DOI:
10.1039/B819336N
(Profile)
Chem. Soc. Rev., 2009,
38, 1521-1529
The master of chemical topology†
When Jean-Pierre Sauvage (Fig. 1) set out on his mission in the early 1980s to devise ways and means to effect the mechanical interlocking of large rings, there was not all that much information or inspiration in the literature from which he could draw a lot of encouragement. The statistical approaches of Wasserman1 and the Harrisons2–4 had produced, at best, minuscule amounts of catenanes and rotaxanes. Also, there were no indications that these first-generation mechanically interlocked molecules (MIMs) had any physical or chemical properties that were worthy of note, let alone bristling with promise. The Möbius strip approach to catenane formation proposed by Frisch and Wasserman,5 and pursued for some time by Walba,6 had a certain amount of fascination for a time but suffered in its implementation from the intervention of bad luck simply because good luck was the exception rather than the rule, i.e., it also relies heavily on statistics. |
| | Fig. 1 Jean-Pierre Sauvage, during a lecture given at Nagoya University in September 2008, manipulating an origami-style catenane made by Junko Furusho. The photograph was taken by Yoshio Furusho. | |
There was, however, another approach, which had been developed by Gottfried Schill in Freiburg, involving the use of conditions-sensitive, reversible covalent bonding (e.g., the formation and cleavage of cyclic acetals under slightly different conditions and dependent upon acid catalysis) to the bringing of the components of MIMs together before completing their constitutions and finally severing the covalent connections between the components to reveal the topological (mechanical) bond(s). Schill produced his blueprint for the use of covalent templation in the synthesis of “Catenanes, Rotaxanes and Knots” in the form of a monograph published7 in 1971. During almost a quarter of a century, Schill demonstrated the application of covalent-directed templated synthesis to the production of [2]catenanes,8,9 [2]rotaxanes9 and [3]catenanes10,11 as well as molecular knots.12 There were two issues that, most unfortunately, diminished the value of Schill’s epic contributions to the chemistry of MIMs. One was that the synthetic chemistry involved numerous steps—commonly in excess of 20—and many of the reactions were daunting to perform and so their efficiencies were not all that high: the net outcomes were quite low overall yields of the catenanes, rotaxanes, and knots themselves. The other issue was the same as the one which had plagued the MIMs obtained by the statistical approach—namely, the lack of any specific interactions between their interlocked components, following cleavage of the covalent templates.
It would be fair to say that by the early 1980s fatigue was beginning to overtake that small part of the chemical community who remained in any way optimistic about the prospect of making property-rich MIMs in quantities in a routine manner. After 15 years of toil and trouble looking for that all-important tipping point, a practical synthesis of catenanes seemed out of reach of synthetic chemists and the community was gradually losing interest in these topological molecules. Then, like a bolt out of the blue, a communication appeared13 in Tetrahedron Letters, late in 1983, in French entitled, “Une Nouvelle Famille de Molecules: Les Metallo-Catenanes,” co-authored by Christiane Dietrich-Buchecker, Jean-Paul Kintzinger and Jean-Pierre Sauvage. It proposed novel strategies for making catenanes, based on the three-dimensional template effect of a transition metal, namely copper(I). One can detect the unbridled enthusiasm of the authors in the short abstract written in English. With some slight editing, it reads—
“A new strategy has been developed for synthesizing catenanes. It is based on a generalized template effect. The first example of a novel class of molecules, the metallo-catenanes, has been obtained in good yield. It contains copper(I) and macrocyclic phenanthroline derivatives.”
It was immediately evident from a cursory reading of this communication that catenanes were at long last easily accessible molecular compounds which could be obtained rather quickly in respectable quantities. The appearance of this short paper marked the birth of molecules which had fascinated chemists for decades, but which were more entities for discussion and laboratory curiosities than real chemical compounds. Jean-Pierre Sauvage started a movement in science. From 1983 to the present, his thinking and strategies have been generalized and used by many others to create a whole new family of topologically fascinating molecules, including higher catenanes and exotic structures based on multiply interlocked ring systems.
