B–N compounds for chemical hydrogen storage†‡
Charles W. Hamiltona, R. Tom Baker*b, Anne Staubitzc and Ian Manners*c
aLos Alamos National Laboratory, Inorganic, Isotope, and Actinide Chemistry, MS J582, Los Alamos, NM 87545, USA. E-mail: chamilton@lanl.gov; Fax: +1 505-667-3502; Tel: +1 505-665-4636
bDepartment of Chemistry, University of Ottawa, 10 Marie Curie, Ottawa, Ontario, Canada K1N 6N5. E-mail: rbaker@uottawa.ca; Tel: +1 613-562-5800 ext. 5698; +1 613-562-5800 ext. 5613
cUniversity of Bristol, School of Chemistry, Cantock’s Close, Bristol, UK BS8 1TS. E-mail: Ian.Manners@bristol.ac.uk; Fax: +44 (0)117 929 0509
First published on 26th November 2008
Abstract
Hydrogen storage for transportation applications requires high volumetric and gravimetric storage capacity. B–N compounds are well suited as storage materials due to their light weight and propensity for bearing multiple protic (N–H) and hydridic (B–H) hydrogens. This critical review briefly covers the various methods of hydrogen storage, and then concentrates on chemical hydrogen storage using B–N compounds. The simplest B–N compound, ammonia borane (H3NBH3), which has a potential 19.6 wt% hydrogen storage capacity, will be emphasised (127 references).
 Charles W. Hamilton | Charles Wayne Hamilton was born in Houston, Texas, USA. He received his BS in Chemistry from Texas A&M University in May of 2001. During his undergrad, he studied nuclear chemistry at the Cyclotron Institute under the direction of Prof. Joseph Natowitz. He then joined the laboratory of Prof. Joseph Sadighi at the Massachusetts Institute of Technology where he studied oxidation catalysis. He received his PhD in 2007, and subsequently accepted a post-doctoral position at Los Alamos National Laboratory where he currently resides. His current research interests include the formation of molecular catalysts for amine borane dehydrogenation. |
 R. Tom Baker | R. Tom Baker received his BSc degree from UBC in 1975 and his PhD from UCLA in 1980 with M. Frederick Hawthorne. After a postdoctoral stint with Philip S. Skell at Penn State, he joined DuPont Central Research in Wilmington, DE, where he applied inorganic and organometallic chemistry and homogeneous catalysis to the nylon, fluoroproducts, and titanium dioxide businesses. He joined the Chemistry Division at Los Alamos National Laboratory in 1996 and worked on multifunctional catalysis approaches to low-temperature hydrocarbon functionalization and chemical hydrogen storage. In 2008, he became a chemistry professor at University of Ottawa and the director of the Centre for Catalysis Research and Innovation. |
 Anne Staubitz | Anne Staubitz obtained a diploma in biochemistry from the University of Tübingen, Germany. For her diploma thesis she worked in the group of Prof. Paul Knochel at the University of Munich with novel Grignard reagents applied to the synthesis of heterocycles. She then did her PhD with Prof. Varinder Aggarwal at the University of Bristol, working on a natural product synthesis. In Ian's group she is working as a postdoctoral fellow on the catalytic dehydrogenation of group 13–group 15 adducts. |
 Ian Manners | Ian Manners received his PhD from the University of Bristol in 1985 in the area of transition metal chemistry. After completing postdoctoral work in Germany in main group chemistry and the USA on polymeric materials, he joined the University of Toronto, Canada in 1990. After 15 years he returned to his Alma Mater to take up a Chair in Inorganic, Macromolecular and Materials Chemistry. His research interests focus on the development of new synthetic reactions in inorganic chemistry and their applications in molecular synthesis, polymer and materials science, supramolecular chemistry, and nanoscience. |
Introduction
An abundant, inexpensive energy supply is an essential component to boost developing economies as well as maintain status quo for established economies. Current energy consumption is based on combustion of carbon-based fuels to water and carbon dioxide (as well as environmentally harmful carbon particulates and sulfur and nitrogen oxides). For many years, climate modelling has shown that increased carbon dioxide concentration in the atmosphere will lead to detrimental environmental effects (recently reiterated in the fourth IPCC report).1 Thus it is vitally important to institute a shift away from carbon-based fuels and toward environmentally-friendly replacements.The transportation sector represented 31(28)% of the EU(US) energy use in 2005(2006) and accounted for 21(34)% of CO2 emissions.2,3 For stationary energy applications, CO2 can be potentially sequestered.4 For portable energy, however, this is impractical and an alternative energy carrier must be used.
Hydrogen has the potential to be a clean, source-independent, energy carrier.5 It has a high energy content per mass compared to petroleum (120 MJ kg−1 for hydrogen versus 44 MJ kg−1 for petroleum). Also, hydrogen can be readily used to run a fuel cell, which greatly increases efficiency over internal combustion engines (∼32% efficiency for diesel-electric; 90% potential efficiency for fuel cell with heat capture, 85% for electric motor, 77% overall efficiency) while eliminating formation of carbon particulates and sulfur and nitrogen oxides.6 Unfortunately, hydrogen has a poor energy content per volume (0.01 kJ L−1 at STP and 8.4 MJ L−1 for liquid hydrogen vs. 32 MJ L−1 for petroleum).
For transportation applications, an energy carrier should have a high energy content in as small a volume as possible to not intrude on passenger space, and as small a mass as possible to maintain fuel efficiency. The US Department of Energy has established a series of targets for hydrogen storage materials so that a vehicle can travel >500 km on a single hydrogen fill.5 Included with this are stringent system volumetric (5.4 MJ L−1 2010, 9.72 MJ L−1 2015) and gravimetric targets (6 wt% 2010, 9 wt% 2015). Evaluation of a hydrogen storage system includes all associated components (tank, valves, regulators, piping, mounting brackets, insulation, added coolants, etc.).
