Photosynthetic energy conversion: natural and artificial†
James
Barber
*
Division of Molecular Biosciences, Faculty of Natural Sciences, Imperial College London, London, UK SW7 2AZ
First published on 10th November 2008
Abstract
Photosystem II (PSII) is the water splitting enzyme of photosynthesis. Its appearance during evolution dramatically changed the chemical composition of our planet and set in motion an unprecedented explosion in biological activity. Powered by sunlight, PSII supplies biology with the ‘hydrogen’ needed to convert carbon dioxide into organic molecules. The questions now are can we continue to exploit this photosynthetic process through increased use of biomass as an energy source and, more importantly, can we address the energy/CO2 problem by developing new photochemical technologies which mimic the natural system? (Critical review, 82 references)
 James Barber | James Barber is the Ernst Chain Professor of Biochemistry at Imperial College London and is a Fellow of The Royal Society of London and Royal Society of Chemistry. He obtained his BSc in chemistry from the University of Wales in 1964 and MSc and PhD in biophysics from the University of East Anglia in 1965 and 1967, respectively. He has been awarded several medals and prizes for his contributions to understanding the molecular processes of photosynthesis with particular focus on Photosystem II, the water splitting enzyme which underpins the energy cycle of biology. |
Introduction
Photosynthetic organisms capture sunlight very efficiently and convert it into organic molecules. These molecules are the building blocks of all living organisms and without photosynthesis life on our planet would not have evolved in the way that we know. It is estimated that currently, photosynthesis produces more than 100 billion tons of dry biomass annually, which would be equivalent to a hundred times the weight of the total human population on Earth at the present time and equal to an average energy storage rate of about 100 TW. Oil, gas and coal are also derived from millions of years of photosynthetic activity. These fossil fuels provide us with most of the energy needed to power our technologies, heat our homes and produce the wide range of chemicals and materials that support everyday life. Ultimately the reserves of fossil fuels will dwindle and then what? Even before this, as a consequence of our ever growing use of oil, gas and coal, we are faced with the problem of increasing levels of carbon dioxide and other greenhouse gases in the atmosphere with implications for global climate change.The success of photosynthesis as an energy generating and storage system stems from the fact that the raw materials and power needed for the synthesis of biomass are available in almost unlimited amounts; sunlight, water and carbon dioxide. At the heart of the photosynthetic process, is the splitting of water by sunlight into oxygen and ‘hydrogen’. The oxygen is released into the atmosphere where it is available for us to breathe and to use for burning fuels to drive our technologies. The ‘hydrogen’ is not normally released into the atmosphere but instead is combined with carbon dioxide to make sugars and other organic molecules of various types. When we burn fuels (fossil, biomass and other biofuels) to release energy, we are simply combining the ‘hydrogen’ stored in these organic molecules with atmospheric oxygen, so completing a cycle started millions of years ago. Similarly, energy is also released from the organic molecules which constitute our food, when they are metabolised within our bodies by the process of respiration. Thus in the biological world, photosynthesis brings about the splitting of water into oxygen and ‘hydrogen’ while respiration is the reverse, combining oxygen and hydrogen in a carefully controlled and highly efficient way so as to create metabolic energy. Therefore, from an energetic view, the synthesis of organic molecules represents a way of storing hydrogen and therefore storing solar energy in the form of chemical bonds (Fig. 1).
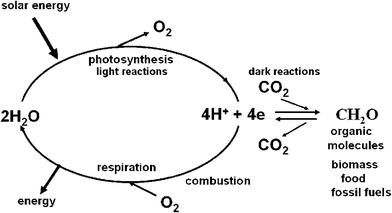 | ||
| Fig. 1 A diagrammatic representation of energy flow in biology. The light reactions of photosynthesis (light absorption, charge separation, water splitting, electron/proton transfer) provides the reducing equivalents in the form of energised electrons (e) and protons (H+) to convert carbon dioxide (CO2) to sugars and other organic molecules which make up living organisms (biomass) including those that provide humankind with food. The same photosynthetic reactions gave rise to fossil fuels formed millions of years ago. The burning of these organic molecules either by respiration (controlled oxidation within our bodies) or by combustion of biomass and fossil fuels to power our technologies, is the reverse to photosynthesis, releasing CO2 and reuniting the stored ‘hydrogen’ with oxygen to form water. In so doing energy is released, energy that originated from sunlight. | ||
In this article I will briefly emphasise the enormity of the energy/carbon dioxide problem that we face within the coming decades and discuss the contributions that could be made by fuels derived directly from photosynthesis (biofuels) and from developing new technologies based on the successful principles of photosynthesis. I will particularly emphasise the possibility of exploiting the vast amounts of solar energy available to split water to produce dioxygen and the reducing equivalents required to generate fuels such as hydrogen gas, alcohols and methane.
Global energy consumption and the enormity of the problem
At the present time, the rate of global energy consumption is approaching about 14 TW,1 with the USA and the extended EU each using about 25% of this. In the future this global value will rise due to industrialization in underdeveloped and developing countries coupled with increasing world population. Based on current projections the global annual energy consumption rate will reach 20 TW by 2030, doubled by 2050 and tripled by the end of the century.2–4 About 85% of the total global energy consumed at present comes from burning fossil fuels with the proportion approaching 90% for developed countries. Oil, gas and coal contribute approximately equally to this demand. The remaining sources of energy are hydroelectric, nuclear, biomass and renewables, such as solar, wind, tide and wave. At present, the use of biomass is a major player and is mainly localised in under-developed regions, such as Africa and India, where wood and other organic matter is used as fuel. Much of this is not strictly renewable since there is no planned regeneration and the trend is towards more use of fossil fuels.The low level of contribution of non-fossil energy sources to present day global energy demand reflects the readily available resources of oil, gas and coal. Even when oil reserves become limiting, there will remain large reservoirs of gas and, particularly, coal to exploit.5 Therefore in the global arena, the problem for the immediate future is not a limitation of fossil fuel reserves but the consequences of its combustion. If the total fossil fuel reserve is burnt then the carbon dioxide level in the atmosphere and oceans would rise to values equivalent to those that existed on our planet long before humankind evolved.6 Despite this concern, it is certain that fossil fuels will continue to be a major source of energy for humankind for some years to come but it is vital that they should be used in such a way as to minimise carbon dioxide release into the atmosphere. Technologies for sequestration of carbon dioxide must be developed.7 Hand in hand with this there will almost certainly be an improvement in the efficiency of energy use and supplementation whenever possible with non-fossil fuel sources. Against this background it seems to me that we must also strive to develop new technologies based on principles which have yet to be revealed from basic studies and in particular those that focus on utilizing the enormous amount of energy available to us as solar radiation.8 The sun provides solar energy to our planet on an annual basis at a rate of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TW. Therefore the energy from one hour of sunlight is equivalent to all the energy humankind currently uses in a year. We do have existing technologies to capture sunlight and produce electricity and the efficiency and robustness of these photovoltaic systems is improving daily.9,10 Compared with the present day price of fossil fuels photovoltaic systems represent an expensive way to generate electricity because of the high cost of their construction. In time these costs will decrease relative to the cost of fossil fuel. Moreover a blending of the principles of photovoltaic systems, especially those using cheap organic or inorganic materials, with concepts derived from natural photosynthetic systems may ultimately provide a long term solution.4,8 In considering this long term solution, let us take a look at the efficiency of the natural photosynthetic process.
