Transition metal alkynyl complexes by transmetallation from Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) CAr)(PPh3) (Ar = C6H5 or C6H4Me-4)†
CAr)(PPh3) (Ar = C6H5 or C6H4Me-4)†
Wan M.
Khairul
ab,
Mark A.
Fox
a,
Natasha N.
Zaitseva
c,
Maryka
Gaudio
c,
Dmitry S.
Yufit
a,
Brian W.
Skelton
d,
Allan H.
White
d,
Judith A. K.
Howard
a,
Michael I.
Bruce
*c and
Paul J.
Low
*a
aDepartment of Chemistry, Durham University, South Road, Durham, UK DH1 3LE
bDepartment of Chemical Sciences, Faculty of Science and Technology, Universiti Malaysia Terengganu, 21030 Kuala Terengganu, Terengganu, Malaysia
cSchool of Chemistry and Physics, University of Adelaide, Adelaide, South Australia 5005, Australia
dChemistry M313, SBBCS, University of Western Australia, Crawley, Western Australia 6009, Australia
First published on 13th November 2008
Abstract
Facile acetylide transfer reactions take place between gold(I) complexes Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAr)(PPh3) (Ar = C6H5 or C6H4Me-4) and a variety of representative inorganic and organometallic complexes MXLn (M = metal, X = halide, Ln = supporting ligands) featuring metals from groups 8–11, to afford the corresponding metal–alkynyl complexes M(CCR)Ln in modest to good yield. Reaction products have been characterised by spectroscopic methods, and molecular structure determinations are reported for Fe(CCC6H4Me-4)(dppe)Cp, Ru(CCC6H4Me-4)(dppe)Cp*, Ru(CCC6F5)(η2-O2)(PPh3)Cp*, Ir(CCC6H4Me-4)(η2-O2)(CO)(PPh3)2, Ni(CCC6H4Me-4)(PPh3)Cp and trans-Pt(CCAr)2L2 (Ar = C6H5, L = PPh3; Ar = C6H4Me-4, L = PPh3, PMe3).
CAr)(PPh3) (Ar = C6H5 or C6H4Me-4) and a variety of representative inorganic and organometallic complexes MXLn (M = metal, X = halide, Ln = supporting ligands) featuring metals from groups 8–11, to afford the corresponding metal–alkynyl complexes M(CCR)Ln in modest to good yield. Reaction products have been characterised by spectroscopic methods, and molecular structure determinations are reported for Fe(CCC6H4Me-4)(dppe)Cp, Ru(CCC6H4Me-4)(dppe)Cp*, Ru(CCC6F5)(η2-O2)(PPh3)Cp*, Ir(CCC6H4Me-4)(η2-O2)(CO)(PPh3)2, Ni(CCC6H4Me-4)(PPh3)Cp and trans-Pt(CCAr)2L2 (Ar = C6H5, L = PPh3; Ar = C6H4Me-4, L = PPh3, PMe3).
Introduction
Transmetallation, defined as the exchange of ligands between two metal centres,1 is one of the essential reaction steps in numerous organometallic catalytic cycles, illustrated effectively by the development of metal-catalysed procedures for the simple synthesis of C–C, C–N and C–O bonds.2 On a preparative scale, transmetallation reactions from Sn,3,4Hg,5,6Cu7–10 and Ag11 have long been known. However, new aspects of this chemistry continue to be developed. For example, despite the prevalence of transmetallation reactions involving alkynyl–copper(I) complexes, and the growing awareness of gold catalysis in which the transfer of ligands from gold to other species is involved,12transmetallation reactions involving aryl–gold(I) complexes have only recently been carried out on a variety of transition metal complexes by van Koten and co-workers.13
Transmetallation reactions involving alkynyl–gold(I) complexes are also scarce. Yam and co-workers have prepared the tetranuclear copper(I)–alkynyl complexes [Cu4(μ3-η1,η1,η2-CCAr′)3(PAr3)4]PF6 from the room temperature transmetallation reaction between [Cu(MeCN)4]PF6 and the appropriate alkynyl–gold(I) polymer [Au(CCAr′)]∞ in the presence of phosphines (PAr3).14 Ferrer and colleagues have observed the ready transfer of 4-pyridylethynyl from [Au(CCpy)2]− salts to rhenium following reaction with [Re(THF)(CO)3(bpy)]OTf.15 The Bruce group has recently found that cross-coupling reactions of Au(CCR)(PPh3) with halo-carbynes and halo-acetylenes can be promoted by Pd(0)/Cu(I) catalyst.16,17 These catalytic reactions, which are carried out at room temperature in ethereal solvents, implicate Au(I)–Pd(II) transmetallation processes, and the AuX(PPh3) by-product can be isolated from the reaction mixture and recycled.
Examples of transmetallation involving the readily prepared gold(I) species Au(CCR)(PR3)18 directly in the preparation of metal alkynyl complexes have been limited to NMR scale experiments. Shaw and co-workers used 31P NMR spectroscopy to follow the acetylide group transfer from Au(I) to Ni(II) during the facile reaction of NiCl2(dppm-P)2 with Au(CCC6H5)(PPh3) (CH2Cl2, ca 20 °C), which resulted in the formation of complexes formulated as [Ni(CCC6H5)2(μ-dppm)2Au]Cl (dppm = Ph2PCH2PPh2).19 Cross and Davidson have used 31P NMR spectroscopy to demonstrate that reactions of cis-PtCl2(PMePh2)2 with Au(CCC6H5)(PPh3) give cis- and/or trans-Pt(CCC6H5)2(PMePh2)2, depending on the conditions used.20 The same authors reported that reactions of cis-PtCl2(CO)(PMePh2) with Au(CCR)(PPh3) (R = Me, C6H5) gave cis-Pt(CCR)2(CO)(PMePh2) as the final product viacis- or trans-Pt(CCR)Cl(CO)(PMePh2).
Our long-standing interest in the preparative chemistry of metal acetylide complexes prompted us to consider the potential application of alkynyl-gold(I) complexes as reagents for the preparation of metal acetylides. Gold(I) phosphine complexes featuring highly conjugated carbon-rich and all-carbon ligands are known,16,21,22 and are generally easier and safer to handle than their analogous protio, lithio, tin, copper, mercury or Grignard derivatives. Convenient transfer of the carbon ligand from gold to other metals would represent a useful addition to the range of available synthetic methods for the preparation of metal complexes containing carbon-rich and all-carbon ligands.16,23
Here, we report transmetallation reactions using the readily available gold complexes Au(CCAr)(PPh3) [Ar = C6H5 (1a), C6H4Me-4 (1b)] and representative inorganic and organometallic compounds MXLn [M = metal, X = halide, Ln = supporting ligands] featuring metals from groups 8–11 (Scheme 1). The corresponding metal–alkynyl complexes M(CCAr)Ln are isolated in their pure form in modest to good yields.
 | ||
| Scheme 1 | ||
Results and discussion
Syntheses
Group 8 metal–alkynyl complexes M(CCR)(L2)Cp′ (M = Fe, Ru, Os; L = PR3; Cp′ = Cp, Cp*) have been prepared on many previous occasions.24–26 The synthesis of such species usually takes advantage of the ready isomerisation of 1-alkynes to vinylidenes that takes place in the coordination sphere of the group 8 metal centre.27 The resulting cationic vinylidene complexes [M{C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)R}(L2)Cp′]+ are acidic, and deprotonation occurs readily to give the corresponding acetylide. Whilst the 1-alkyne–vinylidene–alkynyl conversion has proven to be immensely useful in the preparation of metal alkynyl complexes of the type M(CCR)(L2)Cp′, it is limited to the use of stable 1-alkynes. Alternative synthetic routes to metal alkynyl and poly-ynyl complexes from trimethylsilyl-protected alkynes and poly-ynes based on desilylation/metallation routes have also been developed.28
C(H)R}(L2)Cp′]+ are acidic, and deprotonation occurs readily to give the corresponding acetylide. Whilst the 1-alkyne–vinylidene–alkynyl conversion has proven to be immensely useful in the preparation of metal alkynyl complexes of the type M(CCR)(L2)Cp′, it is limited to the use of stable 1-alkynes. Alternative synthetic routes to metal alkynyl and poly-ynyl complexes from trimethylsilyl-protected alkynes and poly-ynes based on desilylation/metallation routes have also been developed.28
The complexes Fe(CCAr)(dppe)Cp [Ar = C6H5 (2a) and C6H4Me-4 (2b)] were prepared from the reaction between FeCl(dppe)Cp and Au(CCAr)(PPh3) [Ar = C6H5 (1a), C6H4Me-4 (1b)] in the presence of NH4PF6 in refluxing MeOH (see Scheme 2 for a diagrammatic representation of all reactions reported in this work). Although the solvent and salt combination was chosen to promote labilisation of the Fe–Cl bond, the protic conditions also led to formation of the vinylidenes [Fe{CC(H)Ar}(dppe)Cp]PF6. Thus, after reaction, the resulting red solution was treated with DBU and purified by extraction into benzene and preparative TLC to give the iron–alkynyl complexes 2a (43%) or 2b (53%). In addition, a colourless band containing AuCl(PPh3) was also collected (δP 34.2 ppm).29 In the 13C NMR spectra for 2a and 2b, the alkynyl Cβ carbons were identified as broad peaks at 120.7 and 120.3 ppm, respectively. The alkynyl Cα carbon resonances were very broad (unresolved JCP coupling) peaks at 125.3 and 122.0 for 2a and 2b, respectively, with ∼150 Hz width at half height. For a series of related Fe(CCC6H4X-4)(dppe)Cp* (X = NO2, CN, CF, Br, F, H, Me, tBu, OMe, NH2, NMe2) compounds, the Cα and Cβ resonances were reported as triplets with ca. 40 and 3 Hz coupling constants, respectively.30
 | ||
| Scheme 2 | ||
The reactions between 1a or 1b and RuCl(PPh3)2Cp, in the presence of NH4PF6 or NaPF6 in refluxing methanol, resulted in the formation of a red solution, also presumed to contain a vinylidene intermediate, the deprotonation of which by DBU resulted in the precipitation of the target compounds Ru(CCC6H5)(PPh3)2Cp (3a)24 and Ru(CCC6H4Me-4)(PPh3)2Cp (3b)31 as yellow solids in 81 and 73% yields, respectively. As might be reasonably expected, the filtrate recovered from these reactions contained AuCl(PPh3) (δP 34.2 ppm).29 The compounds Ru(CCC6H5)(dppe)Cp* (4a)25 and Ru(CCC6H4Me-4)(dppe)Cp* (4b)31 were obtained from similar reactions of 1a and 1b with RuCl(dppe)Cp*.
