm: −8.3 (s, J(PtP) 3333 Hz). 1H NMR (CDCl3) δH/ppm: 2.98–2.78 (m, 4H), 2.14–1.73 (m, 14H), 1.72–1.46 (m, 12H). 13C NMR (CDCl3) δC/ppm: 29.3 (s), 28.1 (m), 23.1 (m), 22.1 (m), 18.9 (m). FAB mass spectrum: m/z 501 (M+− Cl-1).
Synthesis of [Rh2Cl2(CO)2(μ−L5)2] (5a).
To a solution of L5 (0.034 g, 0.157 mmol) in dichloromethane (3 cm3) was added [RhCl(CO)2]2 (0.030 g, 0.079 mmol) as a solid. After stirring the reaction for 1 h, the solvent was reduced to ca. 1 cm3 and hexane (20 cm3) was added. The resulting precipitate was filtered off, washed with hexane and dried under reduced pressure to afford the desired product as a light brown powder (0.027 g, 0.071 mmol, 45% yield). Elemental analysis, found (calcd for C24H44Cl2O2P4Rh2): C 37.20 (37.67), H 5.27 (5.80). 31P NMR (CD2Cl2) δP/ppm: 27.9 (br d, J(RhP) 108 Hz), 27.0 (d, J(RhP) 113 Hz). 1H NMR (CD2Cl2) δH/ppm: 2.50–1.30 (br m, 22H, CH2). 13C NMR (CD2Cl2) δC/ppm: 32.0–24.4 (m). IRνCO (CH2Cl2): 1968 cm−1. EI mass spectrum: m/z 708 (M+− 2CO).
[Rh2Cl2(CO)2(μ-L6)2] (5b).
Compound 5b was made in a similar fashion to 5a from L6 in 64% yield. Elemental analysis, found (calcd for C28H52Cl2O2P4Rh2): C 41.24 (40.95), H 7.06 (6.38). 31P NMR (CD2Cl2) δP/ppm: 6.7 (d, J(RhP) 117 Hz), 6.0 (d, J(RhP) 117 Hz). 1H NMR (CD2Cl2) δH/ppm: 2.50–1.10 (m, 26H, CH2). 13C NMR (CD2Cl2) δC/ppm: 188.7–187.6 (m, CO), 30.9–20.5 (m). IRνCO (CH2Cl2): 1963 cm−1. EI mass spectrum: m/z 764 (M+− 2CO).
[Rh2Cl2(CO)2(μ-L7)2] (5c).
Compound 5c was made in a similar fashion to 5a from L7 in 88% yield. Elemental analysis, found (calcd for C32H60Cl2O2P4Rh2): C 43.75 (43.80), H 6.98 (6.89). 31P NMR (CD2Cl2) δP/ppm: 18.5 (d, J(RhP) 115 Hz), 17.0 (d, J(RhP) 115 Hz). 1H NMR (CD2Cl2) δH/ppm: 2.60–2.29 (m, 2H), 2.13–1.36 (m, 28H). 13C NMR (CD2Cl2) δC/ppm: 188.8–187.6 (m, CO), 32.5–23.9 (m). IRνCO (CH2Cl2): 1960 cm−1. FAB mass spectrum: m/z 877 (M+), 720 (M+− 2CO).
Hydroformylation catalysis.
The phosphine (0.060 mmol) and [Rh(CO)2(acac)] (3.0 mg, 0.012 mmol) were dissolved in toluene (10 cm3) under nitrogen in a Schlenk tube. The resulting solution was transferred by cannula to a 100 mL autoclave, which had been flushed 3 times with 3 bar CO–H2 (1 : 1). The autoclave was then pressurized with 2 bar of CO–H2 (1 : 1) at room temperature. The reaction mixture was then stirred vigorously with a sparging stirrer and heated to 90 °C over a period of 30 min. After this pre-activation of the catalyst, the 1-octene (10 g) was introduced into the autoclavevia a lock by means of CO–H2 pressure. The pressure was then immediately raised to 10 bar and maintained at this pressure throughout the catalysis by introduction of further CO–H2 (1 : 1) via a pressure regulator. After running the reactions for 4 h, the autoclave was cooled, vented and emptied. Analysis of the reaction products was carried out by GC.