The two strategies proposed by Sauvage for the metal-templated synthesis of compounds he decided to name catenates are recalled from the past in Fig. 2 after the original illustration which appeared in the seminal communication in Tetrahedron Letters and was later to grace the screens and chalk-boards (Fig. 3) in many conference halls all around the world. Strategy B was exploited (Fig. 4) in the first catenate synthesis reported in the 1983 communication, while Strategy A was employed in a one-step synthesis of the same catenate reported (Fig. 5) in a communication14 in the Journal of the American Chemical Society in 1984. Not surprisingly, Strategy B was the more efficient, affording a 42% yield of the catenate in comparison with a yield of 27% when Strategy A was employed. In his second communication, Sauvage showed that the Cu(I) ion could be removed quantitatively from the catenate to give a ligand he referred to as a catenand which could then be used to make other catenates, simply by offering it metal ions, e.g., Li+, Ag+, etc. Although the Strasbourg team’s characterization of the first catenate and the derived catenand using spectroscopic and electrochemical techniques was totally convincing, a third short paper15 in Chemical Communications in 1985 was to illuminate the world with the very first X-ray crystal structures (Fig. 6) of a catenate and a catenand. These illustrations conveyed an important message—namely, that in contrast with many complexing agents (receptors) which are preoriented in a stereoelectronic fashion appropriate to a good matching with a given metal (substrate), the catenand has no geometrical analogy with the corresponding complex (catenate) although the stability and kinetic inertness of the catenate are extremely high. Subsequently, Sauvage and his colleagues demonstrated that the decomplexation rates of the catenates depend a lot on both the topography (the geometry of the coming together of the ligands round the metal) and the topology (the mechanical interlocking of the rings). Moreover, Sauvage, in collaboration with Anne-Marie Albrecht-Gary, identified the so-called catenand effect16 of topological origin in catenanes. When the ligand is composed of two interlocked rings, their unraveling, which is necessary for demetallation, renders this reaction several orders of magnitude slower than those for acyclic analogues of catenates. In keeping with this observation, the introduction of increased rigidity into catenates led to their being even more inert.17 The catenane pioneers are captured (Fig. 7) on film in their laboratory in Strasbourg in 1986.
 |
| | Fig. 2 Two synthetic strategies based on the template effect induced by a transition metal. Functions f and g react to form links (fg). The molecular fragments f–f and g–g interact with the transition metal (m). This metal disposes the fragments perpendicular to one another. | |
 |
| | Fig. 3 Jean-Pierre Sauvage in full flight at the blackboard at the end of his plenary lecture given at the 18th International Symposium on Macrocyclic Chemistry (29 June–2 July) at the University of Twente in The Netherlands in 1993. With reference to Strategy B in Fig. 2, he is illustrating the principle of maximum site occupancy in response to a question from the audience. | |
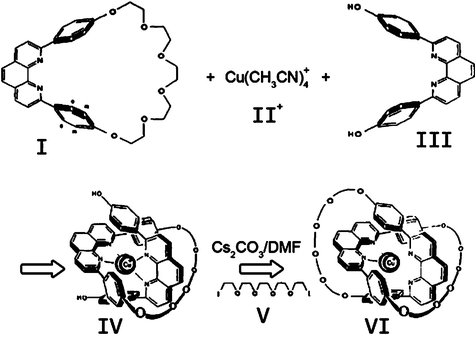 |
| | Fig. 4 Templated synthesis of the copper(I) catenate VI using Strategy B outlined in Fig. 2. | |
 |
| | Fig. 5 The one-step synthesis (Strategy A in Fig. 2) of the catenate, followed by its demetallation to give the corresponding catenand. | |
 |
| | Fig. 6 The solid-state structure of the copper(I) catenate and the catenand derived from it. | |
 |
| | Fig. 7 The Catenane Pioneers. Jean-Pierre Sauvage is standing in the middle with Christiane Dietrich-Buchecker and Sylvie Chardon-Noblat on his right-hand side and Jean-Claude Chambron and Zeinab Saad on his left-hand side. Standing in the background are Abdelaziz Jouaitti and Jean-Paul Collin. Christiane is holding their trophy—the first catenate. | |
Jean-Pierre Sauvage had begun his research career in stellar fashion as the first PhD student under the tutelage of the brilliant young Jean-Marie Lehn. Sauvage obtained his doctorate degree from the University of Strasbourg in 1971 with a thesis describing his research on the synthesis and properties of cryptands and cryptates.18–20 Little did he know it at the time, but these first tiny steps in research into chemistry by the apprentice were to pave the way, with the passage of time, for his mentor to go to Stockholm towards the end of 1987 to collect his share, along with Charles Pedersen and Donald Cram, of the Nobel Prize in chemistry. After spending a year as a postdoctoral scholar at the University of Oxford with Malcolm Green, Sauvage returned to Strasbourg as a CNRS researcher in the Lehn group, working for most of the time in the area of photochemistry. In the period from 1976 until he was invited to create his own “Laboratoire de Chimie Organo-Minérale” in 1980, he was involved in the development of new photochemical systems for the splitting of water21 into hydrogen and oxygen. In 1977, Lehn and Sauvage22 described one of the very first systems to effect the catalytic reduction of water to hydrogen under the influence of light, and in 1979, the opposing redox reaction—namely, the oxidation of water to oxygen—was realised23 employing related reactions and photochemical principles.