The alkynyl–gold transmetallation reaction appears to be sensitive to the nature of the alkynyl moiety, and a reaction between RuCl(PPh3)2Cp* and Au(CCC6F5)(PPh3) (1c) afforded a multitude of products as indicated by separation into several bands upon preparative TLC. One of these bands gave crystals from hexane suitable for a single-crystal X-ray diffraction study (see below) from which the compound was identified as the dioxygen complex Ru(CCC6F5)(η-O2)(PPh3)Cp* (4c). The analogous hydrocarbon complex Ru(CCC6H5)(η-O2)(PPh3)Cp* is known.32
Although complete mechanistic details of the transmetallation reaction have not been elucidated, it is important to note that 1a or 1b can be recovered unchanged after treatment for 1 h in refluxing methanol containing one equivalent of NH4PF6. This observation would appear to discount the possible solvolysis of 1a or 1b to give the free alkyne, followed by the usual vinylidene formation, and supports the intermediacy of the gold reagents in the formation of the new metal–carbon bonds.
In the case of group 9 metals, alkynyl complexes such as Ir(CCR)(CO)(PPh3)2 have been prepared by reactions of Vaska's complex, IrCl(CO)(PPh3)2, or its NCMe derivative, with lithium acetylides or trialkylstannyl acetylenes,3,33 and also from copper-catalysed reactions between IrCl(CO)(PPh3)2 and 1-alkynes carried out in amine solvents or in the presence of NaOMe.8 The complexes Ir(CCR)(CO)(PR′3)2 readily take up oxygen to form the η2-O2 adducts Ir(CCR)(η2-O2)(CO)(PR′3). For example, the CuI-catalysed reaction between W(CCCCH)(CO)3Cp and Vaska's complex in NHEt2 gave {Cp(CO)3W}(μ-CCCC){Ir(CO)(PPh3)2}, which on aerobic work-up gave the η2-O2 adduct.34
In the present work, the complexes Ir(CCAr)(η2-O2)(CO)(PPh3)2 [Ar = C6H5 (5a),33,35 C6H4Me-4 (5b)] were prepared by treating IrCl(CO)(PPh3)2 with 1a or 1b in MeOH at room temperature. The gold by-product AuCl(PPh3) was removed from the crude product by extraction with acetone. Recrystallisation (chloroform/hexane) of the yellow solid that remained after extraction under aerobic conditions afforded yellow crystals of 5a (40%) or 5b (52%). In order to resolve some inconsistencies in literature values relating to the 31P NMR spectra and hence potentially the ν(CO) band positions associated with 16 e acetylide complexes Ir(CCAr)(CO)(PPh3)2,36,37 and also to clarify the sequence of reactions leading to the dioxygen adducts Ir(CCAr)(η2-O2)(CO)(PPh3)2 obtained here, the transmetallation/oxidation reaction sequence was followed by 31P NMR and IR spectroscopy. Under anaerobic conditions, reaction solutions of 1a and Vaska's complex rapidly developed new 31P resonances at 34.2 and 25.2 ppm corresponding to AuCl(PPh3)29 and Ir(CCC6H5)(CO)(PPh3)2, respectively. The ν(CO) band of the acetylide complex (1962 cm−1) is almost coincident with that of Vaska's complex, although the ν(CC) band is distinct (2128 cm−1). Exposure of the reaction solution to air resulted in the rapid disappearance of the δP 25.2 ppm peak and the appearance of a peak at 8.3 ppm corresponding to Ir(CCPh)(η2-O2)(CO)(PPh3)25a, consistent with spectroscopic analysis of the crystallographically characterised sample of 5b. The IR spectrum of 5a is characterised by ν(CO) and ν(CC) bands at 2005 and 2134 cm−1, respectively.
These spectroscopic data from Ir(CCC6H5)(CO)(PPh3)2 observed in situ and authentic samples of 5a can be compared with those reported for Ir(CCC6H5)(CO)(PPh3)2 (δP < 10 ppm, ν(CO)ca. 1960 cm−1) by two independent research groups,38,39 and Ir{CCC(CH2CH2Ph)2Me}(CO)(PPh3)2 (δPca. 22 ppm, ν(CO) 1970 cm−1).40 It appears that the 31P NMR spectra reported previously for Ir(CCC6H5)(CO)(PPh3)2 are actually that of the η2-O2 containing species 5a, although the IR data in the earlier reports are consistent with the proposed 16 e species. It can be concluded that the characteristic 31P resonances for Ir(CCR)(CO)(PPh3)2 and Ir(CCR)(η2-O2)(CO)(PPh3)2 compounds are found at ca. 25 ppm and ca. 5 ppm, respectively. These parameters are consistent with those found for other Ir(R)(CO)(PPh3)2 and Ir(R)(η2-O2)(CO)(PPh3)2 systems.41
Group 10 alkynyl complexes have been prepared on numerous occasions, being among the earliest metal–alkynyl complexes characterised.42 Whilst early synthetic routes took advantage of nucleophilic reactions between alkynyl anions (as lithium salts or Grignard reagents), later developments have given rise to more convenient preparations involving CuI-catalysed reactions between, for example, MCl2(PR3)2 and 1-alkynes.8
The reaction of 1a or 1b with NiBr(PPh3)Cp in THF resulted in the formation of a green solution, from which Ni(CCC6H5)(PPh3)Cp (6a)42 or the new complex Ni(CCC6H4Me-4)(PPh3)Cp (6b) were obtained in 53 and 42% yields, respectively, after preparative TLC and recrystallisation. In addition, a colourless band from preparative TLC that contained the expected by-product AuBr(PPh3) was also collected (δP 36.3 ppm).29
The complexes trans-Pt(CCAr)2(PPh3)2 [Ar = C6H5 (7a), C6H4Me-4 (7b)8] were prepared from room temperature reactions between cis-PtCl2(PPh3)2 and 1a or 1b in methanol, which gave the products as pale cream coloured precipitates, whilst AuCl(PPh3) was recovered from the filtrate.‡ The 31P NMR spectra contain singlet phosphine resonances, each accompanied by platinum satellites, confirming the trans geometry in each case. The complexes trans-Pt(CCAr)2(PMe3)2 [Ar = C6H5 (8a), C6H4Me-4 (8b)] were isolated from similar reactions between cis-PtCl2(PMe3)2 and 1a or 1b in methanol, which gave the products as pale green precipitates. Single crystals of 8b suitable for X-ray diffraction were obtained by recrystallisation (CHCl3/MeOH).
The room temperature reaction of 1a with a large excess of cis-PtCl2(PMe3)2 in the presence of NH4PF6 in methanol resulted in a myriad of products. The solids obtained after removal of the solvent included trans-PtCl(CCC6H5)(PMe3)2 and trans-PtCl(CCC6H5)(PMe3)(PPh3), which were identified by NMR spectrocopy and MS spectrometry. Crystallisation of the solids from MeCN/MeOH gave crystals of a Au(I) salt [Au(PMe3)2]PF6 (9).43 Thus, it appears that phosphine exchange, competitive with the acetylide–halide exchange processes noted above, may also take place under certain conditions.
Given the established utility of copper acetylides in the preparation of many transition metal acetylide complexes, we were interested in the interplay between acetylide ligands and Cu(I) and Au(I) centres. The reaction between 1a or 1b and CuI in THF afforded striking, bright yellow-coloured precipitates of the copper acetylides {Cu(CCAr)}n [Ar = C6H5 (10a), C6H4Me-4 (10b)] in very good yields with AuI(PPh3) recovered from the filtrate as a cream-coloured solid (δP 40.0 ppm).29
Molecular structures
The molecular structures of a number of the compounds prepared in this work (2b, 4b, 4c, 5b, 6b, 7a, 7b and 8b) have been determined by single-crystal X-ray diffraction studies. The crystallographic data are summarised in Table 1.| Complex | 2b | 4b | 4c | 5b | 6b | 7a | 7b | 8b |
|---|---|---|---|---|---|---|---|---|
| Formula | C40H36FeP2·CH2Cl2 | C45H46P2Ru | C36H30F5O2PRu·0.25C6H14 | C46H37IrO3P2·CHCl3 | C32H27NiP | C52H40P2Pt | C54H44P2Pt | C24H32P2Pt |
| MW | 676.94 | 749.83 | 743.2 | 1011.3 | 501.22 | 921.9 | 949.9 | 577.5 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P21/c | P21/n | Pbca | P21/n | P21/c |
| T/K | 120(2) | 120(2) | 100(2) | 120(2) | 120(2) | 120(2) | 120(2) | 120(2) |
| a/Å | 9.5283(2) | 15.4106(3) | 13.027(4) | 9.7735(3) | 14.1937 | 17.9771(3) | 13.5550(2) | 12.3805(5) |
| b/Å | 18.2097(4) | 11.2059(2) | 16.959(5) | 19.5550(4) | 12.6191(4) | 9.5527(1) | 8.5121(1) | 5.7372(2) |
| c/Å | 19.3123(4) | 22.1850(4) | 17.133(6) | 22.3273(6) | 14.9549(4) | 22.9326(3) | 18.0271(3) | 17.1229(7) |
| α/° | 93.20(2) | 90 | 111.86(3) | 90 | 90 | 90 | 90 | 90 |
| β/° | 93.13(2) | 108.33(1) | 106.93(3) | 99.18(2) | 110.05(1) | 90 | 90.30(1) | 105.86(1) |
| γ/° | 97.83(2) | 90 | 93.67(2) | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 3307.85(12) | 3636.75(12) | 3297(2) | 4212.6(2) | 2516.19(13) | 3938.2(1) | 2079.97(5) | 1169.92(8) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 2 | 2 |
| ρ c /g cm−3 | 1.359 | 1.369 | 1.497 | 1.595 | 1.323 | 1.555 | 1.517 | 1.639 |
| μ(MoKα)/mm−1 | 0.662 | 0.551 | 0.59 | 3.5 | 0.853 | 3.7 | 3.5 | 6.1 |
| N t | 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 399 399 |
48050 |
27359 |
37675 |
34844 |
51715 |
25528 |
10382 |
| N (Rint) | 18098 (0.041) |
10598 (0.045) |
11685 (0.089) |
9167 (0.116) | 7062 (0.029) | 5741 (0.045) | 5541 (0.035) | 3307 (0.025) |
| R 1 [I > 2σ(I)] | 0.040 | 0.033 | 0.063 | 0.070 | 0.029 | 0.018 | 0.017 | 0.018 |
| wR2 (all data) | 0.097 | 0.093 | 0.076 | 0.120 | 0.079 | 0.051 | 0.041 | 0.041 |
| GOF | 1.088 | 1.063 | 0.953 | 1.087 | 1.017 | 1.053 | 1.095 | 1.098 |
Crystals of Fe(CCC6H4Me-4)(dppe)Cp (2b), obtained from CH2Cl2/hexane solutions, contain two independent molecules, which differ by the relative orientation of the phenyl rings on the dppe ligand, together with a molecule of CH2Cl2. There are no differences of chemical significance in the two independent molecules, and the geometrical parameters of only one molecule will be used in further discussion here. Although there are many examples of crystallographically characterized arylacetylide complexes of the Fe(dppe)Cp* fragment in both formal Fe(II) and Fe(III) oxidation states,30,44–48 polymetallic compounds45,49 and mixed valence examples,44,50 there are surprisingly few structures of similar compounds containing the Fe(dppe)Cp51,52 or Fe(dppm)Cp53 fragments reported to date.