000 TW. Therefore the energy from one hour of sunlight is equivalent to all the energy humankind currently uses in a year. We do have existing technologies to capture sunlight and produce electricity and the efficiency and robustness of these photovoltaic systems is improving daily.9,10 Compared with the present day price of fossil fuels photovoltaic systems represent an expensive way to generate electricity because of the high cost of their construction. In time these costs will decrease relative to the cost of fossil fuel. Moreover a blending of the principles of photovoltaic systems, especially those using cheap organic or inorganic materials, with concepts derived from natural photosynthetic systems may ultimately provide a long term solution.4,8 In considering this long term solution, let us take a look at the efficiency of the natural photosynthetic process.
Efficiency of photosynthesis
As emphasised in Fig. 1, photosynthesis is a process which converts light energy into the organic molecules of biomass. To estimate the efficiency of this process two main factors must be appreciated.(i) Although photosynthetic organisms can efficiently trap light energy at all wavelengths of visible solar radiation, the energy used for splitting water and reducing carbon dioxide is only equivalent to the red region of the spectrum. Higher energy photons are degraded to heat by internal conversion within the light harvesting pigments to the energy level of ‘red’ photons of about 1.8 eV.
(ii) For every electron/proton extracted from water and used to reduce CO2 the energy of two ‘red’ photons is required. This is accomplished by linking together, in series, two different photosystems, photosystem II (PSII), which uses light to power the extraction of electrons/protons from water, and photosystem I (PSI) which uses light to provide additional energy to the “ PSII-energised” electrons/protons so as to drive the CO2 fixation process (see Fig. 2). Therefore photosynthesis uses the energy of at least 8 ‘red’ photons per O2 molecule released or CO2 molecules fixed. A typical product of carbon fixation is glucose (C6H12O6) whose energy content is 672 kcal mol−1 (2813 kJ mol−1) if burnt in a calorimeter. To make a glucose molecule, the energy of 48 ‘red’ photons is required and assuming a wavelength of 680 nm, corresponding to 42 kcal per quantum mole (176 kJ mol−1), gives the efficiency of conversion at about 30%. Although this is an impressive number, in reality the overall conversion of solar energy to glucose and the very large variety of other organic molecules which constitute biomass is much lower. Energy is lost in degrading shorter wavelength light (e.g. blue light)) to the energy of ‘red’ photons, by saturation processes and more significantly, in driving the enormous number of reactions which occur in photosynthetic organisms to maintain their organisation, metabolism, reproduction and survival.
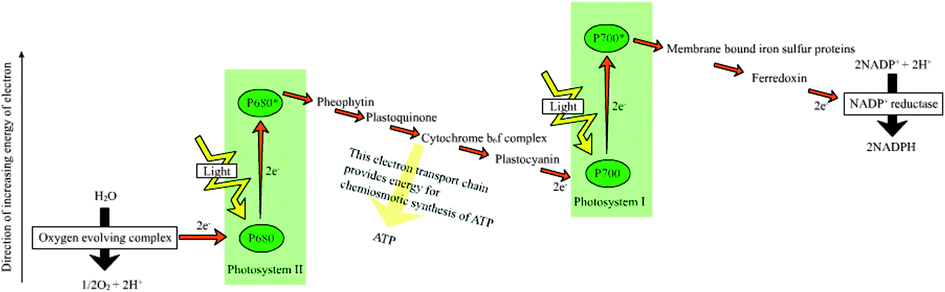 | ||
| Fig. 2 A simplified Z-scheme of the light reactions of photosynthesis taken from http://en.wikipedia.org/wiki/Photosynthesis. For every electron extracted from water and transferred to CO2, the energy of two photons of light is required. One is absorbed by Photosystem II (PSII) which generates a strong oxidising species (P680+), able to drive the water splitting reaction and a reduction of pheophytin (Pheo) and then plastoquinol (Q) to plastoquinol (QH2). The other, Photosystem I (PSI), generates a strong reducing species, NADPH which donates reducing equivalents to CO2 to produce sugars and other organic molecules, and a weak oxidant P700+. Electron and proton flow from QH2 to P700+ is aided by the cytochrome b6f (Cyt b6f) complex and plastocyanin (PC) and results in the release of energy to convert ADP to ATP. The ATP produced is required, along with NADPH, to convert CO2 to sugars. Since the production of O2 requires the splitting of two water molecules, the overall process involves the removal of two electrons per water (as shown) and therefore four photons per PSII and PSI reaction centre. The reduction of oxidised nicotinamide adenine dinucleotide phosphate (NADP+) by PS1 is facilitated by membrane bound iron sulfur proteins (Fx, FA and FB) and soluble ferredoxin (FD). | ||
There are many ways to define and calculate the overall photosynthetic efficiency but the approach adopted by Thorndike11 is attractive since it engulfs the whole range of definitions. He considered free energy stored per photon F:
| F = ηTηRηSηLηOhνO |
In fact, an efficiency of 4.5% is rarely achieved. Only in exceptional cases will dry matter yield exceed 1 or 2%, such as with the intense growing of sugar cane in tropical climates or with optimised culturing of algae. Normally agricultural crops produce yields of biomass at efficiencies less than 1%, even when pampered with ample supplies of fertiliser and water. Environmental conditions, degree of light interception, nutrient and water supply are key factors in reducing the efficiency below the maximum while specific genetic characteristics of particular plant species also dictates growth rates and maximum yields of biomass.
On a global basis the efficiency of photosynthesis is significantly lower than for agricultural and energy crops or algal cultures growing under optimal conditions because of seasonal changes and the existence of large portions of land and oceans on our planet which do not sustain significant levels of photosynthetic activity.13 Thus the 100 TW for the rate of energy storage averaged over a year by photosynthesis represents just 0.1% conversion given that solar energy arrives on our planet at a rate of 100000 TW over the same time period. This energy is stored mainly in wood and fibres of terrestrial trees and plants. A similar amount of photosynthetic activity occurs in the oceans but the fixed carbon is rapidly recycled into the food chain.16 Therefore, an approximate efficiency of global photosynthesis is 0.2% but with only half being stored in biomass. Of course, it was terrestrial biomass that was the major source of energy for humankind prior to the exploitation of fossil fuels. It is not surprising therefore that there is now a growing interest in returning to the use of biomass and biofuels as an alternative to fossil fuels since their production and use is carbon dioxide neutral.