Crystal structure determinations
X-Ray diffraction experiments of 4b, 4c, syn-5a and anti-5c were carried out at 173K on a Bruker SMART diffractometer; an experiment on 4a (as its chloroform solvate) was carried out at 100K on a Bruker SMART APEX diffractometer, all using MoKa radiation (λ = 0.71073 Å). All data collections were performed using a CCD area detector from a single crystal coated in perfluoroalkylether and mounted on a glass fibre. Intensities were integrated29 from several series of exposures measuring 0.3° in ω. Absorption corrections were based on equivalent reflections using SADABS,30 and structures were refined against all Fo2 data with hydrogen atoms riding in calculated positions using SHELXTL.31 Crystal structure and refinement data are given in Table 8. In 4a·CHCl3 the molecular complex has approximate mirror symmetry, as such that the crystal structure may also be approximately described in the space groupPnma, albeit with disorder in the chloroform molecule. The crystal structure refines satisfactorily in P212121 as an inversion twin (Flack parameter = 0.461(5)). Coupled with the fact that there are a significant number of (albeit weak) reflections present in the diffraction data, which should be systematically absent in Pnma, and that the weighted R-factor is lower and residual density map is cleaner in the lower symmetry space group, this suggests that P212121 is the correct choice. In 4c, the ring containing P(4) has three of the carbon atoms disordered over two positions in proportions 0.66/0.34, and the ring containing P(5) has all of the carbon atoms disordered over two positions (ration 0.57/0.43). The use of restraints on carbon–carbon bond lengths was necessary to ensure convergence of the refinement.
| Compound |
4a·CHCl3 |
4b
|
4c
|
5a
|
5c
|
| Colour, habit |
Colourless, block |
Colourless, plate |
Colourless, block |
Yellow, block |
Yellow, plate |
| Size/mm |
0.14 × 0.07 × 0.07 |
0.25 × 0.07 × 0.03 |
0.14 × 0.10 × 0.06 |
0.40 × 0.30 × 0.20 |
0.40 × 0.26 × 0.02 |
| Empirical Formula |
C12H23Cl5P2Pt |
C13H26Cl2P2Pt |
C15H30Cl2P2Pt |
C24H44Cl2O2P4Rh2 |
C32H60Cl2O2P4Rh2 |
|
M
r/g mol−1 |
601.58 |
510.27 |
538.32 |
765.19 |
877.40 |
| Crystal system |
Orthorhombic |
Orthorhombic |
Monoclinic |
Monoclinic |
Orthorhombic |
| Space group |
P212121 |
Pbca
|
Pc
|
P21/c |
P212121 |
|
a/Å |
12.165(2) |
12.723(3) |
34.324(7) |
11.7519(7) |
11.0204(18) |
|
b/Å |
12.425(3) |
11.954(3) |
6.3850(13) |
11.9472(7) |
11.9691(16) |
|
c/Å |
12.614(3) |
22.007(6) |
12.625(3) |
22.4102(13) |
28.877(3) |
|
β/° |
90.00 |
90.00 |
92.05(3) |
91.1220(10) |
90.00 |
|
V/Å3 |
1906.4(7) |
3347.0(15) |
2765.1(10) |
3145.8(3) |
3809.0(9) |
|
Z
|
4 |
8 |
6 |
4 |
4 |
|
μ/mm−1 |
8.217 |
8.878 |
8.065 |
1.442 |
1.202 |
|
T/K |
100 |
173 |
173 |
173 |
173 |
| Total reflections |
21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 202 202 |
20486 |
28297 |
20348 |
25010 |
| Independent reflections |
4334 |
3843 |
12421 |
7198 |
8754 |
|
R
int
|
0.0291 |
0.0602 |
0.0296 |
0.0518 |
0.0811 |
| Final R1 (I > 2σ) |
0.0185 |
0.0292 |
0.0281 |
0.0364 |
0.0444 |
| Final wR2 |
0.0468 |
0.0706 |
0.0693 |
0.0795 |
0.1079 |
| Largest peak, hole (e Å−3) |
1.06, −0.66 |
1.65, −1.61 |
1.76, −1.57 |
0.65, −0.65 |
0.62, −0.95 |
|
ρ
calcd/g cm−3 |
2.096 |
2.025 |
1.940 |
1.616 |
1.530 |
| Flack parameter |
0.461(5) |
— |
-0.001(4) |
— |
−0.01(4) |
Acknowledgements
We thank BASF and the EPSRC for financial support, Johnson-Matthey for a loan of precious metal compounds and the Leverhulme Trust for a Research Fellowship (to PGP).