Complex 2b (Fig. 1) offers a pseudo-octahedral environment at the metal centre, typical of three-legged piano-stool complexes. The Fe(1)–C(1) [1.907(2) Å], C(1)C(2) [1.220(2) Å] and C(2)–C(3) [1.439(2) Å] bond lengths in 2b are indistinguishable from those found in Fe(CCC6H4-Me)(dppe)Cp* [Fe–Cα 1.896(3), CαCβ 1.220(4), Cβ-Cipso 1.440(4) Å],47 but somewhat surprisingly the Fe–P bonds in 2b [2.1687(6) and 2.1714(7) Å], and related compounds,51 are shorter than in the Cp* analogue [2.1829(8) and 2.1924(7) Å]. Since the more electron-rich, Cp*-substituted metal fragment might be expected to induce more effective Fe–P back-bonding, the relatively elongated Fe–P bonds in Fe(CCC6H4-Me)(dppe)Cp* might be taken as an indication of steric interactions between the bulky Cp* and dppe ligands. The Fe(1)–C(1)–C(2)–C(3) fragment in 2b is essentially linear, with small deviations likely to be due to packing effects rather than any profound electronic phenomenon. The plane containing the C(3)–C(8) aromatic ring forms a dihedral angle of 38.6(1)° with the plane of the Cp ring; the significance of the aryl group orientation on the electronic structure of iron aryl acetylide complexes has been discussed elsewhere.48
 | ||
| Fig. 1 A plot of one molecule of Fe(CCC6H4Me-4)(dppe)Cp (2b), showing the atom labelling scheme. Hydrogen atoms in this and other figures are removed for clarity. Selected bond lengths (Å) and angles (°): Fe(1)–C(1) 1.907(2); Fe(1)–P(1) 2.1687(6); Fe(1)–P(2) 2.1714(7); C(1)–C(2) 1.220(2); C(2)–C(3) 1.439(2); P(1)–Fe(1)–P(2) 85.95(2); C(1)–Fe(1)–P(1) 90.19(5); C(1)–Fe(1)–P(2) 84.28(5); Fe(1)–C(1)–C(2) 174.93(15); C(1)–C(2)–C(3) 174.84(17). | ||
The disposition of the tolyl ring with respect to the metal fragment in the ruthenium complex 4b (Fig. 2) is similar to that found in 2b. The Ru(1)–C(1) bond length [2.0205(19) Å] is somewhat longer than in 2b, reflecting the difference in the covalent radii of the two metals,54 but within the range noted for similar compounds, including 4a, noted here and elsewhere.55 Other metric parameters are unremarkable (Fig. 2).
 | ||
| Fig. 2 A plot of one molecule of Ru(CCC6H4Me-4)(dppe)Cp* (4b), showing the atom labelling scheme. Selected bond lengths (Å) and angles (°): Ru(1)–C(1) 2.0205(19); Ru(1)–P(1) 2.2621(5); Ru(1)–P(2) 2.2622(5); C(1)–C(2) 1.211(3); C(2)–C(3) 1.437(3); P(1)–Ru(1)–P(2) 83.15(2); C(1)–Ru(1)–P(1) 79.22(5); C(1)–Ru(1)–P(2) 85.20(6); Ru(1)–C(1)–C(2) 175.5(2); C(1)–C(2)–C(3) 171.9(2). | ||
The molecular structure of Ru(CCC6F5)(η2-O2)(PPh3)Cp* (4c) (Fig. 3) is formally related to that of Ru(CCC6H5)(η-O2)(PPh3)Cp* by replacement of the C6H5group on the acetylide ligand by C6F5. Two molecules of the complex 4c, devoid of crystallographic symmetry, together with one half of a centrosymmetric hexane solvent molecule, comprise the asymmetric unit of the structure. The two independent molecules differ slightly by orientations of one of the phenyl rings and C6F5group. Structural parameters are similar to those found for many other examples of half-sandwich ruthenium alkynyl complexes characterised here and elsewhere. The dioxygen molecule is unsymmetrically attached to Ru [Ru–O(1;2) 2.042(4), 2.039(4); 1.998(4), 2.019(4) Å], probably as a result of differing trans influences of the P and C(1) atoms. The O(1)–O(2) separations are 1.404(4) and 1.400(4) Å. These values are similar to those found in the related non-fluorinated complex Ru(CCC6H5)(η2-O2)(PPh3)Cp*, which has Ru–P 2.327(1), Ru–C(cp) 2.25 (av.), Ru–C(1) 2.022(4), C(1)–C(2) 1.158(5), C(2)–C(21) 1.467(5) and Ru–O 2.032(3), 2.048(3), O–O 1.364(4) Å.32 In both complexes, the Ru–O distances are longer, and the O–O distances shorter, than those found in related cations, such as [Ru(η2-O2)(dppm)Cp*]+, which has Ru–O 2.003(9), 2.002(9), O–O 1.37(1) Å.56
![A projection of one molecule of Ru(CCC6F5)(η2-O2)(PPh3)Cp (4c). Selected bond lengths (Å) and angles (°) for two independent molecules: Ru–P(1) 2.328(2), 2.313(2); Ru–C(Cp) 2.214–2.273(6), 2.202–2.289(6) [<av.> 2.24(3), 2.25(4)]; Ru–C(1) 1.997(6), 2.006(7); C(1)–C(2) 1.187(7), 1.198(7); C(2)–C(3) 1.429(8), 1.455(8); P(1)–Ru–C(1) 85.0(2), 82.2(2); Ru–C(1)–C(2) 178.7(6), 178.8(6); C(1)–C(2)–C(3) 176.0(7), 177.6(7).](/image/article/2009/DT/b809960j/b809960j-f3.gif) | ||
| Fig. 3 A projection of one molecule of Ru(CCC6F5)(η2-O2)(PPh3)Cp (4c). Selected bond lengths (Å) and angles (°) for two independent molecules: Ru–P(1) 2.328(2), 2.313(2); Ru–C(Cp) 2.214–2.273(6), 2.202–2.289(6) [<av.> 2.24(3), 2.25(4)]; Ru–C(1) 1.997(6), 2.006(7); C(1)–C(2) 1.187(7), 1.198(7); C(2)–C(3) 1.429(8), 1.455(8); P(1)–Ru–C(1) 85.0(2), 82.2(2); Ru–C(1)–C(2) 178.7(6), 178.8(6); C(1)–C(2)–C(3) 176.0(7), 177.6(7). | ||
Although dioxygen adducts of Vaska's complex and derivatives are well known, crystallographically characterised examples are surprisingly rare,41,57,58 and to the best of our knowledge, the diyndiyl complex {(PPh3)(CO)(η2-O2)Ir}(μ-CCCC){W(CO)3Cp} is the only example of a dioxygen adduct of a Vaska-type acetylide derivative that has been structurally characterised to date.34 However, given the relatively low precision of the diyndiyl structure, 5b is most conveniently compared with IrCl(η2-O2)(CO)(PPh3)259 and Ir(CCC6H5)(CO)(PPh3)2.39
The structure of the complex 5b (Fig. 4) can be described in terms of a five-coordinate, trigonal bipyramid, if the O2 ligand is assumed to occupy a single coordination site, with C(1), C(4) and the midpoint of the O2 ligand defining the equatorial plane [C(1)–Ir(1)–C(4) 94.5(4), C(1)–Ir(1)–O(0) 137, C(4)–Ir(1)–O(0) 128° (O(0) is the midpoint of the O–O bond)]. Alternatively, the structure may be viewed as a highly distorted octahedron58–60 in keeping with a formal description of the complex in terms of an Ir(III)-peroxo complex. The O–O bond is long [1.492(9) Å] and the Ir–P bonds [Ir(1)–P(1,2) 2.351(2) and 2.350(2) Å] are slightly elongated in comparison with those in the well defined Ir(I) acetylide complex Ir(CCC6H5)(CO)(PPh3)2 [2.286(7)–2.313(9) Å over two independent molecules], and similar to those in IrCl(η2-O2)(CO)(PPh3)2 [2.38(1), 2.36(1) Å]. Thus, whilst formalised descriptions of metal oxidation states in organometallic chemistry are not always especially useful, it seems appropriate to consider 5b and compounds of this type as Ir(III), with a significant distortion from the trigonal bipyramidal geometry towards an octahedral structure.
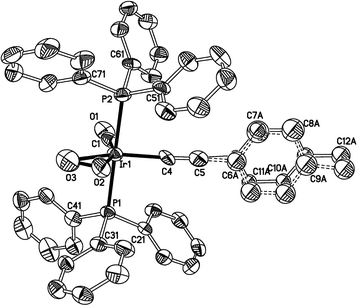 | ||
| Fig. 4 Molecular structure of Ir(CCC6H4Me)(η2-O2)(CO)(PPh3)2 (5b). Selected bond lengths (Å) and angles (°): Ir–P(1) 2.351(2); Ir–P(2) 2.350(2); Ir–C(1) 1.844(10); Ir–C(4) 2.011(10); C(4)–C(5) 1.186(12); C(5)–C(6A) 1.466(16); C(5)–C(6B) 1.484(18); Ir–O(2) 2.030(7); Ir–O(3) 2.036(7); O(2)–O(3) 1.492(9); P(1)–Ir–P(2) 175.58(8); C(1)–Ir–C(4) 94.5(4); C(4)–Ir–O(2) 106.9(3); C(4)–Ir–O(3) 149.8(3); C(1)–Ir–O(3) 115.7(4); Ir–C(4)–C(5) 177.4(7); C(4)–C(5)–C(6A)172.5(12); C(4)–C(5)–C(6B) 178.3(12). | ||
The molecular structures of nickel(II) complexes Ni(CCR)(PPh3)Cp have been studied on previous occasions, with data often interrogated for evidence of the nature of the metal–acetylide bond.61,62 The structure of 6a has been reported by the Humphrey group,61 and unsurprisingly, the structure of 6b (Fig. 5) is similar. The Ni(1)–C(1) bond lengths in 6a [1.856(3), 1.850(3) Å] and 6b [1.8537(14) Å] are indistinguishable, and whilst there is greater variation in the lengths of the Ni(1)–P(1) [6a 2.1350(9), 2.1378(9); 6b 2.1456(3) Å] and C(1)C(2) [6a 1.191(4), 1.193(4); 6b 1.2150(19) Å] bonds, the parameters associated with 6b fall within the ranges defined by many other similar compounds. The Ni(1)–C(1)–C(2)–C(3) moiety is essentially linear with angles at C(1) and C(2) being 175.1(1) and 172.2(1)°, respectively.
 | ||
| Fig. 5 A plot of a molecule of Ni(CCC6H4Me-4)(PPh3)Cp (6b). Selected bond lengths (Å) and angles (°): Ni(1)–P(1) 2.1456(3); Ni(1)–C(1) 1.8537(14); C(1)–C(2) 1.2150(19); C(2)–C(3) 1.4416(18); C(1)–Ni(1)–P(1) 89.10(4); Ni(1)–C(1)–C(2) 175.1(1); C(1)–C(2)–C(3) 172.2(1). | ||
In each of the Pt(II) complexes 7a (Fig. 6) and 7b (Fig. 7), the metal atoms of the molecules are located in special positions at the crystallographic inversion centre, the platinum centres being in square-planar arrangements, with bond lengths comparable to those in many other similar platinum bis(acetylide) complexes.63 The ethynyl moieties are essentially linear [Pt(1)–C(1)–C(2) 172.78(15) (7a); 175.9(2) (7b)°, C(1)–C(2)–C(3) 173.27(18) (7a); 177.32(18) (7b)°] and the acetylenic (CC) and Pt(1)–C(1) bond lengths fall in a narrow range [1.188(2)–1.207(2) and 2.0017(17)–2.0252(17) Å, respectively] typical for these systems.