It was against this background that the fledgling Sauvage group made its rapid and successful foray into the field of chemical topology. The dramatic achievements of the early 80s have been summarised in a couple24,25 of scholarly reviews. Along with catenanes, rotaxanes are now commonplace components of many integrated systems displaying emergent properties in the context of polymers, organic materials, and electron transfer, etc. At present, perhaps the most eye-catching arena utilizing catenanes and rotaxanes is that concerned with controlled dynamic systems—often referred to as “molecular machines” or “motor molecules.” Since the paradigm shift—initiated by Sauvage’s metal template-directed synthesis of a [2]catenane in 1983—lifted the field of chemical topology from imaginary to real, the concept of templation26 has been generalised and used in many different settings to create whole new families of topologically important molecular compounds. There have also been variations on the catenane front, one being a molecular necklace27—consisting of a gigantic macrocycle—no less than a 132-membered ring—with separately interlocked onto it, six very much smaller rings. Many of the early catenates and catenands could be crystallised in the form of good quality single crystals, admirably suitable for X-ray crystallography. At this stage in the development of this brand new chemistry, a picture was worth more than a thousand words. A particularly spectacular example, that of a [3]catenate, is portrayed in Fig. 8. The solid-state structure of this compound is appealing from an aesthetic as well as a chemical perspective.28 The particular catenate is of importance for two additional reasons. One is that the compound and its derivatives are of considerable interest in relation to probing and understanding energy and electron transfer processes. The other reason relates to one of the holy grails in catenane chemistry—the synthesis of a genuine high molecular weight polycatenane. The [3]catenate (Fig. 8) may be regarded as the prototype of future synthetic polymers where the monomer units are the mutually interlocked rings—arranged like the links in a traditional macroscopic chain—of the polymer chain.
![A [3]catenate finds cover on the front of Angewandte Chemie. C. O. Dietrich-Buchecker, J. Guilhem, A.-K. Khemiss, J.-P. Kintzinger, C. Pascard and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1987, 26. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.](/image/article/2009/CS/b819336n/b819336n-f8.gif) |
| | Fig. 8 A [3]catenate finds cover on the front of Angewandte Chemie. C. O. Dietrich-Buchecker, J. Guilhem, A.-K. Khemiss, J.-P. Kintzinger, C. Pascard and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1987, 26. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | |
With catenanes well and truly under his belt, Sauvage quickly turned his considerable expertise to the making of molecular knots. By applying a strategy (Fig. 9) very similar to that described in his first report13 in 1983 on metallo-catenanes, he synthesised (Fig. 10), in collaboration with Christiane Dietrich-Buchecker, the first molecular trefoil knot. The compound, reported29 in 1989, is a small-molecule example of the simplest topologically non-trivial knot (Fig. 11) and, as such, bridges science and art, as well as biology and chemistry. Indeed, many of the mechanically interlocked systems and knots, expressed by the Sauvage group at the small-molecule level, display topological properties similar to those of much larger molecules formed by DNA in the course of recombination and replication processes—all this topology being already recognised by molecular biologists since the early 70s. The first wholly synthetic molecular knot represents a Herculean task accomplished, given the fact that chemists had been hypothesising about molecular knots for several decades before they became a reality in the hands of Sauvage. In Fig. 12, we see the master celebrating his art in song during a visit to Milan in 1990.
 |
| | Fig. 9 The Sauvage strategy for making a molecular trefoil knot using two transition-metal centers as templating species. Two coordinating molecular threads A2 are interlaced on two transition-metal centres, forming a double helix B2. After cyclisation to C2 and demetallation, a knotted system D2 is obtained. | |
 |
| | Fig. 10 Templated syntheses of the first molecular trefoil knot using the strategy outlined in Fig. 9. | |
 |
| | Fig. 11 A trefoil knot graces a front cover. See also the front of this special issue. C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | |
 |
| | Fig. 12 A visit to Milan in 1990 finds Jean-Pierre being auditioned for a supporting role in an upcoming presentation of Carmen at La Scala. | |
As the chemistry of MIMs has evolved with all its associated complexity and diversity, the synthetic protocols for making interlocked and knotted rings have improved out of all recognition compared with the situation that existed back in Sauvage’s pioneering days. Thus, in a collaboration, begun in the late 90s with the Grubbs group at the California Institute of Technology, the original preparation of the [2]catenate has been emulated in spectacular fashion using a methodology based on the ring closing metathesis of terminal olefins.30,31 In a few simple steps starting from commercially available compounds, [2]catenanes can now be obtained in an overall yield of 70% with the key cyclisation step proceeding all but quantitatively in the presence of the Generation I Grubbs catalyst. Likewise, molecular trefoil knots, which could only be isolated in small amounts (50 mg) in the late 80s can nowadays be obtained on the gram scale, thanks to the much better synthetic protocols we have at our disposal today. In collaboration with the von Zelewsky group at the University of Fribourg, ring closing metathesis has been employed extremely successfully in the synthesis of a chiral molecular trefoil knot.32 It has also been possible33 to resolve such a knot—which is unconditionally chiral because a trefoil knot is topologically nontrivial—and even have its absolute configuration determined by X-ray crystallographic means. The chiroptical properties of the resolved enantiomeric molecular trefoil knots are very pronounced! For more on molecular knots, the reader is referred to a couple of splendid reviews34,35 from the Strasbourg group.