References
-
(a)
P. W. N. M. van Leeuwen, Homogeneous Catalysis, Understanding the Art, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2004 Search PubMed;
(b)
Rhodium-Catalysed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000 Search PubMed.
- M. Sparta, K. J. Knut and V. R. Jensen, J. Am. Chem. Soc., 2007, 129, 8487 CrossRef CAS , and references therein.
-
(a)
E. Billig, A. G. Abatjoglou and D. R. Bryant, Union Carbide, U.S. Pat., 4599206 1986 Search PubMed;
(b) G. J. H. Buisman, E. J. Vos, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1995, 409 RSC;
(c) A. van Rooy, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, N. Veldman and A. Spek, Organometallics, 1996, 15, 5681;
(d) G. J. H. Buisman, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1997, 16, 835;
(e) R. Paciello, L. Siggel, H. Kneuper, N. Walker and M. Röper, J. Mol. Catal. A: Chem., 1999, 143, 85 CrossRef CAS;
(f) G. D. Cuny and S. L. Buchwald, J. Am. Chem. Soc., 1993, 115, 2066 CrossRef;
(g) G. Erre, S. Enthaler, K. Junge, S. Gladiali and M. Beller, J. Mol. Catal. A: Chem., 2008, 280, 148 CrossRef CAS , and references therein.
-
(a) S. C. van der Slot, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. Fraanje, K. Goubitz, M. Lutz and A. Spek, Organometallics, 2000, 19, 2504 CrossRef CAS;
(b) M. P. Magee, W. Luo and W. H. Hersh, Organometallics, 2002, 21, 362 CrossRef CAS;
(c) S. Breedon, D. J. Cole-Hamilton, D. F. Foster, G. J. Schwartz and M. Wills, Angew. Chem., Int. Ed., 2000, 39, 4106 CrossRef CAS.
-
T. Mackewitz, W. Ahlers, E. Zeller, M. Röper, R. Paciello, K. Knoll, and R. Papp, BASF, World Pat., 200200669, 2002 Search PubMed.
-
(a) B. Breit and E. Fuchs, Chem. Commun., 2004, 694 RSC;
(b) C. Müller and D. Vogt, Dalton Trans., 2007, 5505 RSC , and references therein.
-
(a)
W. Ahlers, BASF, World Pat., 2001085661, 2001 Search PubMed;
(b)
W. Ahlers, M. Slany, BASF, World Pat., 2001085662, 2001 Search PubMed;
(c)
K. Q. Almeida Lenero, E. Drent, R. van Ginkel, R. I. Pugh, Shell, World Pat., 2005058788, 2005 Search PubMed;
(d) R. A. Baber, M. L. Clarke, K. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, Dalton Trans., 2005, 1079 RSC;
(e) M. L. Clarke and G. J. Roff, Chem.–Eur. J., 2006, 12, 7979;
(f) J. H. Downing, J. Floure, K. Heslop, M. F. Haddow, J. Hopewell, M. Lusi, H. Phetmung, A. G. Orpen, P. G. Pringle, R. I. Pugh and D. Zambrano-Williams, Organometallics, 2008, 27, 3216 CrossRef CAS.
-
(a) A. T. Axtell, C. J. Cobley, J. Klosin, G. T. Whitaker, A. Zanotti-Gerosa and K. A. Abboud, Angew. Chem.,
Int. Ed., 2005, 44, 5834 CrossRef CAS;
(b) M. Jackson and I. C. Lennon, Tetrahedron Lett., 2007, 48, 1831 CrossRef CAS;
(c) T. P. Clarke, C. R. Landis, S. L. Freed, J. Klosin and K. A. Abboud, J. Am. Chem. Soc., 2005, 127, 5040 CrossRef CAS;
(d) A. T. Axtell, J. Klosin and K. A. Abboud, Organometallics, 2006, 25, 5003 CrossRef CAS , and references therein.