![A projection of the molecular structure of centrosymmetric trans-Pt(CCC6H5)2(PPh3)2 (7a). Selected bond lengths (Å) and angles (°): Pt–P(1) 2.3121(4); Pt–C(1) 2.0017(17); C(1)–C(2) 1.207(2); C(2)–C(3) 1.441(2); P(1)–Pt–C(1) 93.27(5); Pt–C(1)–C(2) 172.78(15); C(1)–C(2)–C(3) 173.27(18). [symmetry operations (−x, 1 −y, −z)].](/image/article/2009/DT/b809960j/b809960j-f6.gif) | ||
| Fig. 6 A projection of the molecular structure of centrosymmetric trans-Pt(CCC6H5)2(PPh3)2 (7a). Selected bond lengths (Å) and angles (°): Pt–P(1) 2.3121(4); Pt–C(1) 2.0017(17); C(1)–C(2) 1.207(2); C(2)–C(3) 1.441(2); P(1)–Pt–C(1) 93.27(5); Pt–C(1)–C(2) 172.78(15); C(1)–C(2)–C(3) 173.27(18). [symmetry operations (−x, 1 −y, −z)]. | ||
![The molecular structure of centrosymmetric trans-Pt(CCC6H4Me)2(PPh3)2 (7b). Selected bond lengths (Å) and angles (°): Pt–P(1) 2.2887(4); Pt–C(1) 2.0252(17); C(1)–C(2) 1.188(2); C(2)–C(3) 1.445(2); P(1)–Pt–C(1) 85.90(5); Pt–C(1)–C(2) 175.88(15); C(1)–C(2)–C(3) 177.32(18). [symmetry operations (1 −x, −y, −z)].](/image/article/2009/DT/b809960j/b809960j-f7.gif) | ||
| Fig. 7 The molecular structure of centrosymmetric trans-Pt(CCC6H4Me)2(PPh3)2 (7b). Selected bond lengths (Å) and angles (°): Pt–P(1) 2.2887(4); Pt–C(1) 2.0252(17); C(1)–C(2) 1.188(2); C(2)–C(3) 1.445(2); P(1)–Pt–C(1) 85.90(5); Pt–C(1)–C(2) 175.88(15); C(1)–C(2)–C(3) 177.32(18). [symmetry operations (1 −x, −y, −z)]. | ||
While there are examples of platinum bis(acetylide) complexes known, there is only one previous example of the structurally characterised trans-Pt(CCR)2(PMe3)2 system.64 A comparison of the structural parameters of 8b (Fig. 8) and the Pt moiety in the cyclic heteronuclear complex [{Au{Pt(PMe3)2}2}{1,2-μ-C6H4(CC)2}3] reveals little differences and indicates that the constraining effect at the Pt atom in the cyclic complex is negligible. The change of the phosphine ligand in 7b and 8b has little effect on the overall molecular structure.
![A plot of a molecule of centrosymmetric trans-Pt(CCC6H4Me)2(PMe3)2 (8b). Selected bond lengths (Å) and angles (°): Pt–P(1) 2.2929(6); Pt–C(1) 2.0001(3); C(1)–C(2) 1.214(4); C(2)–C(3) 1.437(3); P(1)–Pt–C(1) 89.55(7); Pt–C(1)–C(2) 178.7(2); C(1)–C(2)–C(3) 175.3(3). [symmetry operations (1 −x, 2 −y, −z)].](/image/article/2009/DT/b809960j/b809960j-f8.gif) | ||
| Fig. 8 A plot of a molecule of centrosymmetric trans-Pt(CCC6H4Me)2(PMe3)2 (8b). Selected bond lengths (Å) and angles (°): Pt–P(1) 2.2929(6); Pt–C(1) 2.0001(3); C(1)–C(2) 1.214(4); C(2)–C(3) 1.437(3); P(1)–Pt–C(1) 89.55(7); Pt–C(1)–C(2) 178.7(2); C(1)–C(2)–C(3) 175.3(3). [symmetry operations (1 −x, 2 −y, −z)]. | ||
Considering the intermolecular interactions in these three platinum complexes, stacking-type π⋯π interactions between tolyl groups were observed only in the structure 7b (Fig. 9) (interplanar distance is equal to 3.64 Å) where they are combined with additional CH(Me)⋯C(sp) close intermolecular contacts (C⋯H 2.83 Å). In molecule 7a, no methyl groups are present while in the structure 8b, trimethylphosphine groups take part in intermolecular CH⋯π contacts and impede parallel arrangement of tolyl groups. It seems that in the case of these Pt complexes, stacking interactions alone are insufficient to determine the packing of molecules in the crystal.
 | ||
| Fig. 9 A fragment of the crystal structure of (7b) showing the stacking interactions between tolyl groups. | ||
The series of acetylide complexes reported here all feature small deviations from linearity along the M–CC–CAr chain, which are consequently gently curved or bowed. In a comprehensive review of polyyne conformation and structure, Szafert and Gladysz have noted a lack of systematic variation in structure within the solid state within a large array of yne-based compounds.65 Given the low bending force constants associated with M–CC and CC–C, crystal packing effects are thought likely to be responsible for the specific conformation adopted by any one compound.
Finally, we note that while our work was in progress, the structure of 9 was independently reported by Horvarth and Raubenheimer.43 The conclusions drawn by these authors are consistent with our own observations from an essentially identical structure determination, and no further comment is necessary.
Conclusions
A series of preparative scale, stoichiometric transmetallation reactions involving alkynyl–gold(I) complexes, Au(CCR)(PPh3) (R = Ph, C6H4Me-4), and inorganic or organometallic compounds MXLn (M = metal, X = halide, Ln = supporting ligands), have afforded the corresponding metal alkynyl complexes M(CCR)Ln, (35–90% yields), with representative examples featuring metals from groups 8–11 being described for the first time. The alkynyl products were fully characterised by the usual spectroscopic methods and molecular structural analyses in several cases. Looking ahead, we note that easily prepared phosphine–gold(I) complexes featuring highly conjugated carbon-rich and all-carbon ligands are known, and which are usually easier and safer to handle than their analogous protio, lithio, tin, copper, mercury or Grignard derivatives. The AuX(PPh3) by-products can be isolated from the reaction mixture and recycled. The transmetallation process has not been as successful with Au(CCC6F5)(PPh3) based on one exploratory reaction here. Further investigations are desirable with other Au(CCAr)(PR3) complexes to assess the generality of the alkynyl–gold transmetallation process and also to understand the alkynyl–gold transmetallation mechanism.
Experimental
All reactions were carried out under an atmosphere of nitrogen using standard Schlenk techniques. Reaction solvents were purified and dried using an Innovative Technology SPS-400 system, and degassed before use. No special precautions were taken to exclude air or moisture during work-up. Preparative TLC was performed on 20 × 20 cm glass plates coated with silica gel (0.5 mm thick, Merck GF-254). The reagents FeCl(dppe)Cp,66RuCl(PPh3)2Cp,67RuCl(PPh3)2Cp*,32RuCl(dppe)Cp*,68NiBr(PPh3)Cp,69cis-PtCl2(PPh3)2,70cis-PtCl2(PMe3)2,71IrCl(CO)(PPh3),72 Au(CCC6H5)(PPh3) (1a),18 Au(CCC6H4Me-4)(PPh3) (1b)18 and Au(CCC6F5)(PPh3) (1c)18 were prepared by literature methods. Other reagents were purchased and used as received.
IR spectra were recorded from dichloromethane solutions in a cell fitted with CaF2 windows, from KBr discs, or from Nujol mulls between NaCl plates using a Nicolet Avatar spectrometer. NMR spectra were obtained with Bruker Avance and Varian Mercury spectrometers from CDCl3 solutions and referenced against solvent resonances (1H, 13C) or external H3PO4 (31P). The 31P chemical shift values for the gold starting materials 1a and 1b are at 43.4 ppm. Mass spectra were recorded using Thermo Quest Finnigan Trace MS-Trace GC or Thermo Electron Finnigan LTQ FT mass spectrometers.
Preparation of Fe(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CC6H5)(dppe)Cp (2a)
CC6H5)(dppe)Cp (2a)
A suspension of FeCl(dppe)Cp (50 mg, 0.09 mmol), 1a (51 mg, 0.09 mmol) and NH4PF6 (15 mg, 0.09 mmol) in MeOH (10 ml) was heated at reflux for ca. 1 h, the progress of the reaction being monitored by TLC (hexane–acetone = 7 : 3). The resulting clear orange solution was treated with 2–3 drops of DBU and the reaction mixture was taken to dryness. The crude solid was extracted with benzene, and the extracts purified by preparative TLC (hexane–acetone = 7 : 3). The red band was collected and afforded 2a (24 mg, 43%). IR (CH2Cl2) ν/cm−1: (CC) 2060. 1H NMRδ/ppm: 2.23 (m, 2H, dppe), 2.63 (m, 2H, dppe); 4.25 (s, 5H, Cp), 6.49 (2H, d, phenyl C2H of CCC6H5), 6.81 (1H, t, C4H), 6.92 (2H, dd, C3H), 7.26 (m, 10H, dppe), 7.41 (m, 6H, dppe), 7.94 (m, 4H, dppe). 31P{1H} NMRδ/ppm: 107.5 (s, dppe). 13C{1H} NMRδ/ppm: 28.6 (m, dppe CH2); 79.3 (Cp); 120.7 (Cβ); 123.1 (C4), 125.3 (br, Cα), 127.5 (C3); 127.8, 128.1 (dd, 3JCP,5JCCP∼5 Hz, Cm,m′); 129.0, 129.4 (Cp,p′); 130.1 (C1); 130.5 (C2); 132.0, 134.0 (dd, 2JCP,4JCCP∼5, Co,o′); 138.2, 142.5 (m, Ci,i′). Found: C 75.2, H 5.6%. C39H34P2Fe requires: C 75.5, H 5.5%. ES-MS: m/z 621 [M + H]+, 519 [M − CCPh]+. High resolution calculated for FeC39H34P2: [M]+: 620.14797; found: 620.14757.