Biomass
Wood and other forms of biomass can be used to generate heat, electricity, biogas (mainly methane and carbon dioxide), syngas (hydrogen and carbon monoxide) and other biofuels (mainly bioethanol and biodiesel). Biomass is the end product of photosynthesis and, as stated above, represents energy conversion efficiencies in the region of 0.1 to 1.0% depending on its origin. Many organisations consider “biomass power” as an increasingly attractive option to replace fossil fuels, including the European Union, U.S. Department of Energy (USDOE) and many national government departments and agencies, major companies and utilities in countries like Brazil, Finland, Sweden, UK and elsewhere. Currently the global use of biomass is equivalent to about 1.4 TW. In the US, biomass surpasses hydroelectric power as a source of renewable energy in providing over 3% of the country’s energy consumption, corresponding to about 0.1 TW. However, a recent joint report from the US Departments of Energy (USDOE) and Agriculture (USDA)17 has concluded that biomass and biofuels could provide the US with about 30% of its present total energy needs. This would be achieved by utilising non-food producing agricultural land and maximising on forestry outlets to generate 1.3 billion tons of dry biomass annually, corresponding to about 1 TW. This optimistic projection also relies on plant breeding and genetic engineering strategies to produce new cultivars for high yields of biomass requiring minimum input of fertilizers, water and pesticides. Moreover, improved technologies will be required to maximise on the use of biomass including those for producing liquid biofuels to replace gasoline, such as second generation bioethanol. Also such calculations must take into consideration the energy costs of maintaining “energy farms”, harvesting the biomass, transporting to a central location and conversion into a usable form.The major biomass-derived fuel is ethanol produced from the fermentation of sugars or starches.18 Two countries, Brazil and US are major producers of bioethanol, with a total combined production being equivalent to about 0.02 TW. Brazil has invested significantly in producing ethanol from sugar cane which is used as a substitute or as an additive to gasoline.19–21 Similarly the US has recently invested heavily in the production of bioethanol from corn starch. Although the production and use of ethanol, as well as other biofuels, has been heavily subsidised, improved technologies and the rising cost of petroleum means that these types of fuels are now competing favourably with gasoline and, with the introduction of new technologies to exploit lignocellulose as a source of sugar for fermentation, could become major players.22,23
For millennia, biomass was the only primary energy source available to humankind. For the last two centuries, however, energy demand has outpaced biomass production. Although biomass and its products such as bioethanol, can still contribute to this demand in different ways and to different extents depending on climate and available landmass, it is hard to see how it could match the present level of global fossil fuel consumption or to cope with increasing demands for energy in the future. Using the best known plants for energy production to achieve 20 TW corresponds to almost three times all cultivatable land currently used for agriculture globally. To reduce this to a reasonable level so as not to seriously compete with food production would require the biomass crops to have solar energy conversion efficiencies close to the theoretical maximum of about 4.5%. Nevertheless, if a new generation of energy crops could be produced by plant breeding or genetic engineering which are environmentally robust requiring minimal inputs of water and fertilizer and can convert solar energy at efficiencies well above 1%, biomass could make a significant contribution to global energy demand. New and improved technologies to convert it into useable fuel will also have to emerge if this source of energy is to become available at a significant level. A particular challenge is to derive new and efficient methods to release sugars from the lignocellulose which constitutes the majority of the biomass and to improve fermentation technologies to produce bioethanol.22,23
Finally we should remind ourselves that plant biomass is not just a store of energy but is also a source of complex chemical molecules that constitute our food and which provides us with a wide range of valuable materials such as timber, linen, cotton, oils, rubbers, sugars, starches etc. The increased use of ‘designer’ plants to produce high value chemicals for the chemical and pharmaceutical industries should not be underestimated.24
Although it may be possible to engineer plants and other types of photosynthetic organisms (algae) as energy converting ‘machines’ and ‘chemical factories’ the overall efficiency of solar energy conversion will rarely exceed 1% and will usually be much less. However, the efficiency of the early photochemical and chemical reactions of photosynthesis, which are not directly involved in biomass production, is significantly higher. Because of this there are alternative and complementary approaches for utilizing solar energy. For example, it may be possible to develop a highly efficient, artificial, molecular-based solar energy converting technology which exploits the principles of the “front-end” of natural photosynthesis. Indeed, our knowledge of the natural process is sufficient to provide a blue-print for the design and assembly of such devices.
Photosynthesis and the water splitting reaction
As emphasised above, photosynthesis has produced most of the energy that fuels human society and sustains life on our planet. The process is underpinned by the light driven water splitting reaction that occurs in PSII of plants, algae and cyanobacteria (Fig. 2). Solar energy is absorbed by chlorophyll and other pigments and is transferred efficiently to the PSII reaction centre where charge separation takes place. This initial conversion of light energy into electrochemical potential occurs in the reaction centre of PSII with a maximum thermodynamic efficiency of about 70% and generates a radical pair state P680˙+Pheo˙− where P680 is a chlorophyll a molecule and Pheo is a pheophytin a molecule (chlorophyll molecule without a Mg ion ligated into its tetrapyrrole head group). The redox potential of P680˙+ is very oxidising, estimated to be more than +1 V while that of Pheo˙− is about −0.6 V. The latter is sufficiently negative that, in principle, it could drive the formation of hydrogen. Instead the reducing equivalent is passed along an electron transport chain to PSI (see Fig. 2), where it is excited by the energy of a second ‘red’ photon absorbed by a chlorophyll molecule, known as P700, to give a redox potential of −1 V or more. In this way sufficient energy is accumulated to drive the fixation of carbon dioxide, which not only requires the generation of the reduced ‘hydrogen carrier’, nicotinamide adenine dinucleotide phosphate (NADPH) but also the energy rich molecule adenosine triphosphate (ATP) formed by the release of some energy during electron transfer from PSII to PSI (in the form of an electrochemical potential gradient of protons) (Fig. 2 and 3). The P680˙+ species generated in PSII drives the splitting of water at the oxygen evolving centre (OEC). It does so by extracting electrons from a catalytic centre composed of a cluster of four manganese (Mn) ions and a calcium ion (Ca2+). The splitting of water into dioxygen and reducing equivalents is a four electron process and therefore PSII must absorb four photons (4hν) to drive this reaction.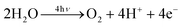
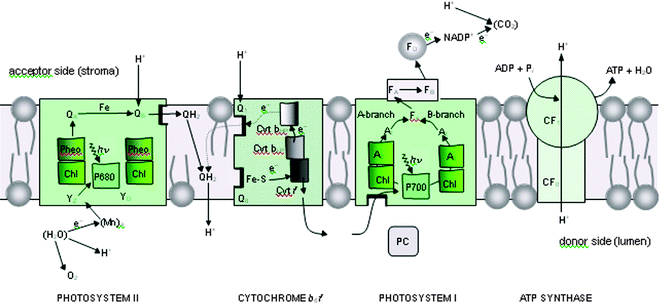 | ||
| Fig. 3 Schematic diagram of the electron–proton transport chain of oxygenic photosynthesis in the thylakoid membrane, showing how photosystem I (PSI) and photosystem II (PSII) work together to use absorbed light to oxidise water and reduce NADP+, in an alternative representation to the Z-scheme shown in Fig. 2. The diagram also shows how the proton gradient generated by the vectorial flow of electrons across the membrane is used to convert ADP to ATP at the ATP synthase complex (CF0–CF1). In both PSI and PSII, the redox-active cofactors are arranged around a pseudo-two-fold axis. In PSII, primary charge separation and subsequent electron flow occurs along one branch of the reaction centre. However, in the case of PSI, it is likely that electron flow occurs up both branches as shown. Electron flow through the cytochrome b6f complex also involves a cyclic process known as the Q cycle. YZ = tyrosine; P680 = primary electron donor of PSII composed of chlorophyll (Chl); Pheo = pheophytin; QA and QB = plastoquinone; Cyt b6f = cytochrome b6f complex, consisting of an Fe–S Rieske centre, cytochrome f (Cyt f), cytochrome b low- and high-potential forms (Cyt bLP and Cyt bHP), plastoquinone binding sites, Q1 and Q0; PC = plastocyanin; P700 = primary electron Chl donor of PSI; A0 = Chl; A1 = phylloquinone (Q); Fx, FA and FB = Fe–S centres, FD = ferredoxin; FNR = ferredoxin NADP reductase; NADP+ = oxidised nicotinamide adenine dinucleotide phosphate. YD = symmetrically related tyrosine to YZ but not directly involved in water oxidation, and QH2 = reduced plastoquinone (plastoquinol), which acts as a mobile electron/proton carrier from PSII to the cytochrome b6f complex. With the exception of the mobile electron carriers, PQ/PQH2 , PC and FD, the remaining redox active cofactors are bound to multisubunit protein complexes which span the membrane depicted as coloured boxes. Similarly CF0–CF1 is a multisubunit membrane spanning complex. Modified from ref. 13. | ||
The reducing equivalents leave PSII in the form of plastoquinol (QH2) while the dioxygen is released into the atmosphere.
| 4H+ + 4e− + Q → O2 + 2QH2 |
The light-driven transfer of electrons and protons from H2O to CO2, involves a number of redox active cofactors located in the PSII and PSI protein complexes (see Fig. 3 and its legend for specific details). The transfer of reducing equivalents between PSII and PSI is aided by a third membrane protein complex known as the cytochrome b6f (Cyt b6f) as detailed in Fig. 3. The three complexes, PSI, PSII and Cyt b6f are located in the photosynthetic membrane such that electron flow from water to NADP+ is vectorial leading to the generation of a proton gradient (see Fig. 3). This gradient is used chemiosmotically by a fourth complex, CF0–CF1 to drive its ATPsynthase activity to convert ADP to ATP and thus provide chemical energy for the CO2 reduction process (for details see ref. 25).