When it comes to making MIMs quantitatively, there is nothing quite like thermodynamic control provided the building blocks and the reaction conditions are chosen with kid gloves. By teaming up with the Fujita group at the University of Tokyo, Sauvage has been able to make [2]catenanes36,37 and a doubly interlocked [2]catenane,38 quantitatively in all cases. By using palladium(II) (Fujita chemistry) in conjunction with copper(I) (Sauvage chemistry), catenanes can now be obtained routinely in 100% yields from these two metals and organic fragments as long as the coordination chemistry is exquisitely well planned—which was, of course, the case when the minds of Sauvage and Fujita combined to address the challenge. These successes apart, it should be added that the Sauvage mind has never shown signs of closure to new ideas and opportunities. So despite his remarkable feats in catenane chemistry using Fujita-style chemistry employing thermodynamics to control reactions according to his needs, he was not averse to using a kinetically controlled reaction par excellence—namely, the copper(I) catalysed 1,3-dipolar cycloaddition reaction between azides and alkynes (click chemistry) established and popularized by Barry Sharpless—to effect the mild and efficient stoppering of copper(I) complexed pseudorotaxanes for the preparation of their related rotaxanes.39,40
With international recognition beginning to blossom for the master of chemical topology (Fig. 13) at the outset of the 90s, Sauvage and his team designed and synthesised a series of multi-component second-row transition-metal complexes containing, for example, ruthenium(II) and osmium(II) that are able to undergo long-range charge separation under the action of a light signal. In collaboration with a group of Italian photochemists and photophysicists, including Balzani at the University of Bologna, Flamigni and Barigelletti at the CNR Institute in Bologna for Photochemistry and High Energy Radiation, and Scandola at the University of Ferrara, the Strasbourg team investigated many such complexes containing a terpyridine ligand in the central core of the molecule and second- and third-row transition metals such as iridium(III). This highly cited seminal work41–44 is considered nowadays as particularly significant and important by the inorganic chemistry community. It demonstrates that a strict geometrical control over the spatial arrangements of the various photo- and electroactive components of transition metal-based models of the photosynthetic reaction centre found in nature is possible and can lead to well-defined photochemical and photophysical properties, culminating in very long-lived charge-separated states by analogy with the natural photosynthetic reaction centre. The research has been the subject of three major reviews by the Strasbourg team (Fig. 14), firstly in Chemical Reviews45 in 1994, in Chemical Society Reviews46 in 2000, and last year in Accounts of Chemical Research.47 The 1994 Chemical Reviews is Sauvage’s most highly cited article in the chemical literature having chalked up over 800 citations at the time of writing this profile. More international recognition came Jean-Pierre’s way in 1994 with the award of the prestigious Prelog Medal (Fig. 15) by the Eidgenössische Technische Hochschule Zürich.
 |
| | Fig. 13 International recognition for the master of chemical topology in 1991. Top Left: Reed Izatt announcing the award of the first International Izatt-Christensen Award in Macrocyclic Chemistry at the 16th International Symposium on Macrocyclic Chemistry (1–6 September) held at the University of Sheffield. Top Right: Jean-Pierre receives the award from Reed Izatt. Bottom Left: Jean-Pierre with Norma and Fraser Stoddart. Bottom Right: Jean-Pierre with Fraser Stoddart and Stéphane Guillerez, a graduate student with the former in Strasbourg and a postdoctoral scholar with the latter in Sheffield. Subsequently, the Izatt-Christensen Prize has been awarded annually to the following scientists—1992 Eiichi Kimura, Hiroshima University, Hiroshima Japan; 1993 Fraser Stoddart, University of Sheffield, United Kingdom; 1994 Daryle Busch, University of Kansas, Lawrence, Kansas; 1995 David Reinhoudt, University of Twente, Enschede, The Netherlands; 1996 George Gokel, Washington University School of Medicine, St. Louis, Missouri; 1997 Alan Sargeson, Australian National University, Canberra, Australia; 1998 Seiji Shinkai, Kyushu University, Fukuoka, Japan; 1999 Fritz Vögtle, University of Bonn, Bonn, Germany; 2000 Jerry Atwood, University of Missouri, Columbia, Missouri; 2001 Jonathan Sessler, University of Texas, Austin, Texas; 2002 David Gutsche, Texas Christian University, Fort Worth, Texas; 2003 Jeremy Sanders, University of Cambridge, Cambridge, United Kingdom; 2004 Makoto Fujita, University of Tokyo, Tokyo, Japan; 2005 Kenneth Raymond, University of California, Berkeley, California; 2006 Roeland Nolte, University of Nijmegen, Nijmegen, The Netherlands; 2007 David Leigh, University of Edinburgh, Edinburgh, United Kingdom; 2008 Akira Harada, Osaka University, Osaka, Japan; 2009 Omar Yaghi, University of California, Los Angeles, California. | |
 |