Preparation of Fe(CCC6H4Me-4)(dppe)Cp (2b)
Complex 2b (62 mg, 54%) was formed from FeCl(dppe)Cp (100 mg, 0.18 mmol), 1b (103 mg, 0.18 mmol) and NH4PF6 (30 mg, 0.18 mmol) in MeOH (20 mL) using a similar procedure as described for 2a. Crystals of 2b suitable for X-ray crystallography were grown from slow diffusion of hexane into a CH2Cl2 solution. IR (CH2Cl2) ν/cm−1: (CC) 2067. 1H NMRδ/ppm: 2.16 (s, 3H, CH3), 2.21 (m, 2H, dppe), 2.60 (m, 2H, dppe), 4.23 (s, 5H, Cp), 6.38 (2H, d, tolyl C2H), 6.72 (2H, dd, C3H), 7.26 (m, 10H, dppe), 7.40 (m, 6H, dppe), 7.93 (m, 4H, dppe). 31P{1H} NMRδ/ppm: 107.3 (s, dppe). 13C{1H} NMRδ/ppm: 21.0 (s, CH3), 28.3 (m, dppe CH2), 78.9 (Cp), 120.3 (Cβ), 122.0 (br, Cα), 126.9 (C1), 127.5, 127.8 (dd, 3JCP,5JCCP∼5 Hz, Cm,m′), 128.0 (C3), 128.6, 129.0 (Cp,p′), 130.0 (C2), 132.3 (C4), 131.7, 133.7 (dd, 2JCP,4JCCP∼5, Co,o′), 138.1, 142.3 (m, Ci,i′). Found: C 75.6, H 5.7%. C40H36P2Fe requires: C 75.7, H 5.7%. ES-MS(+): m/z 635 [M + H]+.
Preparation of Ru(CCC6H5)(PPh3)2Cp (3a)
A suspension of RuCl(PPh3)2Cp (100 mg, 0.14 mmol), 1a (77 mg, 0.14 mmol) and NH4PF6 (22 mg, 0.14 mmol) in MeOH (10 mL) was heated at reflux point for 30 min to form a bright red solution. Addition of 2–3 drops of DBU caused a yellow precipitate to form, which was collected by filtration, washed with cold MeOH (3 mL), and air-dried to afford 3a as a yellow solid (88 mg, 81%) and characterised by comparison of spectroscopic data with those reported elsewhere.24,31
Preparation of Ru(CCC6H4Me-4)(PPh3)2Cp (3b)
Complex 3b (84 mg, 74%) was formed from 1b and RuCl(PPh3)2Cp, using a similar procedure to that described for 3a, and identified by comparison with an authentic sample.31
Preparation of Ru(CCC6H5)(dppe)Cp* (4a)
A suspension of RuCl(dppe)Cp* (100 mg, 0.15 mmol), 1a (84 mg, 0.15 mmol) and NH4PF6 (24 mg, 0.15 mmol) in MeOH (10 mL) was heated at reflux point for 1 h to form a bright red solution, which was treated with 2–3 drops of DBU and allowed to stir for a further 1 h. The yellow precipitate that formed over this time was collected by filtration, washed with cold MeOH (3 mL), and air-dried to afford 4a as a yellow solid (56 mg, 51%). Characterisation data for 4a were identical to that reported elsewhere.31,55
Preparation of Ru(CCC6H4Me-4)(dppe)Cp* (4b)
Complex 4b was prepared (63 mg, 56%) from 1b and RuCl(dppe)Cp* in a manner identical to that described for 4a, and characterised by comparison of spectroscopic data with that reported elsewhere.31
Formation of Ru(CCC6F5)(η2-O2)(PPh3)Cp* (4c)
A mixture of RuCl(PPh3)2Cp* (50 mg, 0.063 mmol) and 1c (86.6 mg, 0.127 mmol) was heated in refluxing benzene (13 mL) for 2.5 h to give a red-brown solution. Conventional work-up gave numerous unidentified products, appearing as multi-coloured bands on preparative TLC plates, together with AuCl(PPh3) (5 mg). One yellow band afforded yellow crystals (from hexane) (1.1 mg, 2.4%), identified by a single-crystal X-ray diffraction structure determination as Ru(CCC6F5)(η2-O2)(PPh3)Cp* (4c). The small amount obtained precluded further characterisation.
Preparation of Ir(CCC6H5)(η2-O2)(CO)(PPh3)2 (5a)
A suspension of IrCl(CO)(PPh3)2 (100 mg, 0.13 mmol) and 1a (73 mg, 0.13 mmol) were stirred at room temperature in THF (12 mL) for ca. 6 h to give an orange-brown solution. The progress of the reaction was monitored by TLC and 31P NMR [26.0 for IrCl(CO)(PPh3)2 and 25.2 ppm for Ir(CCC6H5)(CO)(PPh3)2]. When the reaction was adjudged complete, 7 mL of the solvent was removed to give a cloudy brown solution. Acetone (5 mL) was added to the solution and the mixture stirred for 10 min followed by filtration to give a yellow solid that was recrystallised (CHCl3/hexane) to afford small pale yellow crystals of 5a (71 mg, 62%). IR (CH2Cl2) ν/cm−1: (CC) 2134; (CO) 2005, (O–O) 883. 1H NMRδ/cm−1: 2.23 (s, 3H, Me), 6.24 (d, 2H, JHH = 8 Hz, C6H4), 6.81 (d, 2H, JHH = 8 Hz, C6H4), 7.39 (m, 12H, PPh3), 7.45 (m, 6H, PPh3), 7.59 (m, 12H, PPh3). 31P{1H} NMRδ/ppm: 8.3 (s). 13C{1H} NMRδ/ppm: 67.8 (t, 2JCP 11 Hz, Cα), 108.5 (s, Cβ), 125.1 (C4), 127.3 (C3), 128.1 (dd, 3JCP, 5JCP∼5 Hz, Cm), 128.1 (m, Ci), 128.2 (C1), 130.8 (Cp), 131.3 (C2), 134.7 (dd, 2JCP, 4JCP∼5 Hz, Co), 167.1 (s, CO). ES-MS: m/z 879 [M + H]+, 920 [M + H + MeCN]+.
Preparation of Ir(CCC6H4Me-4)(η2-O2)(CO)(PPh3)2 (5b)
A suspension of IrCl(CO)(PPh3)2 (100 mg, 0.13 mmol) and 1b (74 mg, 0.13 mmol) were stirred at room temperature in THF (12 mL) for ca. 6 h to give an orange solution. After the same work-up as described above for 5a, the yellow solid was recrystallised from CHCl3/hexane to yield yellow crystals of 5b (46 mg, 40%) suitable for X-ray crystallography. IR (CH2Cl2) ν/cm−1: (CC) 2128; (CO) 2008, (O–O) 834. 1H NMRδ/ppm: 2.23 (s, 3H, Me), 6.24 (d, 2H, JHH = 8 Hz, C6H4), 6.81 (d, 2H, JHH = 8 Hz, C6H4), 7.39 (m, 12H, PPh3), 7.45 (m, 6H, PPh3), 7.59 (m, 12H, PPh3). 31P{1H} NMRδ/ppm: 8.2 (s). 13C{1H} NMRδ/ppm: 21.1 (s, Me), 66.0 (t, 2JCP 11 Hz, Cα), 109.1 (s, Cβ), 125.0 (C1), 128.1 (C3), 131.1 (C2), 134.8 (C4), 128.1 (dd, 3JCP, 5JCP∼5 Hz, Cm), 128.1 (m, Ci), 130.7 (Cp), 134.6 (dd, 2JCP, 4JCP∼5 Hz, Co), 167.1 (s, CO). Found: C 61.5, H 4.1%. C46H37P2O3Ir requires: C 62.0, H 4.2%. ES-MS: m/z 893 [M + H]+, 934 [M + H + MeCN]+. High resolution calculated for IrC46H38O3P2: [M + H]+ 893.19199, found 893.19242.
Preparation of Ni(CCC6H5)(PPh3)Cp (6a)
A solution of NiBr(PPh3)Cp (100 mg, 0.21 mmol) in THF (10 mL), was treated with 1a (120 mg, 0.21 mmol) and the mixture allowed to stir at ambient temperature in the dark, the progress of the reaction being monitored by TLC (hexane–acetone, 8 : 2) and by 31P NMR [34.2 ppm for AuBr(PPh3)]. The solution colour became pale brown after 1 h. After 4 h, the solution was distinctly green. After stirring for 6 h, the reaction was adjudged complete and the mixture was taken to dryness, followed by purification by preparative TLC. A green band was isolated, which was recrystallised (chloroform/MeOH) to afford green crystals of 6a (55 mg, 53%). IR (CH2Cl2) ν/cm−1: (CC) 2097 cm−1. 1H NMRδ/ppm: 5.25 (s, 5H, Cp), 6.64 (d, 2H, phenyl C2H at CPh), 6.91 (t, 1H, C4H), 6.93 (m, 2H, C3H), 7.38 (m, 9H, PPh3), 7.74 (m, 6H, PPh3). 31P NMRδ/ppm: 41.8 (s, PPh3). 13C NMRδ/ppm: 85.8 (d, 2JPC = 50 Hz, Cα), 92.5 (d, 3JPC = 2 Hz, Cp), 119.7 (d, 3JPC = 2 Hz, Cβ), 124.5 (C4), 127.2 (C3), 128.0 (C1), 128.1 (d, 3JPC = 10 Hz, Cm), 130.1 (d, 4JPC = 2 Hz, Cp), 130.9 (C2), 133.8 (d, 2JPC = 10 Hz, Co), 134.0 (d, 2JPC = 49 Hz, Ci). ES-MS: m/z 995 [2M + Na]+, 509 [M + Na]+, 486 [M]+, 385 [M − CCPh]+.
Preparation of Ni(CCC6H4Me)(PPh3)Cp (6b)
The modestly air- and light-sensitive green complex 6b was prepared (44 mg, 42%) from 1b and NiBr(PPh3)Cp using the method described for 6a. IR (CH2Cl2) ν/cm−1: (CC) 2100. 1H NMRδ/ppm: 2.17 (s, 3H, CH3), 5.25 (s, 5H, Cp), 6.56 (d, 2H, tolyl C2H), 6.74 (d, 2H, C3H), 7.40 (m, 9H, PPh3), 7.76 (m, 6H, PPh3). 31P NMRδ/ppm: 41.7 (s, PPh3). 13C NMRδ/ppm: 21.2 (CH3), 83.5 (d, 2JPC = 50 Hz, Cα), 92.6 (d, 3JPC = 2 Hz, Cp), 119.7 (d, 3JPC = 2 Hz, Cβ), 125.2 (C1), 128.0 (C3), 128.1 (d, 3JPC = 10 Hz, Cm), 130.1 (d, 4JPC = 2 Hz, Cp), 130.7 (C2), 133.9 (d, 2JPC = 10 Hz, Co), 134.0 (d, 2JPC = 47 Hz, Ci), 134.1 (C4). ES-MS: m/z 501 [M + 1]+. Found: C 76.7, H 5.5%. C32H27PNi requires: C 76.7, H 5.5%.