In many ways, the photosystems of photosynthesis, including those of anoxygenic photosynthetic bacteria (organisms that do not split water) are highly efficient molecular photovoltaic nanomachines in that they use light energy to bring about electrical charge separation across a membrane of high dielectric strength.26 The organisation of the electron carriers and other cofactors in these nanomolecular devices are optimised to facilitate forward energy storing reactions and minimising backward and wasteful energy releasing reactions. There is considerable information about these photosynthetic photosystems which indicates that they are structurally and functionally very similar.27,28 Indeed, there are aspects of their design which could be incorporated into an ‘artificial photosynthetic’ device and are similar to existing photovoltaic systems, particularly the dye-sensitised photoelectrochemical solar cells developed by Graetzel and his colleagues.10
Similarly the light harvesting systems associated with the photosystems of different types of photosynthetic organisms have common principles for capturing solar energy across the whole of the visible spectrum and facilitating efficient energy transfer to the associated reaction centres with minimum losses of energy. Again detailed spectroscopic and structural studies have revealed the molecular basis of these systems, details which could also be adopted for designing light concentrating systems for a new generation of solar energy converting technologies.29 However it is the water splitting reaction of PSII which holds the greatest promise for developing new technologies for converting solar radiation into usable energy, particularly in generating hydrogen or “high energy electrons” for reducing carbon dioxide. In this way PSII is unique when compared with all other types of photosystems which are far more limited in the redox chemistry they catalyse.
Photosystem II
The photosynthetic water splitting reaction appeared on our planet about 2.5 billion years ago and was the ‘big bang of evolution’ since for the first time living organisms had available an inexhaustible supply of ‘hydrogen’ (in the form of reducing equivalents) to convert carbon dioxide into organic molecules. From that moment, living organisms on Earth could prosper and diversify on an enormous scale; biology had solved its energy problem and PSII established itself as the “engine of life”.30Clearly, using solar energy to split water to produce hydrogen or “high energy” electrons is also the perfect solution for humankind. In principle, the technology exists today to do this. Electricity can be generated by photovoltaic solar cells and used to carry out the electrolysis of water. With a solar cell efficiency of 10% and 65% efficiency for the electrolytic system, the overall efficiency would be 6.5%. Electrolysis relies on platinum or other catalysts for gas evolution, which are in limited supply and therefore expensive. At present very little hydrogen is generated by electrolysis because of the lower price of electricity generated by conventional means. Similarly, the cost of photovoltaic solar cells marginalises this route for using solar energy to produce hydrogen directly from water. But perhaps a bio-inspired water splitting catalyst can be devised which works along similar chemical principles employed by the OEC of PSII.
Because of the importance of understanding the chemistry of the water splitting reaction of PSII there has been a wide range of techniques employed to probe the molecular mechanisms involved and to investigate the structure of the catalytic centre (see various articles in refs. 31 and 32), being particularly spurred by the recent structural analyses of PSII by X-ray absorption spectroscopy33–36 and X-ray crystallography.37–40 These studies, coupled with quantum mechanical analyses have provided refinement of the structure of the OEC41–43 allowing detailed schemes to be formulated for the water splitting chemistry mechanism leading to O–O bond formation.44–50
Structure of PSII
The first complete and refined crystal structure of PSII was reported by Ferreira et al.39 which revealed considerable information about the organisation of the Mn4Ca-cluster and the details of its protein environment. Similarly the protein environments of the other cofactors involve in light absorption and charge separation where revealed. In this work, PSII was isolated as a dimeric complex from a cyanobacterium called Thermosynechococcus elongatus. Each monomer contained 19 different protein subunits with the reaction centre, composed of the D1 and D2 proteins, at its heart (see Fig. 4). A crystal structure published later of cyanobacterial PSII had one extra peripheral subunit40 possibly reflecting different biochemical procedures for its isolation.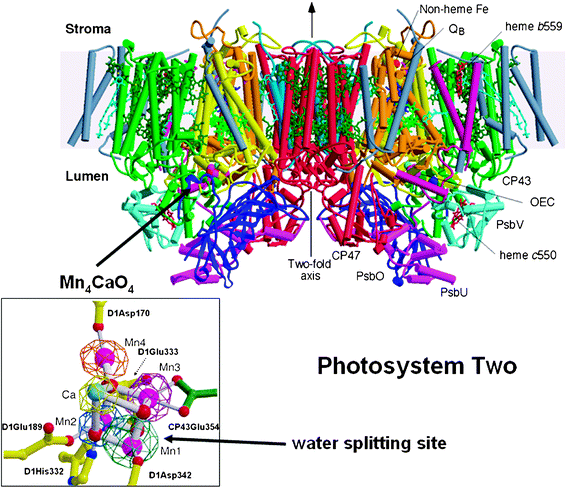 | ||
| Fig. 4 Side view of the structure of Photosystem II, the water splitting enzyme of photosynthesis. This structure was determined by X-ray crystallography.39 The complex is embedded in the thylakoid membrane spanning between their lumenal and stromal surfaces. It is composed of two monomers related to each other by a two-fold axis. Each monomer contains 19 different protein subunits with 16 being located within the membrane matrix and having σ-helices (depicted by cylinders). In total there are 35 transmembrane helices. The D1- and D2-proteins that compose the reaction centre are shown in yellow and orange, respectively. 57 cofactors were assigned to the structure, including 36 chlorophyll a molecules. Of particular importance was characterisation of the water splitting site consisting of a cubane-like organisation of a Mn3CaO4-cluster with a fourth Mn linked to the cubane by mono-μ-oxo bridges (see insert where Mn ions are shown in magenta, calcium in turquoise and oxygen atoms in red). This catalytic centre is located on the lumenal side of the complex and is stabilised and shielded by three extrinsic proteins, PsbO, PsbU and PsbV. Also shown in the insert are the amino acids which provide ligands to the metal cluster. Figure modified from ref. 39. | ||
The crystal structure of PSII determined by Ferreira et al. (2004) confirmed earlier models37,38 for the organisation of the cofactors involved in primary and secondary charge separation in the reaction centre although these earlier studies did not provide details about their protein environments. However the most important outcome of the Ferreira et al. structural study, was the suggestion that three Mn ions and a Ca ion of the OEC form a cubane-like structure with the four metal ions linked by oxo-bridges. It was proposed that a fourth Mn ion is linked to the cubane by mono-μ-oxo bridges via one of the oxo groups of the cubane (see Fig. 4 inset). Surrounding the Mn4Ca-cluster are a number of amino acid residues that either provide ligands to the metal ions or act to facilitate hydrogen bonding networks which almost certainly play a key role in the deprotonation of the substrate water molecules. A nearby tyrosine (residue 161 of the D1 protein), hydrogen bonded to a histidine (D1His190), is the redox active cofactor YZ which functions as an intermediate electron carrier between the Mn4Ca-cluster and P680˙+ (see Fig. 3). Most of the amino acids identified in the OEC belong to the D1 protein although another PSII protein, known as CP43, also provides key residues. All these amino acids are fully conserved in all known amino acid sequences of the D1 and CP43 proteins whether they are from prokaryotic cyanobacteria or eukaryotic algae and higher plants. We can therefore assume based on current knowledge, that there is no variation on a theme for this catalytic centre, which is able to carry out one of the most oxidative and difficult reactions of biology.