| | Fig. 14 A part of Team Sauvage in 2001. Standing—Christiane Dietrich-Buchecker, Benoît Colasson, Christine Hamann, Jean-Marc Kern, Valérie Heitz, Jean-Claude Chambron, Kimoon Kim. Sitting—Louise Mechin, Isabelle Dixon, Emma Schofield and Jean-Pierre Sauvage. Thanks to Isabelle Dixon and Valérie Heitz for providing this photograph. | |
 |
| | Fig. 15 The Master with the maestro in Zürich in 1994. Top—Jean-Pierre Sauvage receives the Prelog Medal from Vladimir Prelog himself. Bottom—The two of them sitting side-by-side. | |
Inevitably the Sauvage fixation with photochemistry and photophysics has spilled over not only into his love—namely, that aspect of chemical topology that is tied up with knots, rotaxanes, and catenanes, or MIMs for short—but also into another Sauvage preserve, that of porphyrin chemistry. Multiporphyrin molecules, including oblique bis-porphyrins,48,49 porphyrin-stoppered rotaxanes,50–56 and other transition metal-assembled multicomponent systems,57,58 have been designed, made and investigated as models for the photosynthetic reaction center. The original13 copper(I)-templated synthesis of catenanes has been generalized (Fig. 16) and has afforded a large collection and variety of porphyrin-containing catenanes57,58 and rotaxanes50–56 which have been the subject of a 1998 concept article in Chemistry—A European Journal59 and reviews in Structure and Bonding60 in 2006, as well as in Chemical Society Reviews61,62 in 1999 and earlier this year (2009). In association with photophysicists—in particular with Harriman and co-workers at the University of Texas at Austin and with Flamigni at the CNR in Bologna—Sauvage and his group in Strasbourg have shown that ultrafast electron transfer takes place by means of a super-exchange mechanism between a photoexcited electron donor and an acceptor in a manner which is reminiscent of the natural photosynthetic reaction centre. They have also found that long-range energy transfer processes are operative in transition metal-assembled multiporphyrins. The importance of porphyrinic catenanes and rotaxanes lies in the fact that they contain mechanically linked chromophores with no “real” chemical bond between the electron donor and the acceptor. A particular example (Fig. 17) is that of a rotaxane whose ring incorporates a gold porphyrin (electron acceptor) and whose threaded dumbbell bears two zinc porphyrins (electron donors) at the ends. Photoactive catenanes and rotaxanes combine the flexibility of topologically non-trivial molecules with an ability to undergo complete rearrangement (Fig. 18) rather easily during the electron and energy transfer of porphyrins and metallo-porphyrins. As such, they offer the opportunity to induce large amplitude motions in MIMs activated with light.
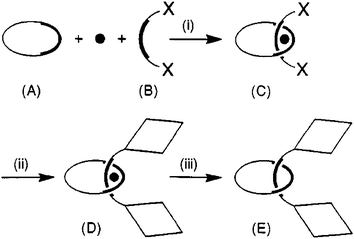 |
| | Fig. 16 Schematic representation of the principle of transition-metal macrocyclic chelate (A), a metal cation (black moon) and an open chelate (B). The latter bears at its extremities functions X which can be employed for anchoring or constructing stoppers (represented as diamonds). (i) The threading step affords the pseudorotaxane (C). (ii) The stoppering step affords the metal-complexed rotaxane (D). (iii) Removal of the metal template releases the free rotaxane (E). | |
![(a) Through-space electron transfer (represented by an arrow) in a [2]rotaxane from a zinc porphyrin stopper in its singlet excited state (*) to the gold porphyrin covalently attached to the ring in the rotaxane. (b) The same rotaxane complexing a copper(i) cation.](/image/article/2009/CS/b819336n/b819336n-f17.gif) |
| | Fig. 17 (a) Through-space electron transfer (represented by an arrow) in a [2]rotaxane from a zinc porphyrin stopper in its singlet excited state (*) to the gold porphyrin covalently attached to the ring in the rotaxane. (b) The same rotaxane complexing a copper(I) cation. | |
 |
| | Fig. 18 Control of the mutual arrangement (relative geometry) between the gold porphyrin (hatched diamond) and the zinc porphyrins (empty diamonds) by complexation/decomplexation of a metal centre (black circle) within/from the central coordination site. | |
One of the most challenging fields of research in chemistry today is that of triggering and controlling motion within molecules, particularly MIMs. This internal movement of components in MIMs can be actuated chemically, electrochemically or photochemically. Bi- and multi-stable [2]catenanes60–62 and [2]rotaxanes,63–67 which can be made to undergo relative motions internally have been elaborated in an incremental manner by the Sauvage group from as early as 1994. In a [2]catenane, one ring can be induced60–62 to circumrotate (glide) with respect to the other ring under the action of an electrochemical signal—and the motion is reversible because of the redox nature of the chemistry. In metallo-[2]rotaxanes, dumbbells have been made to pirouette67 with respect to the ring, once again by employing redox chemistry. An important aspect of harnessing the movements that occur internally in MIMs in a device setting is to be able to locate the molecules on surfaces without impairing their solution properties. It is therefore encouraging to know that the Sauvage group has managed68,69 to locate switchable molecules, based on the Cu(I)/Cu(II) redox couple, at gold surfaces.