-
(a) A. G. Orpen and N. G. Connelly, Chem. Commun., 1985, 1310 RSC;
(b) A. G. Orpen and N. G. Connelly, Organometallics, 1990, 9, 1206 CrossRef CAS.
-
J. L. V. Winkle, R. C. Morris, and R. F. Mason, Shell, Ger. Pat., 1909620, 1969 Search PubMed.
-
(a) R. P. J. Bronger, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2003, 22, 5358 CrossRef CAS;
(b) L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1999, 18, 4765 CrossRef CAS;
(c) L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 336 CrossRef CAS;
(d) T. Hayashi, M. Tanaka and I. Ogata, J. Mol. Catal., 1979, 6, 1 CAS;
(e) C. F. Hobbs and W. S. Knowles, J. Org. Chem., 1981, 46, 4422 CrossRef CAS;
(f)
T. J. Devon, G. W. Philips, T. A. Puckette, J. L. Stavinoha and J. J. Vanderbilt, Eastman Kodak, U.S. Pat., 5332846, 1994 Search PubMed;
(g)
W. A. Hermann, R. B. Manetsberger, G. P. Albanese, H. Bahrmann, and P. Lappe, Ger. Patent, DE 4 333 307 (1994) to Hoechst;
(h) M. beller, B. Cornils, C. D. Frohning and C. W. Kohlpainter, J. Mol. Catal. A: Chem., 1995, 104, 17 CrossRef CAS.
- R. A. Baber, M. F. Haddow, A. J. Middleton, A. G. Orpen, P. G. Pringle, A. Haynes, G. L. Williams and R. Papp, Organometallics, 2007, 26, 713 CrossRef CAS.
-
(a) R. Doherty, M. F. Haddow, Z. A. Harrison, A. G. Orpen, P. G. Pringle, A. Turner and R. L. Wingad, Dalton Trans., 2006, 4310 RSC;
(b) M. R. Eberhard, E. Carrington-Smith, E. E. Drent, P. S. Marsh, A. G. Orpen, H. Phetmung and P. G. Pringle, Adv. Synth. Catal., 2005, 347, 1345 CrossRef CAS;
(c) M. R. Eberhard, K. M. Heslop, A. G. Orpen and P. G. Pringle, Organometallics, 2005, 24, 335 CrossRef CAS;
(d) J. H. Downing, V. Gee and P. G. Pringle, Chem. Commun., 1997, 1527 RSC;
(e) D. W. Norman, C. A. Carraz, D. J. Hyett, P. G. Pringle, J. B. Sweeney, A. G. Orpen, H. Phetmung and R. L. Wingad, J. Am. Chem. Soc., 2008, 130, 6840 CrossRef CAS;
(f) M. J. Baker, K. N. Harrison, A. G. Orpen, P. G. Pringle and G. S. Shaw, J. Chem. Soc., Chem. Commun., 1991, 803 RSC;
(g) M. J. Baker and P. G. Pringle, J. Chem. Soc., Chem. Commun., 1991, 1292 RSC;
(h) M. J. Baker and P. G. Pringle, J. Chem. Soc., Chem. Commun., 1993, 314 RSC;
(i) C. Claver, E. Fernandez, A. Gillon, K. Heslop, D. J. Hyett, A. Martorell, A. G. Orpen and P. G. Pringle, J. Chem. Soc., Chem. Commun., 2000, 961 RSC;
(j) R. A. Baber, J. G. de Vries, A. G. Orpen, P. G. Pringle and K. von der Luehe, Dalton Trans., 2006, 4821 RSC.
- W. E. Hill, M. Q. Islam, T. S. Webb and C. A. McAuliffe, Inorg. Chim. Acta, 1988, 146, 111 CrossRef CAS.
- R. W. Alder, C. Ganter, M. Gil, R. Gleiter, C. J. Harris, S. E. Harris, H. Lange, A. G. Orpen and P. N. Taylor, J. Chem. Soc., Perkin Trans. 1, 1998, 10, 1643 Search PubMed.