Preparation of trans-Pt(CCC6H5)2(PPh3)2 (7a)
To a stirred aliquot of MeOH (15 mL), cis-PtCl2(PPh3)2 (141 mg, 0.18 mmol) and 1a (200 mg, 0.36 mmol) were added to form a pale yellow suspension. The reaction mixture was allowed to stir for ca. 15 h, after which time it was filtered and the precipitate collected and washed with acetone to give a yellow solid, which was recrystallised (CH2Cl2/hexane) to afford pale yellow crystals of 7a (86 mg, 59%). IR (nujol) ν/cm−1: (CC) 2107. 1H NMRδ/ppm: 6.28 (m, 4H, phenyl C2H at CCPh), 6.89 (m, 2H, C4H), 6.91 (m, 4H, C3H), 7.37 (m, 18H, PPh3), 7.81 (m, 12H, PPh3). 1P{1H} NMR (CDCl3, 80.96 MHz) δ/ppm: 19.7 (s + d, 1JPtP = 2640 Hz, PPh3). 13C{1H} NMRδ/ppm: 109.7 (t, 2JCP∼16 Hz, Cα), 113.2 (Cβ), 124.5 (C4), 127.0 (C3), 127.8 (dd, 3JCP/5JCP∼5 Hz, Cm), 128.5 (C1), 130.8 (C2), 130.1 (Cp), 131.4 (dd, 1JCP/3JCP∼29 Hz Ci), 135.1 (dd, 2JCP/4JCP∼5 Hz, Co). Found: C 68.0, H 4.4%. C52H40P2Pt requires: C 67.8, H 4.4%. ES-MS: m/z 922 [M + H]+, 944 [M + Na]+. High resolution calculated for PtC52H41P2: [M + H]+ 922.23257, found 922.23318, calculated for PtC52H40P2Na: [M + Na]+ 944.21452, found 944.21662.
Preparation of trans-Pt(CCC6H4Me)2(PPh3)2 (7b)
A reaction similar to that described for 7a between cis-PtCl2(PPh3)2 (200 mg, 0.25 mmol) and 1b (291 mg, 0.51 mmol) gave 7b as a pale yellow precipitate, which was recrystallised (CH2Cl2/MeOH) to afford pale yellow crystals (83 mg, 35%). IR (nujol) ν/cm−1: (CC) 2106. 1H NMRδ/ppm: 2.17 (s, 6H, Me), 6.16 (d, JHH∼7 Hz, C6H4), 6.70 (d, JHH∼7 Hz, C6H4), 7.36 (m, 18H, PPh3), 7.80 (m, 12H, PPh3). 1P{1H} NMRδ/ppm: 19.7 (s + d, 1JPtP = 2660 Hz, PPh3). 13C{1H} NMRδ/ppm: 21.2 (Me), 127.7 (dd, 3JCP, 5JCP∼5 Hz, Cm), 127.9 (C3), 130.1 (C2), 130.8 (Cp), 131.9 (dd, 1JCP, 3JCP∼29 Hz, Ci), 135.1 (dd, 2JCP, 4JCP∼5 Hz Co), other 13C peaks were not observed due to poor solubility. Found: C 68.8, H 4.6%. C54H44P2Pt requires: C 68.3, H 4.7%. ES-MS: m/z 950 [M + H]+, 972 [M + Na]+, 982 [M + H + MeOH]+. High resolution calculated for 194PtC54H45P2: [M]+ 949.256611, found 949.26387.
Preparation of trans-Pt(CCC6H5)2(PMe3)2 (8a)
To a stirred aliquot of MeOH (15 mL), trans-PtCl2(PMe3)2 (100 mg, 0.24 mmol) and 1a (269 mg, 0.48 mmol) were added under nitrogen to afford a pale green suspension, which was stirred at rt for ca. 15 h, filtered and washed with cold MeOH (3 mL) and hexane (3 mL), and recrystallised (CHCl3/MeOH) to afford pale yellow crystals of 8a (99 mg, 73%). IR (nujol) ν/cm−1: (CC) 2108. 1H NMRδ/ppm: 1.77 (dd, 2JPH, 4JPH∼4 Hz, and Pt satellites, 3JPt–H∼30 Hz, PMe3), 1.80 (dd, 3JPt–H∼30 Hz), 2.29 (s, 6H, Me), 7.03 (d, JHH = 8 Hz, C2H of tolyl), 7.23 (d, JHH = 8 Hz, C3H). 31P{1H} NMRδ/ppm: −19.4 (s + d, 1JPtP = 2300 Hz, PMe3). 13C{1H} NMRδ/ppm: 15.3 (dd, 2JCP,4JCP∼20 Hz, and Pt satellites, 2JPt–C∼81 Hz, PMe3), 107.2 (t, 2JCP = 15 Hz, Cα), 108.5 (Cβ), 125.3 (C4), 127.9 (C3), 128.2 (C1), 130.9 (C2). Found: C 47.1, H 5.1%. C24H32P2Pt requires: C 48.1, H 5.1%. ES-MS: m/z 1162 [2M + Na]+; 550 [M + H]+.
Preparation of trans-Pt(CCC6H4Me-4)2(PMe3)2 (8b)
Complex 8b was prepared from 1b (275 mg, 0.48 mmol) and PtCl2(PMe3)2 and isolated as pale yellow-green crystals (93 mg, 67%). IR (nujol) ν/cm−1: (CC) 2108. 1H NMRδ/ppm: 1.77 (dd, 2JPH, 4JPH∼4 Hz, and Pt satellites, 3JPt–H∼30 Hz, PMe3), 1.80 (dd, 3JPt–H∼30 Hz), 2.29 (s, 6H, Me), 7.03 (d, JHH = 8 Hz, C2H of tolyl), 7.23 (d, JHH = 8 Hz, C3H). 31P{1H} NMRδ/ppm: −19.4 (s + d, 1JPtP = 2310 Hz, PMe3). 13C{1H} NMRδ/ppm: 15.4 (dd, 2JCP,4JCP∼20 Hz, and Pt satellites, 2JPt–C∼83 Hz, PMe3), 21.3 (s, Me), 106.0 (t, 2JCP = 15 Hz, Cα), 108.4 (Cβ), 125.2 (C1), 128.6 (C3), 130.9 (C2), 135.0 (C4). Found: C 49.3, H 5.6%. C24H32P2Pt requires: C 49.9, H 5.6%. ES-MS: m/z 1177 [2M + Na]+, 632 [M + Na + MeOH]+, 578 [M + H]+. High resolution calculated for PtP2C24H33: [M + H]+ 578.16997, found 578.17067.
Isolation of Au(PMe3)2[PF6] (9)
A suspension of PtCl2(PMe3)2 (200 mg, 0.48 mmol), 1a (130 mg, 0.23 mmol) and NH4PF6 (78 mg, 0.48 mmol) in MeOH (15 mL) was allowed to stir at rt for ca. 8 h to give a white precipitate, which was collected by filtration, washed with a small quantity of cold MeOH (3 mL) followed by hexane (3 mL) and dried in vacuo. Crystallisation of the precipitate from MeCN/MeOH gave white crystals of 9 (14 mg, 0.028 mmol, 12%) suitable for X-ray crystallography. 1H NMRδ/ppm: 1.70. 31P{1H} NMRδ/ppm: 5.0 (s), −144.1 (septet, JPF = 710 Hz). 13C{1H} NMRδ/ppm: 16.4 (br). ES-MS: m/z 721 [2M + Na]+, 349 [M]+ C6H18AuP2 = [M]+. High resolution calculated for C6H18AuP2: [M]+ 349.05439, found 349.05424.Preparation of {Cu(CCC6H5)}n (10a)
An aliquot of THF (25 mL) was rapidly stirred and treated with CuI (100 mg, 0.50 mmol) and 1a (280 mg, 0.50 mmol) to form a white suspension, which gradually changed to yellow after ca. 2 h. The reaction was allowed to stir for ca. 24 h to ensure complete reaction. The yellow suspension was filtered and the solid was washed with hexane (5 mL) to give a bright yellow solid (71 mg, 86%) identified as {Cu(CCC6H5)}n10a. IR (KBr) ν/cm−1: (CC) 1940 (vw), 1481 (m), 742 (s), 679 (s), 520 (m), 511 (m).73 Found: C 58.1, H 3.1%. C8H5Cu requires: C 58.4, H 3.0%.
Preparation of {Cu(CCC6H4Me-4)}n (10b)
A stirred suspension of CuI (200 mg, 1.05 mmol) in THF (60 mL) was treated with 1b (600 mg, 1.05 mmol) to form a white suspension, which gradually changed to yellow after ca. 2 h. Using a similar work-up procedure as described for 10a, a yellow solid (175 mg, 94%) was isolated and identified as {Cu(CCC6H4Me-4)}n (10b). IR (KBr) ν/cm−1: (CC) 1937 (w), 1504 (s), 808 (s), 526 (m), 520 (m). Found: C 63.6, H 4.1%. C9H7Cu requires: C 60.5, H 3.9%.
Structure determinations†
Single-crystal X-ray data were collected using an Oxford Diffraction Xcalibur CCD (11), Rigaku R-AXIS Spider IP (2b and 5b), Bruker Smart CCD 1 K (6b and 8b) and 6 K (4b, 7a, 7b and 9) diffractometers at low temperature and using monochromatic MoKα radiation, λ = 0.71073 Å. All structures were solved by direct methods and refined by full matrix least squares refinement on F2. Anisotropic displacement parameters were refined for the non-hydrogen atoms, excepting the disordered ones in the structures 5b and 9; hydrogen atom treatment followed a riding model. The CCC6H4Me-4 fragment of molecule 5b is disordered over two equally occupied positions, which correspond to two different directions of the flexing in the alkyne moiety. The inclinations of the planar aryl rings are also slightly different. The disorder does not affect the geometry of rest of the molecule. Neutral atom complex scattering factors were employed within the SHELXL-97 program.74 Pertinent results are given in the tables and figures, the latter showing non-hydrogen atoms with 50% probability amplitude displacement ellipsoids.
Acknowledgements
We gratefully acknowledge funding from the EPSRC in the form of a Visiting Fellowship (MIB) and research grants (PJL, JAKH). WMK gratefully acknowledges funding from the Human Resources Development in Science and Technology Programme, Ministry of Science, Technology and Innovation, Malaysia.References
- (a) L. Hegedus, Transition Metals in the Synthesis of Complex Organic Molecules, University Science Books, Mill Valley, California, 1994 Search PubMed; (b) C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, Germany, 3rd edn, 2006 Search PubMed.
- (a) F. Diederich and P. J. Stang, Metal-Catalysed Cross-Coupling Reactions, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed; (b) E.-I. Negishi and A. De Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, New York, 2002 Search PubMed; (c) L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms, Elsevier, Oxford, 2005 Search PubMed; (d) F. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS; (e) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (f) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS; (g) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467 CrossRef CAS; (h) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1979, 101, 4992 CrossRef CAS; (i) J. Louie and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 11598 CrossRef CAS; (j) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866 RSC; (k) S. Baba and E. Negishi, J. Am. Chem. Soc., 1976, 98, 6729 CrossRef CAS.