With structural information available, realistic chemical schemes are now being formulated for the water splitting reaction and the formation of molecular oxygen. It has been known for some time that there are at least five intermediate states leading to the formation of dioxygen, known as S-states. The sequential advancement from S0 to S4 is driven by each photochemical turnover of the PSII reaction centre as depicted in the S-state cycle (Fig. 5)
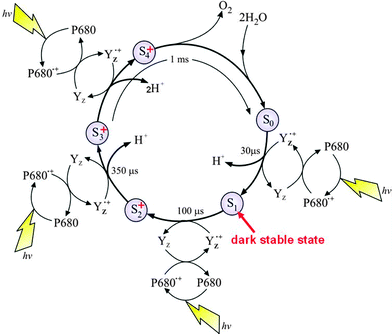 | ||
| Fig. 5 The S-state cycle showing how the absorption of four photons of light (hν) by P680 drives the splitting of two water molecules and formation of O2 through a consecutive series of five intermediates (S0, S1, S2, S3 and S4). Protons (H+) are released during this cycle except for the S1 to S2 transition. Electron donation from the Mn4Ca-cluster to P680˙+ is aided by the redox active tyrosine YZ. Also shown are half-times for the various steps of the cycle. | ||
The progression through the S-states to S4 results in the storing of four oxidising equivalents, which are reduced in the final step (S4 to S0) by four electrons derived from two substrate water molecules with the concomitant formation of dioxygen.
As explained above, the processes underpinning this catalytic S-state cycle are initiated by the absorption of visible light by P680 and the crystal structure indicates that this primary electron donor is composed of four closely located Chls designated PD1, PD2 ChlD1 and ChlD2 in Fig. 6. Excited P680 (depicted as P680* in Fig. 2) is probably delocalised over all four Chls and that initial electron donation to the primary electron acceptor, pheophytin a (Pheo D1) occurs from ChlD1. The formation of the primary radical pair takes place in a few picoseconds and probably involves “hole” migration to PD1 to form PD1˙+Pheo˙−. Stabilisation of the PD1˙+Pheo˙− state is accomplished by electron transfer from Pheo˙− to a plastoquinone acceptor QB in the microsecond to millisecond time domain according to its redox state. This terminal quinone electron acceptor is bound to the D1 protein while the intermediate plastoquinone molecule QA, which facilitates the electron transfer from Pheo˙− to QB is bound to the D2 protein of the reaction centre. A non-heam iron (Fe) is located midway between QA and QB, having four histidine ligands provided equally by the D1 and D1 proteins coupled with bidentate ligation of a bicarbonate ion. Unlike QB, QA plastoquinone is tightly bound to the D2 protein, functions as a single electron acceptor and does not undergo protonation. The QB plastoquinone, however, accepts two electrons and is fully protonated prior to its departure from the reaction centre via the hydrophobic lipid phase of the membrane and in this way enter the electron/proton transfer chain as described in Fig. 2 and 3.
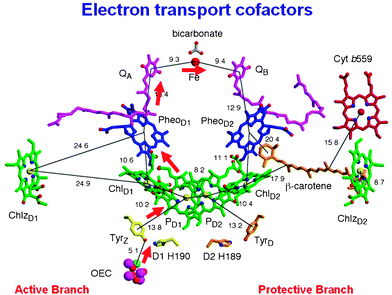 | ||
| Fig. 6 Organisation of the electron transfer cofactors that make up the reaction centre of Photosystem II as revealed by X-ray crystallography.39 Excitation of the reaction centre via the chlorophylls (Chl) shown in green leads to the reduction of pheophytin (Pheo) resulting in the formation of the radical pair P680˙+Pheo˙−. The radical cation of P680 is localized on PD1 while the radical anion is located on PheoD1. The electron on PheoD1˙− is rapidly donated to a firmly bound plastoquinone QA (shown in purple) and then transferred to a second plastoquinone QB (also shown in purple). This electron transfer is aided by the presence of a non-haem iron (Fe) located mid-way between them. When the QB plastoquinone has been doubly reduced and protonated the resultant plastoquinol (PQH2) diffuses from the QB-binding site into the lipid matrix of the membrane, P680˙+ is reduced by a redox active tyrosine (TyrZ or YZ) which then extracts electrons from the Mn4Ca-cluster that constitutes the oxygen-evolving centre (OEC). These electron transfer processes occur mainly on the D1-side of the reaction centre as shown by the red arrow. Some of the symmetrically related cofactors located on the D2-side (PheoD2) are non-functional. Other cofactors shown on the D2-side, however, are functionally active and seem to play a role in protecting PSII against photoinduced damage, the haem of cytochrome b559 (Cyt b559 shown in red), the β-carotene molecule (shown in brown) and ChlZD2. Figure modified from ref. 39. | ||
On the oxidising side of the PSII reaction centre, which is localised towards the lumenal surface of the thylakoid membrane, P680˙+ provides the oxidising potential to split water with the redox active tyrosine YZ (TyrZ) aiding the electron/proton removal from the OEC.
As can be in Fig. 6 the electron transfer giving rise to the reduction of QB and oxidation of water is biased to the D1-side of the reaction centre. However other side reactions can occur under some conditions on the D2-side which involve the P680˙+ driven oxidation of high potential cytochrome b559 (Cyt b-559), a β-carotene molecule and a Chla molecule (ChlZ).51–54 These side reactions occur on the tens of millisecond time scale and therefore do not compete with the electron transfer pathway leading to water oxidation. Indeed, they probably only occur when the rate of water oxidation becomes limited and thus provides a protective mechanism against the detrimental reactions resulting from the very high redox potential of the long lived P680 radical cation.