-
(a)
B. Testa, Principles of Organic Stereochemistry, Marcel Dekker, New York, 1979 Search PubMed;
(b)
F. A. L. Anet, Conformational Analysis of Medium-Sized Heterocycles, ed. R. S. Glass, VCH, New York, 1988 Search PubMed;
(c) G. Favini, J. Mol. Struct., 1983, 93, 139 CrossRef.
-
(a) C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS;
(b) K. A. Bunten, L. Chen Fernandez and A. J. Poë, Coord. Chem. Rev., 2002, 233–234, 41 CrossRef CAS , and references therein.
- B. E. Mann, C. Masters and B. L. Shaw, J. Chem. Soc. A, 1971, 1104 RSC.
-
(a) A. R. Sanger, J. Chem. Soc., Chem. Commun., 1975, 893 RSC;
(b) A. L. Balch and B. Tulyathan, Inorg. Chem., 1977, 16, 2840 CrossRef CAS.
-
(a) A. Roodt, S. Otto and G. Steyl, Coord. Chem. Rev., 2003, 245, 121 CrossRef CAS;
(b) M. L. Clarke, G. L. Halliday, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc. Dalton Trans., 2002, 1093 RSC.
-
(a) A. R. Sanger, J. Mol. Catal., 1977, 3, 221;
(b) M. Tanaka, T. Hayashi and I. Ogata, Bull. Chem. Soc. Jpn., 1977, 50, 2351 CAS;
(c) C. U. Pittman, Jr. and A. Hirao, J. Org. Chem., 1978, 43, 640 CrossRef CAS;
(d) M. Matsumoto and M. Tamura, J. Mol. Catal., 1982, 16, 195 CAS;
(e) M. Matsumoto and M. Tamura, J. Mol. Catal., 1982, 16, 209 CAS;
(f) W. Chen, Y. Xu and S. Liao, J. Mol. Catal. A: Chem., 1998, 129, 153 CrossRef CAS;
(g) M. Diéguez, M. M. Pereira, A. M. Masdeu-Bultó, C. Claver and J. C. Bayón, J. Mol. Catal. A: Chem., 1999, 143, 111 CrossRef CAS;
(h) A. Caiazzo, R. Settambolo, L. Pontorno and R. Lazzaroni, J. Organomet. Chem., 2000, 599, 298 CrossRef CAS.
- There is a small decrease in n-selectivity in the order L5 > L6 > L7, which, as pointed out by a reviewer, would be the expected trend with increasing donation of the ligand and this is consistent with the thesis of this article.
- This trend with bite angle is observed for bis equatorially bound ligands;
(a) P. W. N. M. van Leeuwen, P. C. J. Kamer and J. N. H. Reek, Pure Appl. Chem., 1999, 71, 1443;
(b) Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890 RSC.
-
(a) C. P. Casey, E. L. Paulinsen, E. W. Beuttenmueller, B. R. Proft, L. M. Petrovich, B. A. Matter and D. R. Powell, J. Am. Chem. Soc., 1997, 119, 11817 CrossRef CAS;
(b) C. P. Casey, E. L. Paulinsen, E. W. Beuttenmueller, B. R. Proft, B. A. Matter and D. R. Powell, J. Am. Chem. Soc., 1999, 121, 63 CrossRef CAS;
(c) T. V. RajanBabu and T. A. Ayers, Tetrahedron Lett., 1994, 35, 4295 CrossRef CAS.
- M. L. Clarke, D. Ellis, K. L. Mason, A. G. Orpen, P. G. Pringle, R. L. Wingad, D. A. Zaher and R. T. Baker, Dalton Trans., 2005, 1294 RSC.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521 CrossRef CAS.
- J. A. McCleverty and G. Wilkinson, Inorg. Synth., 1990, 28, 212.
-
Bruker SAINT v6.02A (4b, 4c, syn-5a, anti-5c) or v6.28A (4a), Siemens Analytical X-ray Instruments Inc., Madison, WI, 1994 or 1998 Search PubMed.
-
G. M. Sheldrick, SADABS v2.03 (4b, syn-5a), v2.05 (4a) or v2.10 (4c, anti-5c), University of Göttingen, Germany Search PubMed.
-
SHELXTL program system version v5.1, Bruker Analytical X-ray instruments Inc., Madison, WI, 1998 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 