In the realm of potential molecular machinery, the work of the Sauvage group on chemically and electrochemically switchable rotaxane dimers ([2]daisy chains) is of particular note and significance. The manner in which the two linear portions of these MIMs can glide along beside each other in a controlled manner is surely a property that can be expressed on a macroscopic level: artificial muscles come to mind, as the Sauvage group remind us from time to time in their delightful discussions70–74 of their research.
When one considers the level of world-wide activity today in the field of MIMs, there is no doubt, with the aid of hindsight, that Jean-Pierre Sauvage began a revolution in the chemical sciences a quarter of a century ago that could well spill over into the arenas of information technology and healthcare, to mention only two possible beneficiaries, in relatively short measure.
In common with many great scientists, Sauvage identified a big problem to which he brought a solution by a sequence of well-reasoned approaches made credible by a constant series of elegant demonstrations from his research laboratory. In the early 80s, he ventured into a chemical desert that had been all but forsaken by his fellow chemists and created a chemical oasis in next-to-no time. He nurtured this oasis through its early days with so much loving care and attention to detail that, before he knew it, he was being joined left and right and centre by chemical disciples from near and far. What is perhaps most remarkable of all is that the master of chemical topology orchestrated the creation of this Chemical Garden of Eden while remaining very much the gentleman of scientific etiquette—and very much a Frenchman—whom all respect for his style and charm, just as much as they do for his chemistry and his creativity therein.
Aside from the Izatt-Christensen Award (1991) and the Prelog Medal (1994), Jean-Pierre has had many other honours and prizes bestowed upon him. He was elected a Correspondent of the French Academy of Sciences on 26 March 1990 and a Member on 24 November 1997. Fig. 19 captures some of the precious moments relating to his becoming a Member of the French Academy of Sciences.
 |
| | Fig. 19 The French Academy of Sciences opens its doors fully in 1998 to the master of chemical topology. Bottom Left: Jean-Pierre uncorking a bottle of champagne at the celebration marking the announcement of his election to Membership. Carmen Sauvage can be spotted in the background. Top Left: Jean-Pierre standing outside the Institut de France in Paris with his son Julien. Top Right: Jean-Pierre in conversation with Academy Vice-President Guy Ourisson. Bottom Right: Jean-Pierre receives the medal of the Institut de France from Guy Ourisson. | |
I would like to thank David Amabilino for orchestrating the acquisition of most of the photographs displayed in this profile. According to David, they were variously supplied by Carmen Sauvage and by Isabelle Dixon and Valérie Heitz, and also by Yoshio Furusho. Some of this piece is based on a document that Jean-Pierre gave me access to for other reasons. I hope he will not hold it against me that I plagiarized some phrases and sentences from it. In my defence, I would claim to be following some advice given to me in 1997 by Donald Cram when I arrived at UCLA. He told me that I should always supply material to anyone who cares to write about my research for the simple reason that I should know more about my own research than anyone else! I am sure that Jean-Pierre knows a lot more about his research than I do, although I would claim to be much better informed—and hugely impressed—after writing this profile. I thank Robert Eagling, the editor of Chemical Society Reviews, for going right up to the wire with me. We both hope that the result will leave Jean-Pierre and his many students, research associates, colleagues, friends, and family happy at the outcome. I look forward to writing the next installment of science Sauvage-style.
References
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS.
- I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89, 5723–5724 CrossRef CAS.
- I. T. Harrison, J. Chem. Soc., Chem. Commun., 1972, 231–232 RSC.
- I. T. Harrison, J. Chem. Soc., Perkin Trans. 1, 1974, 301–304 RSC.
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS.
- D. M. Walba, Tetrahedron, 1985, 41, 3161–3212 CrossRef CAS.
- G. Schill, Catenanes, Rotaxanes and Knots, Academic Press, New York, NY, 1971 Search PubMed.
- W. Vetter and G. Schill, Tetrahedron, 1967, 23, 3079–3093 CrossRef CAS.
- G. Schill, N. Schweickert, H. Fritz and W. Vetter, Chem. Ber., 1988, 121, 961–970 CAS.
- G. Schill and C. Zürcher, Chem. Ber., 1977, 110, 2046–2066 CrossRef CAS; G. Schill and C. Zürcher, Chem. Ber., 1977, 110, 3964–3979 CrossRef.