- B. Cetinkaya, M. F. Lappert, J. McMeeking and D. E. Palmer, J. Chem. Soc., Dalton Trans., 1973, 1202 RSC.
- (a) L. Medei, L. Orian, O. V. Semeikin, M. G. Peterleitner, N. A. Ustynyuk, S. Santi, C. Durante, A. Ricci and C. Lo Sterzo, Eur. J. Inorg. Chem., 2006, 2582 CrossRef CAS; (b) M. S. Khan, S. J. Davies, A. K. Kakkar, D. Schwartz, B. Lin, B. F. G. Johnson and J. Lewis, J. Organomet. Chem., 1992, 424, 87 CrossRef CAS; (c) M. S. Khan, A. K. Kakkar, S. L. Ingham, P. R. Raithby, J. Lewis, B. Spencer, F. Wittmann and R. H. Friend, J. Organomet. Chem., 1994, 472, 247 CrossRef CAS; (d) S. J. Davies, B. F. G. Johnson, J. Lewis and P. R. Raithby, J. Organomet. Chem., 1991, 414, C51 CrossRef CAS.
- (a) R. J. Cross and M. F. Davidson, Inorg. Chim. Acta, 1985, 97, L35 CrossRef CAS; (b) A. F. Hill and R. P. Melling, J. Organomet. Chem., 1990, 396, C22 CAS; (c) R. D. Dewhurst, A. F. Hill, A. D. Rae and A. C. Willis, Organometallics, 2005, 24, 4703 CrossRef CAS; (d) J. P. Collman and J. W. Kang, J. Am. Chem. Soc., 1967, 89, 844 CrossRef CAS; (e) A. F. Hill and J. D. E. T. Wilton-Ely, Organometallics, 1997, 16, 4517 CrossRef CAS; (f) R. B. Bedford, A. F. Hill, A. R. Thompsett, A. J. P. White and D. J. Williams, Chem. Commun., 1996, 1059 RSC; (g) G. B. Deacon and D. L. Wilkinson, Inorg. Chim. Acta, 1988, 142, 155 CrossRef CAS; (h) G. B. Deacon and A. J. Koplick, J. Organomet. Chem., 1978, 146, C43 CrossRef CAS; (i) G. B. Deacon, A. J. Koplick, W. D. Raverty and D. G. Vince, J. Organomet. Chem., 1979, 182, 121 CrossRef CAS; (j) G. B. Deacon, A. J. Koplick and T. D. Tuong, Aust. J. Chem., 1982, 35, 941 CAS; (k) G. B. Deacon and R. H. Newham, Aust. J. Chem., 1985, 38, 1757 CAS; (l) G. B. Deacon, S. Nickel, P. MacKinnon and E. R. T. Tiekink, Aust. J. Chem., 1990, 43, 1245 CAS; (m) G. Lin, R. McDonald and J. Takats, Organometallics, 2000, 19, 1814 CrossRef CAS; (n) G. R. Clark, S. V. Hoskins and W. R. Roper, J. Organomet. Chem., 1982, 234, C9 CrossRef CAS; (o) M. A. Gallop, T. C. Jones, C. E. F. Rickard and W. R. Roper, J. Chem. Soc., Chem. Commun., 1984, 1002 RSC.
- For a discussion of the hazards associated with the use of mercuric acetylides see: R. D. Dewhurst, A. F. Hill and M. K. Smith, Organometallics, 2006, 25, 2388 Search PubMed.
- (a) M. I. Bruce, O. M. Abu Salah, R. E. Davis and N. V. Ragavan, J. Organomet. Chem., 1974, 64, C48 CrossRef CAS; (b) O. M. Abu Salah and M. I. Bruce, J. Chem. Soc., Chem. Commun., 1972, 858 RSC; (c) O. M. Abu Salah and M. I. Bruce, J. Chem. Soc., Dalton Trans., 1975, 2311 RSC; (d) O. M. Abu Salah and M. I. Bruce, J. Chem. Soc., Dalton Trans., 1974, 2302 RSC; (e) O. M. Abu Salah and M. I. Bruce, Aust. J. Chem., 1976, 29, 531 CAS; (f) M. I. Bruce, N. N. Zaitseva, B. W. Skelton, N. Somers and A. H. White, Aust. J. Chem., 2003, 56, 509 CrossRef CAS.
- (a) K. Sonogashira, T. Yatake, Y. Tohda, S. Takahashi and N. Hagihara, J. Chem. Soc., Chem. Commun., 1977, 291 RSC; (b) K. Sonogashira, Y. Fujikura, T. Yatake, N. Toyoshima, S. Takahashi and N. Hagihara, J. Organomet. Chem., 1978, 145, 101 CrossRef CAS.
- M. I. Bruce, M. G. Humphrey, J. G. Matisons, S. K. Roy and A. G. Swincer, Aust. J. Chem., 1984, 37, 1955 CAS.
- O. M. Abu Salah and M. I. Bruce, Aust. J. Chem., 1976, 29, 73.
- (a) O. M. Abu Salah and M. I. Bruce, Aust. J. Chem., 1977, 30, 2639 CAS; (b) D. Miguel and V. Riera, J. Organomet. Chem., 1985, 293, 379 CrossRef CAS.
- (a) E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 333 RSC; (b) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef; (c) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2005, 44, 6990 CrossRef CAS; (d) M. Haruta, Nature, 2005, 437, 1098 CrossRef CAS.
- (a) M. Contel, M. Stol, M. A. Casado, G. P. M. van Klink, D. D. Ellis, A. L. Spek and G. van Koten, Organometallics, 2002, 21, 4556 CrossRef CAS; (b) M. Robitzer, I. Bouamaïed, C. Sirlin, P. A. Chase, G. van Koten and M. Pfeffer, Organometallics, 2005, 24, 1756 CrossRef CAS; (c) M. Stol, D. J. M. Snelders, H. Kooijman, A. L. Spek, G. P. M. van Klink and G. van Koten, Dalton Trans., 2007, 2589 RSC.
- C.-L. Chan, K.-L. Cheung, W. H. Lam, E. C.-C. Cheng, N. Zhu, S. W.-K. Choi and V. W.-W. Yam, Chem.–Asian J., 2006, 1, 273 CrossRef CAS.
- M. Ferrer, L. Rodríguez, O. Russell, J. C. Lima, P. Gómez-Sal and A. Martín, Organometallics, 2004, 23, 5096 CrossRef CAS.
- M. I. Bruce, M. E. Smith, N. N. Zaitseva, B. W. Skelton and A. H. White, J. Organomet. Chem., 2003, 670, 170 CrossRef CAS.
- (a) M. I. Bruce, B. W. Skelton, A. H. White and N. N. Zaitseva, J. Organomet. Chem., 2003, 683, 398 CrossRef CAS; (b) M. I. Bruce, P. A. Humphrey, G. Melino, B. W. Skelton, A. H. White and N. N. Zaitseva, Inorg. Chim. Acta, 2005, 358, 1453 CrossRef CAS; (c) A. B. Antonova, M. I. Bruce, B. G. Ellis, M. Gaudio, P. A. Humphrey, M. Jevric, G. Melino, B. K. Nicholson, G. J. Perkins, B. W. Skelton, B. Stapleton, A. H. White and N. N. Zaitseva, Chem. Commun., 2004, 960 RSC.
- M. I. Bruce, E. Horn, J. G. Matisons and M. R. Snow, Aust. J. Chem., 1984, 37, 1163 CAS.
- X. L. R. Fontaine, S. J. Higgins, C. R. Langrick and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1987, 777 RSC.
- (a) R. J. Cross and M. F. Davidson, J. Chem. Soc., Dalton Trans., 1986, 411 RSC; (b) R. J. Cross, M. F. Davidson and A. J. McLennan, J. Organomet. Chem., 1984, 265, C37 CrossRef CAS.
- (a) M. I. Bruce, B. C. Hall, B. W. Skelton, M. E. Smith and A. H. White, J. Chem. Soc., Dalton Trans., 2002, 995 RSC; (b) M. I. Bruce, N. N. Zaitseva, P. J. Low, B. W. Skelton and A. H. White, J. Organomet. Chem., 2006, 691, 4273 CrossRef CAS.
- (a) C.-M. Che, H.-Y. Chao, V. M. Miskowski, Y. Li and K.-K. Cheung, J. Am. Chem. Soc., 2001, 123, 4985 CrossRef CAS; (b) W. Lu, H.-F. Xiang, N. Zhu and C.-M. Che, Organometallics, 2002, 21, 2343 CrossRef CAS; (c) W. Lu, N. Zhu and C.-M. Che, J. Organomet. Chem., 2003, 670, 11 CrossRef CAS.
- (a) P. J. Low and M. I. Bruce, Adv. Organomet. Chem., 2002, 48, 71 CrossRef; (b) M. I. Bruce and P. J. Low, Adv. Organomet. Chem., 2004, 50, 179 CrossRef CAS.
- M. I. Bruce and R. C. Wallis, Aust. J. Chem., 1979, 32, 1471 CAS.
- C. Bitcon and M. W. Whiteley, J. Organomet. Chem., 1987, 336, 385 CrossRef CAS.
- (a) For a representative selection of recent references see: C. E. Powell and M. G. Humphrey, Coord. Chem. Rev., 2004, 248, 725 Search PubMed; (b) N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586 CrossRef CAS; (c) M. I. Bruce, K. Costuas, B. G. Ellis, J.-F. Halet, P. J. Low, B. Moubaraki, K. S. Murray, N. Ouddaï, G. J. Perkins, B. W. Skelton and A. H. White, Organometallics, 2007, 26, 3735 CrossRef CAS; (d) M. I. Bruce, K. Costuas, J.-F. Halet, K. A. Kramarczuk, P. J. Low, B. K. Nicholson, G. J. Perkins, R. L. Roberts, B. W. Skelton, M. E. Smith and A. H. White, Dalton Trans., 2007, 5387 RSC; (e) K. Costuas, F. Paul, L. Toupet, J.-F. Halet and C. Lapinte, Organometallics, 2004, 23, 2053 CrossRef CAS; (f) F. Paul, L. Toupet, J.-Y. Thépot, K. Costuas, J.-F. Halet and C. Lapinte, Organometallics, 2005, 24, 5464 CrossRef CAS; (g) M. A. Fox, B. Le Guennic, R. L. Roberts, T. E. Baines, D. S. Yufit, D. Albesa-Jové, F. Hartl, J. A. K. Howard and P. J. Low, J. Am. Chem. Soc., 2008, 130, 3566 CrossRef CAS.
- M. I. Bruce, Chem. Rev., 1991, 91, 197 CrossRef CAS.
- M. I. Bruce, B. C. Hall, B. D. Kelly, P. J. Low, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1999, 3719 RSC.
- R. C. Bott, G. A. Bowmaker, R. W. Buckley, P. C. Healy and M. C. S. Perera, Aust. J. Chem., 1999, 52, 271 CAS.