New developments
The model proposed by Ferreira et al.39 for the OEC has recently been analysed in considerable depth using quantum mechanical and molecular mechanical (QM/MM) analyses.41,42 The calculations assumed that the carbonate ion, tentatively identified as a ligand in the OEC is replaced by a chloride ion in the active S1 state of the water splitting catalytic cycle and that the assigned protein ligands were complemented by water and hydroxyl ligands to satisfy the coordination requirements of the five metal ions. Even with these adjustments the calculated model for the metal cluster was remarkably similar to that proposed by Ferreira et al.39 and confirmed that it is a chemically stable structure. Despite, the good correlation between the Ferreira et al. structural model of the OEC and theoretical calculations there are inconsistencies with distance and angular information derived from EXAFS. Indeed recent polarised EXAFS studies conducted on single crystals of PSII isolated from T. elongatus (Yano et al. 2006) gave at least four different arrangements for the Mn4Ca-cluster while more recent crystal structures reported40 suggest yet another organisation. Radiation damage during the collection of X-ray diffraction data has been implied as being the cause for inconsistencies between the different models.55 Recently Barber and Murray56 have attempted to rationalise the existing data to provide a series of working models of the OEC. The differences between them are not large having the same amino acid environments as first defined by Ferreira et al.39Fig. 7C shows one such model in which the fourth Mn (Mn4) is linked to the Mn3CaO4-cubane via one of its Mn rather than by a bridging oxygen of the cubane.57 As a consequence this new arrangement has one rather than two mono-μ-oxo bonds and is more compatible with EXAFS analyses.58 The adjustment of the linkage between Mn4 and the cubane requires a repositioning of the other Mn ions and therefore changes in the amino acid ligation pattern as emphasised by comparing Fig. 7B and 7D. Despite uncertainty about the precise organisation of the Mn4Ca-cluster, the most recent simulation of the polarised EXAFS data by Sproviero et al.43 continues to provide support for their QM/MM derived model of the OEC41,42 and therefore for the original model proposed by Ferreira et al.39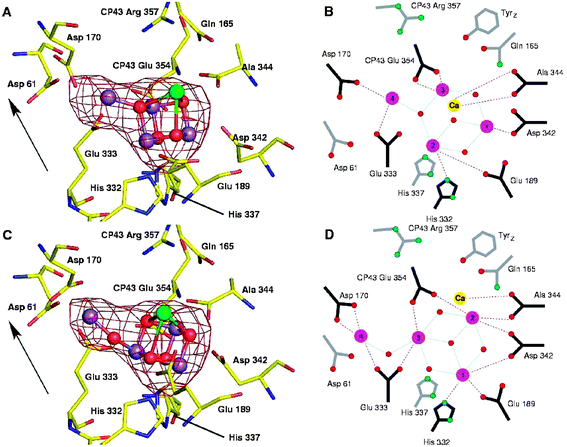 | ||
| Fig. 7 The water splitting site of PSII showing the Mn4Ca-cluster positioned within the Mn-anomalous difference map of Ferreira et al.39 (A) Based on the model of Ferreira et al. (39). (B) Schematic representation of the amino acid ligation pattern for model in (A) with distance less than 3 Å shown by connecting lines. (C) Remodelling the water splitting site using the native electron density maps of Ferreira et al.39 and Loll et al.40 and Mn-anomalous difference map of Ferreira et al.,39 keeping the Mn3CaO4 cubane of Ferreira et al. but with the fourth Mn linked to it via a single 3.3 Å mono-μ-oxo bridge (D) Schematic representation of the amino acid ligation pattern for model in (C) with distance less than 3 Å shown by connecting lines. The Mn-anomalous difference map is shown in red and contoured at 5σ with the fitting of the metal ions into this density by real-space refinement using the molecular graphics programme. Arrow indicates the direction of the normal to the membrane plane (taken from refs. 56 and 57). | ||
Mechanism of water splitting and dioxygen formation
Although the geometry of the Mn4Ca-cluster and its exact ligand field characteristics are not yet known precisely, either for its relaxed S1-state or for higher S-state conditions, the models available do provide a basis for developing chemical mechanisms for water oxidation and dioxygen formation. The Mn ion outside the cubane (Mn4) is adjacent to the Ca2+. Their positioning towards the side chains of several key amino acids, including the redox active YZ suggests that they provide the ‘catalytic’ surface for binding the two substrate water molecules and their subsequent oxidation. One well championed mechanism45,46,59,60 suggests that the substrate water, associated with Mn4, is deprotonated during the S-state cycle and converted to a highly electrophilic oxo (see Fig. 8A). This mechanism is dependent on Mn4 being converted to a high oxidation state (possible Mn(V)) during progression to the S4-state just prior to O–O bond formation. The other three Mn ions are progressively driven into high valency states (Mn(IV) and act as a further “oxidising battery” for the Mn(V)-oxo species in the S4-state (see Fig. 8A). In this way the reactive oxo is electron deficient, so much so that it makes an ideal target for a nucleophilic attack by the oxygen of the second substrate water bound within the coordination sphere of the Ca2+ (see Fig. 8A). An alternative mechanism proposed by Siegbahn, which is based on in depth density function theory (DFT) calculations,49 suggests that the deprotonated water molecule on Mn4 forms an oxyl radical and that this attacks an oxo-ligand of the Mn3CaO4-cubane (see Fig. 8B.)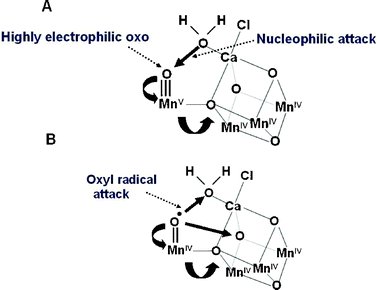 | ||
| Fig. 8 Two different mechanisms for the final step of the S-state cycle when the dioxygen bond of O2 is formed. (A) Mechanism 1. The very high oxidation state of the Mn-cluster, particularly the Mn ion outside the Mn3CaO4-cubane, leads to a highly electron deficient oxo (after deprotonation of water molecules during the S-state cycle). Nucleophilic attack by the hydroxide of the second substrate water within the coordination sphere of Ca2+ leads to O2 formation. (B) Mechanism 2. The formation of a oxyl radical after deprotonation of the substrate water molecule ligated to the Mn outside the cubane leads to a radical attack of an oxo-ligand within the Mn3CaO4-cubane. | ||
Artificial photosynthesis
While some progress has been made in mimicking photosynthesis in artificial systems, researchers have not yet developed components that are both efficient and robust for incorporation into a working system for solar fuel production. To date the main focus of research has been to design and synthesise molecular systems consisting of electron donors and acceptors and mimic light driven charge separation which occurs in photosynthetic reaction centres.61,62 These studies have allowed rigorous analyses of the dependencies of electron transfer efficiency on the physical and chemical properties (donor/acceptor distance and orientation, free energy of the reaction and electronic interactions) of the system thus providing basic rules for their construction, including the desirability for multi-step charge separation.63The bio-inspired systems employ chromophores to absorb light energy which are analogous with the photosynthetic pigments, such as chlorophyll. Often, however, the chromophores are directly engaged in electron transfer processes64 and in this way act like redox active chlorophylls within photosynthetic reaction centres, for example P680 and P700. Having an antenna, or light harvesting array, without carrying out charge separation itself, is an engineering design adopted by natural photosynthesis to maximise solar energy absorption. Covalently linked arrays of light harvesting chromophores that funnel energy to a central site where charge separation can occur have been demonstrated in artificial systems.65,66 However the synthesis of such arrays requires significant effort and it would be desirable to create self-assembly, functional antenna arrays using robust dyes such as those used as pigments in industrial paints.
Despite the fact that PSII contains the chemical machinery to split water into oxygen and “hydrogen” no artificial photochemical system has been built to mimic this reaction. However the insights gleaned from the recent structural determination of PSII have major implications for the design of an artificial catalytic system for using solar energy to release hydrogen from water.67 The hydrogen produced could be used directly as a source of energy but could also be used, as it is in photosynthesis, to reduce carbon dioxide to other types of fuels such as methane. Artificial catalysts designed to reproduce the reactions of PSII may also have to incorporate other bio-inspired features, particularly the employment of light harvesting systems which can capture the energy of all the colours of the visible solar spectrum and efficiently use this energy to drive the chemistry of water splitting. Our understanding of natural photosynthetic light harvesting systems and how they are coupled to reaction centres is at an advanced level such that the basic principles of their construction and operation are now well established.