- G. Schill, K. Rissler, H. Fritz and W. Vetter, Angew. Chem., Int. Ed. Engl., 1983, 22, 889–891 CrossRef.
- G. Schill, G. Doerjer, E. Logemann and H. Fritz, Chem. Ber., 1979, 112, 3606–3615.
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef CAS.
- M. Cesario, C. O. Dietrich-Buchecker, J. Guilhem, C. Pascard and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1985, 244–247 RSC.
- A.-M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1985, 107, 3205–3209 CrossRef.
- A.-M. Albrecht-Gary, C. O. Dietrich-Buchecker, Z. Saad, J.-P. Sauvage and J. Weiss, J. Chem. Soc., Chem. Commun., 1986, 1325–1327 RSC.
- B. Dietrich, J.-M. Lehn and J.-P. Sauvage, Tetrahedron Lett., 1969, 2885–2888 CrossRef CAS; B. Dietrich, J.-M. Lehn and J.-P. Sauvage, Tetrahedron Lett., 1969, 2889–2892 CrossRef.
- B. Dietrich, J.-M. Lehn, J.-P. Sauvage and J. Blanzat, Tetrahedron, 1973, 29, 1629–1645 CrossRef CAS.
- B. Dietrich, J.-M. Lehn and J.-P. Sauvage, Tetrahedron, 1973, 29, 1647–1658 CrossRef CAS.
- J.-M. Lehn, J.-P. Sauvage and R. Ziessel, Nouv. J. Chim., 1980, 4, 623–627 Search PubMed.
- J.-M. Lehn, J.-P. Sauvage and R. Ziessel, Nouv. J. Chim., 1977, 1, 449–451 Search PubMed.
- J.-M. Lehn, J.-P. Sauvage and R. Ziessel, Nouv. J. Chim., 1979, 3, 423–427 Search PubMed.
- J.-P. Sauvage, Nouv. J. Chim., 1985, 9, 299–310 Search PubMed.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Chem. Rev., 1987, 87, 795–810 CrossRef CAS.
- D. H. Busch and N. A. Stephenson, Coord. Chem. Rev., 1990, 100, 119–154 CrossRef CAS.
- F. Bitsch, C. O. Dietrich-Buchecker, A.-K. Khemiss, J.-P. Sauvage and A. van Dorsselaer, J. Am. Chem. Soc., 1991, 113, 4023–4025 CrossRef CAS.
- C. O. Dietrich-Buchecker, J. Guilhem, A.-K. Khemiss, J.-P. Kintzinger, C. Pascard and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1987, 26, 661–664 CrossRef.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef.
- B. Mohr, M. Weck, J.-P. Sauvage and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 1308–1310 CrossRef CAS.
- M. Weck, B. Mohr, J.-P. Sauvage and R. H. Grubbs, J. Org. Chem., 1999, 64, 5463–5471 CrossRef.
- L.-E. Perret-Aebi, A. von Zelewsky, C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2004, 43, 4482–4485 CrossRef CAS.
- G. Rapenne, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1996, 118, 10932–10933 CrossRef.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, New J. Chem., 1992, 16, 277–285 Search PubMed.
- C. O. Dietrich-Buchecker, B. X. Colasson and J.-P. Sauvage, Top. Curr. Chem., 2005, 249, 261–283 CAS.
- C. O. Dietrich-Buchecker, N. Geum, A. Hori, M. Fujita, S. Sakamoto, K. Yamaguchi and J.-P. Sauvage, Chem. Commun., 2001, 1182–1183 RSC.
- C. O. Dietrich-Buchecker, B. Colasson, M. Fujita, A. Hri, N. Geum, S. Sakamoto, K. Yamaguchi and J.-P. Sauvage, J. Am. Chem. Soc., 2003, 125, 5717–5725 CrossRef.
- F. Ibukuro, M. Fujita, K. Yamaguchi and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 11014–11015 CrossRef CAS.
- P. Mobian, J.-P. Collin and J.-P. Sauvage, Tetrahedron Lett., 2006, 47, 4907–4909 CrossRef CAS.
- S. Durot, P. Mobian, J.-P. Collin and J.-P. Sauvage, Tetrahedron, 2008, 64, 8496–8503 CrossRef CAS.
- J.-P. Collin, S. Guillerez, J.-P. Sauvage, F. Barigelletti, L. DeCola, L. Flamigni and V. Balzani, Inorg. Chem., 1991, 30, 4230–4238 CrossRef CAS.
- F. Barigelletti, L. Flamigni, V. Balzani, J.-P. Collin, J.-P. Sauvage, A. Sour, E. C. Constable and A. M. W. Cargill Thompson, J. Am. Chem. Soc., 1994, 116, 7692–7699 CrossRef CAS.