- R. Denis, L. Toupet, F. Paul and C. Lapinte, Organometallics, 2000, 19, 4240 CrossRef CAS.
- M. A. Fox, R. L. Roberts, W. M. Khairul, F. Hartl and P. J. Low, J. Organomet. Chem., 2007, 692, 3277 CrossRef CAS.
- M. I. Bruce, B. C. Hall, N. N. Zaitseva, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 1793 RSC.
- C. A. Reed and W. R. Roper, J. Chem. Soc., Dalton Trans., 1973, 1370 RSC.
- M. I. Bruce, B. C. Hall, P. J. Low, M. E. Smith, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 300–302, 633 Search PubMed.
- R. H. Walter and B. F. G. Johnson, J. Chem. Soc., Dalton Trans., 1978, 381 RSC.
- C. S. Chin, M. Oh, G. Won, H. Cho and D. Shin, Polyhedron, 1999, 18, 811 CrossRef CAS.
- K. Osakada, M. Kimura and J.-C. Choi, J. Organomet. Chem., 2000, 602, 144 CrossRef CAS.
- C. S. Chin, M. Oh, G. Won, H. Cho and D. Shin, Polyhedron, 1999, 18, 811 CrossRef CAS.
- K. Osakada, M. Kimura and J.-C. Choi, J. Organomet. Chem., 2000, 602, 144 CrossRef CAS.
- M. Itazaki, C. Yoda, Y. Nishihara and K. Osakada, Organometallics, 2004, 23, 5402 CrossRef CAS.
- A. Antiñolo, I. Fonseca, A. Ortiz, M. J. Rosales, J. Sanz-Aparicio, P. Terreros and H. Torrens, Polyhedron, 1999, 18, 959 CrossRef CAS.
- (a) H. Yamazaki and N. Hagihara, Bull. Chem. Soc. Jpn., 1964, 37, 907 CrossRef CAS; (b) H. Yamazaki, T. Nishido, Y. Matsumoto, S. Sumida and N. Hagihara, J. Organomet. Chem., 1966, 6, 86 CrossRef CAS; (c) R. Nast, Coord. Chem. Rev., 1982, 47, 89 CrossRef CAS.
- U. E. I. Horvarth and H. G. Raubenheimer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m567 CrossRef.
- S. I. Ghazala, F. Paul, L. Toupet, T. Roisnel, P. Hapiot and C. Lapinte, J. Am. Chem. Soc., 2006, 128, 2463 CrossRef CAS.
- S. I. Ghazala, N. Gauthier, F. Paul, L. Toupet and C. Lapinte, Organometallics, 2007, 26, 2308 CrossRef CAS.
- (a) J. Coumarcel, G. Le Gland, L. Toupet, F. Paul and C. Lapinte, J. Organomet. Chem., 2003, 670, 108 CrossRef; (b) F. Paul, L. Toupet, J.-Y. Thepot, K. Costuas, J.-F. Halet and C. Lapinte, Organometallics, 2005, 24, 5464 CrossRef CAS; (c) M. P. Cifuentes, M. G. Humphrey, J. P. Morrall, M. Samoc, F. Paul, C. Lapinte and T. Roisnel, Organometallics, 2005, 24, 4280 CrossRef CAS.
- K. Costuas, F. Paul, L. Toupet and C. Lapinte, Organometallics, 2004, 23, 2053 CrossRef CAS.
- F. Paul, G. da Costa, A. Bondon, N. Gauthier, S. Sinbandhit, L. Toupet, K. Costuas, J.-F. Halet and C. Lapinte, Organometallics, 2007, 26, 874 CrossRef CAS.
- (a) K. M.-C. Wong, S. C.-F. Lam, C.-C. Ko, N. Zhu, V. W.-W. Yam, S. Roue, C. Lapinte, S. Fathallah, K. Costuas, S. Kahlal and J.-F. Halet, Inorg. Chem., 2003, 42, 7086 CrossRef CAS; (b) Y. Tanaka, T. Ozawa, A. Inagaki and M. Akita, Dalton Trans., 2007, 928 RSC; (c) J. A. Shaw-Taberlet, S. Sinbandhit, T. Roisnel, J. R. Hamon and C. Lapinte, Organometallics, 2006, 25, 5311 CrossRef CAS; (d) T. Weyland, C. Lapinte, G. Frapper, M. J. Calhorda, J.-F. Halet and L. Toupet, Organometallics, 1997, 16, 2024 CrossRef CAS.
- (a) T. Weyland, K. Costuas, L. Toupet, J.-F. Halet and C. Lapinte, Organometallics, 2000, 19, 4228 CrossRef CAS; (b) F. de Montigny, G. Argouarch, K. Costuas, J.-F. Halet, T. Roisnel, L. Toupet and C. Lapinte, Organometallics, 2005, 24, 4558 CrossRef CAS.
- M. H. Garcia, M. P. Robalo, A. R. Dias, M. T. Duarte, W. Wenseleers, G. Aerts, E. Goovaerts, M. P. Cifuentes, S. Hurst, M. G. Humphrey, M. Samoc and B. Luther-Davis, Organometallics, 2002, 21, 2107 CrossRef CAS.
- C. J. McAdam, A. R. Manning, B. H. Robinson and J. Simpson, Inorg. Chim. Acta, 2005, 358, 1673 CrossRef CAS.
- M. P. Gamasa, J. Gimeno, E. Lastra, M. Lanfranchi and A. Tiripicchio, J. Organomet. Chem., 1991, 405, 333 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- F. Paul, B. G. Ellis, M. I. Bruce, L. Toupet, T. Roisnel, K. Costuas, J.-F. Halet and C. Lapinte, Organometallics, 2006, 25, 649 CrossRef CAS.
- G. Jia, W. S. Ng, H. S. Chu, W.-T. Wong, N.-T. Yu and I. D. Williams, Organometallics, 1999, 18, 3597 CrossRef CAS.
- (a) H.-H. Wang, L. H. Pignolet, P. E. Reedy, Jr, M. M. Olmstead and A. L. Balch, Inorg. Chem., 1987, 26, 377 CrossRef CAS; (b) D. G. Ho, R. Ismail, N. Franco, R. Gao, E. P. Leverich, I. Tsyba, N. N. Ho, R. Bau and M. Selke, Chem. Commun., 2002, 570 RSC; (c) J. A. McGinnety, R. J. Doedens and J. A. Albers, Inorg. Chem., 1967, 6, 2243 CrossRef CAS.
- (a) M. S. Weininger, I. F. Taylor, Jr. and E. L. Amma, J. Chem. Soc. D, 1971, 1172 RSC; (b) M. S. Weininger, E. A. H. Griffith, C. T. Sears and E. L. Amma, Inorg. Chim. Acta, 1982, 60, 67 CrossRef CAS.
- S. J. La Placa and J. A. Ibers, J. Am. Chem. Soc., 1965, 87, 2581 CrossRef CAS.
- L. Vaska, Acc. Chem. Res., 1976, 9, 175 CrossRef CAS.
- I. R. Whittall, M. G. Humphrey and D. C. R. Hockless, Aust. J. Chem., 1998, 51, 219 CrossRef.
- (a) C. J. McAdam, A. R. Manning, B. H. Robinson and J. Simpson, Inorg. Chim. Acta, 2005, 358, 1673 CrossRef CAS; (b) J. F. Gallagher, P. Butler, R. D. A. Hudon and A. R. Manning, J. Chem. Soc.,Dalton Trans., 2002, 75 RSC; (c) R. H. Naulty, M. P. Cifuentes, M. G. Humphrey, S. Houbrechts, C. Boutton, A. Persoons, G. A. Heath, D. C. R. Hockless, B. Luther-Davies and M. Samoc, J. Chem. Soc., Dalton Trans., 1997, 4167 RSC; (d) P. Butler, J. F. Gallagher, A. R. Manning, H. Mueller-Bunz, C. J. McAdam, J. Simpson and B. H. Robinson, J. Organomet. Chem., 2005, 690, 4545 CrossRef CAS.
- (a) M. Mayor, C. von Hanisch, H. B. Weber, J. Reichert and D. Beckmann, Angew. Chem., Int. Ed., 2002, 41, 1183 CrossRef CAS; (b) A. J. Deeming, G. Hogarth, M. Lee, M. Saha, S. P. Redmond, H. Phetmung and A. G. Orpen, Inorg. Chim. Acta, 2000, 309, 109 CrossRef CAS; (c) M. Ravera, R. D'Amato and A. Guerri, J. Organomet. Chem., 2005, 690, 2376 CrossRef CAS; (d) K. Campbell, R. McDonald, M. J. Ferguson and R. R. Tykwinski, J. Organomet. Chem., 2003, 683, 379 CrossRef CAS; (e) T. L. Schull, J. G. Kushmerick, C. H. Patterson, C. George, M. H. Moore, S. K. Pollack and R. Shashidhar, J. Am. Chem. Soc., 2003, 125, 3202 CrossRef CAS; (f) R. D'Amato, A. Furlani, M. Colapietro, G. Portalone, M. Casalboni, M. Falconieri and M. V. Russo, J. Organomet. Chem., 2001, 627, 13 CrossRef CAS.
- J. Vicente, M.-T. Chicote, M. M. Alvarez-Falcón and P. G. Jones, Chem. Commun., 2004, 2658 RSC.
- (a) S. Szafert and J. A. Gladysz, Chem. Rev., 2003, 103, 4175 CrossRef CAS; (b) S. Szafert and J. A. Gladysz, Chem. Rev., 2006, 106, PR1 CAS.
- M. J. Mays and P. L. Sears, J. Chem. Soc., Dalton Trans., 1973, 1873 RSC.
- M. I. Bruce, C. Hameister, A. G. Swincer and R. C. Wallis, Inorg. Synth., 1990, 18, 270.
- M. I. Bruce, B. G. Ellis, P. J. Low, B. W. Skelton and A. H. White, Organometallics, 2003, 22, 3184 CrossRef CAS.
- E. Hernandez and P. Royo, J. Organomet. Chem., 1985, 291, 387 CrossRef CAS.
- G. B. Kaufmann and L. A. Teter, Inorg. Synth., 1963, 7, 245.
- D. L. Packett, C. M. Jensen, R. L. Cowan, C. E. Strouse and W. C. Trogler, Inorg. Chem., 1985, 24, 3578 CrossRef CAS.
- K. Vrieze, J. P. Collman, C. T. Sears, Jr. and M. Kubota, Inorg. Synth., 1968, 11, 101 CAS.
- I. A. Garbusova, V. T. Alexanjan, L. A. Leites, I. R. Golding and A. M. Sladkov, J. Organomet. Chem., 1973, 54, 341 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † CCDC reference numbers 660622 (4c) and 691297–691304. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b809960j |
| ‡ In the process of crystallization of the complex 7b, crystals of previously unknown chloroform di-solvate of trans-PtCl2(PPh3)2 were isolated and structurally studied.† |
| This journal is © The Royal Society of Chemistry 2009 |