The challenge is to have a molecular arrangement in the artificial catalyst which will use light energy to split water, allow the dioxygen bond to form and concomitantly provide reducing potential for hydrogen gas production or CO2 reduction. It has been demonstrated that catalysts based on Mn are capable of water splitting and the generation of dioxygen occurs when a strong oxidant is used to drive the Mn into high oxidation states; believed to be Mn(V).68,69 Complementing this was the earlier discovery that ruthenium based catalysts, such as the “blue dimer” can photooxidise water to dioxygen.70–72
Light driven water splitting can also be accomplished using semiconductor-based photocatalysis as first demonstrated by Fujishima and Honda in 1972.73 These workers initially used TiO2 but it was necessary to replace TiO2 by SrTiO3 in order to produce both hydrogen and oxygen. Since these reactions are driven by high energy UV radiation they are of limited use for fuel generation. Current efforts are being made to dope these types of semiconductors with dyes which can carry out photo-driven redox reactions using visible light.74 Combining photovoltaic technology with appropriate electrochemical catalysts is a complementary approach. For example, recently Kanan and Nocera75 have described a self-assembling catalyst composed of cobalt and phosphate ions which can efficiently produce molecular oxygen from water at neutral pH with a low over potential akin to that which operates in the OEC of PSII. The next step will be to couple this oxygen producing system to another catalyst which will use the electrons and protons derived from the water splitting reaction to produce hydrogen gas or reduce carbon dioxide. In the case of the former, considerable progress is being made, in part by mimicking the natural hydrogenase enzymes found in a wide variety of microorganisms.76–78 The reduction of carbon dioxide, however, is more difficult because multielectron reactions are required and the emergence of catalysts for generating useful carbon fuels will require considerable effort.79,80
Conclusions
It is anticipated that the global demand for energy will more than double by the mid-century and perhaps more than triple by the end of the century. Satisfying this demand will be necessary in order to achieve vibrant technological progress, economic growth and most importantly, political stability over the coming decades. Already we are faced with the prospect of catastrophic climate change due to the release of carbon dioxide into the atmosphere brought about by the burning of fossil fuels. In the short term we must exploit all technologies known to us to produce energy while at the same time reduce carbon dioxide emission. The nature of the mix will vary between different countries depending on their resources and populations with some dominating (e.g. geothermal in Iceland, biomass in Brazil, etc). Coupled with this challenge is to use energy more efficiently. Here again we can learn from Nature. In biology the ‘combustion’ of fuel (food) is accomplished isothermally by highly efficient and subtle biological reactions involving a host of clever enzymes. For example, when the ‘hydrogen’ of glucose is combined with oxygen during the process of respiration to produce water and carbon dioxide, 38 ATP molecules are made. ATP is the energy currency of cells. Since ATP stores 12 kcal mol−1 (50 kJ mol−1) of usable energy and the energy content of glucose is 672 kcal mol−1 (2813 kJ mol−1) the efficiency of energy conversion is 68%. It therefore seems to me that mankind should follow biology’s example and strive to develop new technologies that are as energy efficient as those in biology. In a sense the invention of the World Wide Web is an example of how new technologies based on microelectronics has provided a way to communicate throughout the globe which is far more energy efficient compared with conventional mailing. This significant advance and many other similar examples were unthinkable a few years back and give optimism for the development of new technologies which will generate energy and use it with maximum efficiency.For the long term we will have very few options to replace fossil fuels and satisfy the increased energy demands of a global population of 10 billion. Renewables such as hydropower, wind, wave, geothermal and biomass will not be able to supply energy equivalent to 20 TW even when taken together.4,81 Nuclear fission is a short term solution but in the long run will probably not be a realistic option. Nuclear fusion is a possibility but the construction of a working reactor is proving problematic. Nevertheless we must continue to research this technology with the hope that it will come on stream sooner or later. But there is another nuclear reaction which is already up and running, namely the Sun. Our sun is the champion of energy sources: delivering more energy to the earth in an hour than we currently use in a year from fossil, nuclear and all renewable sources combined. Its energy supply is inexhaustible in human terms, and its use is harmless to our environment and climate.
The enormous untapped potential of solar energy is an opportunity which should be addressed with urgency. Biology chose this energy source and there is no reason why the chemical reactions devised by photosynthetic organisms cannot be mimicked by ingenuity of humans. We already have a considerable knowledge base and emerging nanotechnologies to exploit. With a concerted input of the talents of scientists trained in different disciplines it should be possible to move the technologies of solar energy conversion forward. The recognition of a Manhattan- or Apollo-like initiative to develop new sustainable energy technologies in response to the CO2 problem as suggested by Hoffert et al.2,82 should be the driver for encouraging basic and applied research in this area.
Abbreviations used
| ATP | Adenosine triphosphate |
| CF0–CF1 | ATPsynthase complex |
| Chl | Chlorophyll |
| CP43 | Chlorophyll binding PsbC protein of PSII |
| Cyt b6f | Cytochrome b6f |
| DFT | Density Functional Theory |
| EXAFS | Extended X-ray absorption fine structure |
| OEC | Oxygen Evolving Centre |
| PSI | Photosystem I |
| PSII | Photosystem II |
| Pheo | Pheophytin |
| P680 | Primary electron donor chlorophyll in PSII |
| P700 | Primary electron donor chlorophyll in PSI, NADP+, nicotinamide adenine dinucleotide phosphate |
| QM/MM | Quantum Mechanical and Molecular Mechanical |
Acknowledgements
I wish to thank the Biotechnology and Biological Science Research Council (BBSRC) for the financial support that led to the determination of the structure of PSII. I have drawn on information from a wide range of sources but I would particularly like to acknowledge that my thinking has been significantly influenced by discussion with Professors Nathan Lewis and Harry Gray of Caltech and Professor Daniel Nocera at MIT, all of whom are strong advocates of using solar energy as an inexhaustible and non-polluting source of energy for the long term benefit of humankind.References
- Energy Information Administration 2005 Annual Energy Outlook, US Dept of Energy, Washington DC.
- M. T. Hoffert, K. Caldeira, A. K. Jain, E. F. Haites, L. D. Harvey, S. D. Potter, M. E. Schlesinger, T. M. L. Wigley and J. J. Wuebbles, Nature, 1998, 395, 881 CrossRef CAS.
- Special Report on Emissions Scenarios, Intergovernmental Panel on Climate Change, ed. N. Nakicenovic and R. Swart, Washington, DC, 2000, pp. 48–55 Search PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 43, 15729 CrossRef.
- United Nations Development Program 2003 World Energy Assessment Report: Energy and the Challenge of Sustainability (United Nations, New York).
- Intergovernmental Panel on Climate Change 2001, Synthesis Report Summary for Policymakers (Intergovernmental Panel on Climate Change, Washington, DC) Third Assessment Report.
- Carbon Dioxide Capture and Storage, ed. B. Metz, O. Davidson, X. deConinck, H. M. Loos, L. Meyer, Intergovernmental Panel on Climate Change, Washington, DC, 2005 Search PubMed.
- Solar Energy Utilization Workshop, Basic Science Needs for Solar Energy Utilization, US Dept of Energy, Washington, DC, 2005.
- S. E. Shaheen, D. S. Ginley and G. E. Jabbour, MRS Bull., 2005, 30, 10–19 CAS.
- D. P. Hagberg, J.-H. Yum, H.-J. Lee, F. De Angelis, T. Marinado, K. M. Karlsson, R. Humphry-Baker, L.-C. Sun, A. Hagfeldt, M. Graetzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2008, 130, 6259 CrossRef CAS.
- E. H. Thorndike, Energy and the Environment, Pub Addison-Wesley, Reading Mass, USA, 1996 Search PubMed.