- F. Barigelleti, L. Flamigni, M. Guardigli, A. Juris, M. Beley, S. Chodorowski-Kimmes, J.-P. Collin and J.-P. Sauvage, Inorg. Chem., 1996, 35, 136–142 CrossRef CAS.
- J.-P. Collin, I. M. Dixon, J.-P. Sauvage, J. A. G. Williams, F. Barigelletti and L. Flamigni, J. Am. Chem. Soc., 1999, 122, 5009–5016 CrossRef.
- J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. DeCola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- I. M. Dixon, J.-P. Collin, J.-P. Sauvage, L. Flamigni, S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385–391 RSC.
- L. Flamigni, J.-P. Collin and J.-P. Sauvage, Acc. Chem. Res., 2008, 41, 857–871 CrossRef CAS.
- S. Chardon-Noblat, J.-P. Sauvage and P. Mathis, Angew. Chem., Int. Ed. Engl., 1989, 28, 593–595 CrossRef.
- A.-M. Brun, A. Harriman, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8657–8663 CrossRef CAS.
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1992, 1131–1133 RSC.
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1993, 115, 12378–12384 CrossRef CAS.
- D. B. Amabilino, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1996, 118, 3285–3286 CrossRef CAS.
- N. Solladie, J.-C. Chambron, C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1996, 35, 906–909 CrossRef CAS.
- N. Solladie, J.-C. Chambron and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 3684–3692 CrossRef CAS.
- L. Flamigni, N. Armaroli, F. Barigelletti, J.-C. Chambron, J.-P. Sauvage and N. Solladie, New J. Chem., 1999, 23, 1151–1158 RSC.
- M. Andersson, M. Linke, J.-C. Chambron, J. Davidsson, V. Heitz, J.-P. Sauvage and L. Hammarström, J. Am. Chem. Soc., 2000, 122, 3526–3527 CrossRef CAS.
- M. Linke, N. Fujita, J.-C. Chambron, V. Heitz and J.-P. Sauvage, New J. Chem., 2001, 25, 790–796 RSC.
- M. Beyler, V. Heitz and J.-P. Sauvage, Chem. Commun., 2008, 5396–5398 RSC.
- J.-C. Chambron and J.-P. Sauvage, Chem.–Eur. J., 1998, 4, 1362–1366 CrossRef CAS.
- L. Flamigni, V. Heitz and J.-P. Sauvage, Struct. Bonding, 2006, 121, 217–261 CAS.
- M.-J. Blanco, M. C. Jiménez, J.-C. Chambron, M. Linke and J.-P. Sauvage, Chem. Soc. Rev., 1999, 28, 293–305 RSC.
- J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422–442 RSC.
- A. Livoreil, C. O. Dietrich-Buchecker and Jean-Pierre Sauvage, J. Am. Chem. Soc., 1994, 116, 9399–9400 CrossRef CAS.
- D. J. Cárdenas, A. Livoreil and J.-P. Sauvage, J. Am. Chem. Soc., 1996, 118, 11980–11981 CrossRef CAS.
- A. Livoreil, J.-P. Sauvage, N. Armaroli, V. Balzani, L. Flamigni and B. Ventura, J. Am. Chem. Soc., 1997, 119, 12114–12124 CrossRef CAS.
- J. P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS.
- S. Bonnet, J.-P. Collin, M. Koizumi, P. Mobian and J.-P. Sauvage, Adv. Mater., 2006, 18, 1239–1250 CrossRef CAS.
- L. Raehm, C. Hamann, J.-M. Kern and J.-P. Sauvage, Org. Lett., 2000, 2, 1991–1994 CrossRef CAS.
- D. Payer, S. Rauschenbach, N. Malinowski, M. Konuma, C. Virojanadara, U. Starke, C. O. Dietrich-Buchecker, J.-P. Collin, J.-P. Sauvage, N. Lin and K. Kern, J. Am. Chem. Soc., 2007, 129, 15662–15667 CrossRef CAS.
- M. C. Jimenez-Molero, C. Dietrich-Buchecker and J.-P. Sauvage, Chem. Commun., 2003, 1613–1616 RSC.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS.
- B. X. Colasson, C. Dietrich-Buchecker, M. C. Jimenez-Molero and J.-P. Sauvage, J. Phys. Org. Chem., 2002, 15, 476–483 CrossRef CAS.
- C. Dietrich-Buchecker, M. C. Jimenez-Molero, V. Sartor and J.-P. Sauvage, Pure Appl. Chem., 2003, 75, 1383–1393 CrossRef CAS.
- E. Baranoff, F. Barigelletti, S. Bonnet, J.-P. Collin, L. Flamigni, P. Mobian and J.-P. Sauvage, Struct. Bonding, 2007, 123, 41–78 CAS.
Footnote |
| † Dedicated to Professor Jean-Pierre Sauvage on the occasion of his 65th birthday. |
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here.