- D. A. Walker, Energy Plants and Man, Pub Packard Publishing Ltd, Chichester UK, 1977 Search PubMed.
- M. D. Archer and J. Barber, in: Molecular to Global Photosynthesis, ed. M. D. Archer and J. Barber, Pub Imperial College Press, London UK, 2004, pp. 1–41 Search PubMed.
- J. R. Bolton, Solar Power and Fuels, Academic Press, New York, USA, 1977 Search PubMed.
- J. R. Bolton, in: The Chemical Conversion and Storage of Solar Energy, ed. J. B. King, R. R. Hautala and C. R. Kutal, Humana Press, Clifton, NJ, USA, 1979, pp. 31–50 Search PubMed.
- P. G. Falkowsky and J. A. Raven, Aquatic Photosynthesis, Blackwell, Oxford, 1997 Search PubMed.
- USDA/DOE Report 2005 A billion to feedstock supply for a bioenergy and bioproducts industry.
- A. V. Bridgewater and K. Maniatis, in Molecular to Global Photosynthesis, ed. M. D. Archer and J. Barber, Imperial College Press, London, UK, 2004, pp. 521–611 Search PubMed.
- H. S. Geller, Ethanol fuel from sugar cane in Brazil, Annu. Rev. Energy, 1985, 10, 135 Search PubMed.
- J. Goldemberg, Ethanol for a Sustainable Energy Future Science, 2004, 315, 808 Search PubMed.
- J. Goldemberg, S. T. Coelho, O. Lucon and P. M. Nastari, Biomass Bioenergy, 2004, 26, 301 CrossRef.
- M. D. Bullard, in Molecular to Global Photosynthesis, ed. M. D. Archer and J. Barber, Imperial College Press, London, UK, 2004, pp. 453–519 Search PubMed.
- C. Somerville, S. Bauer, G. Brininstool, M. Facette, T. Hamann, J. Milne, E. Osborne, A. Paredez, S. Persson, T. Raab, S. Vorwerk and H. Youngs, Science, 2004, 306, 2206 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, J. Frederick, J. P. Hallett, D. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford UK, 2002 Search PubMed.
- J. Barber and B. Andersson, Nature, 1994, 370, 31 CrossRef CAS.
- W. D. Schubert, O. Klukas, W. Saenger, H. T. Witt, P. Fromme and N. Krauss, J. Mol. Biol., 1998, 280, 297 CrossRef CAS.
- K. -H. Rhee, E. P. Morris, J. Barber and W. Kühlbrandt, Nature, 1998, 396, 283 CrossRef CAS.
- H. van Amerongen, L. Valkunas and R. van Grondelle, Photosynthetic Excitons, World Scientific, Singapore 2000 Search PubMed.
- J. Barber, Photosystem II: The engine of life, Biophys. Q. Rev., 2003, 36, 71 Search PubMed.
- T. J. Wydrzynski and K. Satoh, Photosystem II: The Light-Driven Water Plastoquinone Oxidoreductase, in Vol. 22 Advances in Photosynthesis and Respiration, Springer, Dordrecht, The Netherlands, 2005, pp. 1–786 Search PubMed.
- J. Barber and A. W. Rutherford, Revealing how nature uses sunlight to split water, Philos. Trans. R. Soc. London, Ser. B, 2003, 363, 1123.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821 CrossRef CAS.
- K. Sauer, J. Yano and V. K. Yachandra, Coord. Chem. Rev., 2008, 252, 318 CrossRef CAS.
- J. Yano and V. K. Yachandra, Inorg. Chem., 2008, 47, 1711 CrossRef CAS.
- Y. Pushkar, J. Yano, K. Sauer, A. Boussac and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1879 CrossRef CAS.
- A. Zouni, H. T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger and P. Orth, Nature, 2001, 409, 739 CrossRef CAS.
- N. Kamiya and J. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 98 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS.
- B. Loll, J. Kern, W. Saenger and J. Biesiadka, Nature, 2005, 438, 1040 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Chem. Theory Comput., 2006, 2, 1119 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, Curr. Opin. Struct. Biol., 2007, 17, 173 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130, 3428 CrossRef CAS.
- J. P. McEvoy and G. W. Brudvig, Phys. Chem. Chem. Phys., 2004, 6, 4754–4763 RSC.
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6, 4764–4771 RSC.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS.
- P. E. M. Siegbahn and M. Lundberg, Photochem. Photobiol. Sci., 2005, 4, 1035 RSC.
- P. E. M. Siegbahn, Chem.–Eur. J., 2006, 12, 9217 CrossRef CAS.
- P. E. M. Siegbahn, Inorg. Chem., 2008, 47, 1779 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, Coord. Chem. Rev., 2008, 252, 395 CrossRef CAS.
- D. H. Stewart and G. W. Brudvig, Biochim. Biophys. Acta, 1998, 1367, 63 CrossRef CAS.
- P. Faller, A. Pascal and A. W. Rutherford, Biochemistry, 2001, 40, 6431 CrossRef CAS.
- A. Telfer, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 1431 CrossRef CAS.
- C. A. Tracewell and G. W. Brudvig, Biochemistry, 2003, 42, 9127 CrossRef CAS.
- J. Yano, J. Kern, K. D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12047 CrossRef CAS.
- J. Barber and J. W. Murray, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1129 CrossRef CAS.
- J. Barber and J. W. Murray, Coord. Chem. Rev., 2008, 252, 233 CrossRef.
- V. K. Yachandra, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 1347 CrossRef CAS.
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209 CAS.
- V. L. Pecoraro, M. J. Baldwin, M. T. Caudle, M. W. -Y. Hsieh and N. A. Law, Pure Appl. Chem., 1998, 70, 925 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36 RSC.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 427, 607.
- L. Hammerstrom and S. Styring, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1283 CrossRef.
- S. Kirstein and S. Daehne, Int. J. Photoenergy, 2006 Search PubMed , Article ID 20363, 21 pp..
- J. L. Allwood, A. K. Burrell, D. L. Officer, S. M. Scott, K. Y. Wild and K. C. Gordon, Chem. Commun., 2000, 747 RSC.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS.
- J. Limberg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524 CrossRef CAS.
- R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2008, 47, 1815 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727 CrossRef CAS.
- I. Romero, M. Rodriguez, C. Sens, J. Mola, M. R. Kollipara, L. Francas, E. Mas-Marza, L. Escriche and A. Llobet, Inorg. Chem., 2008, 47, 1824 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- L. Schmidt-Mende, W. M. Campbell, Q. Wang, K. W. Jolley, D. L. Officer, M. K. Nazeeruddin and M. Graetzel, ChemPhysChem, 2005, 6, 1253 CrossRef.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS.
- F. A. Armstrong and S. P. J. Albracht, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 937 CrossRef CAS.
- C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L. -S. Wang, G. Sawers and C. J. Pickett, Nature, 2004, 433, 610.
- S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. -M. Liu and C. J. Pickett, J. Am. Chem. Soc., 2004, 126, 16988 CrossRef CAS.
- E. Fujita, in The 2001 McGraw-Hill Yearbook of Science & Technology, ed. M. D. Licker, McGraw-Hill Book Co, New York. NY, pp. 71–74 Search PubMed.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. Dubois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS.
- N. S. Lewis, Chemical Challenge in Renewable Energy, Lecture 2005, text on http://www.cce.caltech.edu:16080/faculty/lewis/research.htm.
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981 CrossRef CAS.
Footnote |
| † Part of the renewable energy theme issue. |
| This journal is © The Royal Society of Chemistry 2009 |