There are currently four leading methods to store hydrogen: physical means, sorbents, metal hydrides, and so-called chemical hydrides. All four will be briefly summarised, and then chemical hydrides with B–N bonds will be discussed in greater detail. Details of the potential of ammonia borane as a hydrogen storage material have been presented recently.7
Physical hydrogen storage
Safely containing hydrogen at high enough pressures requires strong tanks. The tanks must also be lightweight to maintain a high gravimetric capacity. Carbon-fibre reinforced composite tanks are light and capable of storing hydrogen at pressures up to 700 bar (4.7 MJ L−1). These tanks are nonconformable (normally cylinder shaped), which makes them difficult to adapt to a car design. There is also an energy penalty for gas compression (15–20% of the lower heating value for hydrogen).5 General Motors recently demonstrated the Chevy Sequel, which is able to achieve 482 km (300 miles) on a single hydrogen fill using compressed tanks to store the hydrogen fuel.8 Honda recently released the FCX Clarity, which is capable of 435 km (270 miles) on a single fill, and has recently become available for lease in California, USA.9 Both cars use efficient polymer electrolyte membrane fuel cells. While these fuel cells allow for the large range, they currently require expensive platinum catalysts and suffer from limited durability. Although these specific designs may not yet be practical for world-wide use, their demonstration is an important step to show the viability of hydrogen-fuelled cars.While liquid hydrogen has a higher density than compressed gas, maintaining the low temperatures requires extra components that adversely affect system volumetric and gravimetric storage capacity. Boil-off control is also a significant problem, with tanks losing approximately 1% per day. Finally, significant energy needs to be input to liquefy hydrogen (30% of the lower heating value for hydrogen).5
Sorbents
A wide range of nanoporous materials have been studied as potential hydrogen storage materials. The advantage of these sorbent materials lies in their ready reversibility. This is also their largest disadvantage, however, as they contain hydrogen by weak physisorption forces (van der Waals), which are normally less than 1.4 kcal mol−1. Hence, low temperatures (normally liquid nitrogen temperature, 77 K) are necessary to obtain reasonable hydrogen uptake.10Carbon-based materials such as nanotubes, nanofibres, solid foams, and activated carbon have been extensively studied. There was initially some confusion about the hydrogen uptake potential of these materials due to the various methods of measuring hydrogen uptake. Recently, the results have been more consistent, with several methods being employed in the same study.11 The maximum adsorption is ∼5 material wt% hydrogen at 77 K; with addition of tanks and cooling systems, the system storage capacity will be much lower.
Zeolite structures have also been examined.12 Zeolites are microporous structures of hydrated aluminate and silicate that are more easily prepared than nanotubes and offer greater control over pore size. However, they consist of atoms that are heavier than carbon, which limits gravimetric capacity. An emerging field is using zeolite structures as a template to form carbon networks.13 Templating carbon onto zeolites is difficult because zeolite structures can be disordered. Nonetheless, recent results demonstrated hydrogen uptake for zeolite-like carbon materials of 6.9 wt% at 77 K and 20 bar.
Organic polymers with intrinsic microporosity (PIMs) are another alternative.14 PIMs are made of rigid monomers to maintain microporosity (Fig. 1a), yielding gravimetric storage capacity of up to 3.0 wt% at 77 K and 15 bar.15 Increasing pore size (and thus surface area) should result in a higher storage capacity.
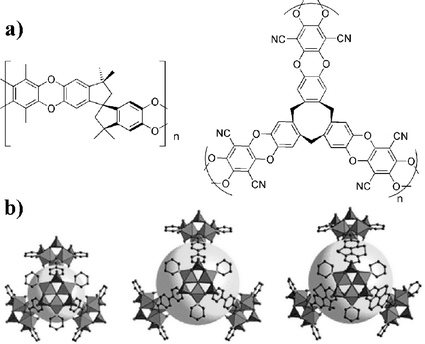 | ||
| Fig. 1 (a) Some examples of PIMs made up of rigid monomers. (b) Different linkers result in different pore sizes for MOFs.17 | ||
Metal–organic frameworks (MOFs) have also recently come into focus as potential hydrogen storage materials.16 These are three-dimensional polymers of metal atoms linked by bridging ligands (Fig. 1b).17 MOFs are readily synthesised by solution methods and then activated by heating to remove solvent molecules. Pore sizes can be rationally designed by choosing the metal and the linker. However, with large linkers, these tend to form interpenetrated structures which can decrease pore size. Similar to carbon nanotubes, MOFs rely primarily on physisorption to bind hydrogen and low temperatures (typically 77 K) are required.
To make these materials more practical, the temperature of effective adsorption must be raised (ideally to ambient temperatures). By adding in metal atoms or other materials capable of chemisorption, the energy of binding can be adjusted. This combination of chemisorption and physisorption could potentially increase the temperature of effective uptake. There are several recent examples of increasing the enthalpy of hydrogen adsorption for zeolites,18 MOFs,19 and carbon materials.20 However, the inclusion of extra elements decreases material gravimetric capacity.
Metal hydrides
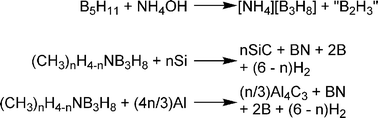
Metal hydrides rely on stronger chemical interactions than sorbents, and this results in materials that store hydrogen at higher temperatures. Heating is required to release the hydrogen after uptake. Since most metal hydrides are dense powders, achieving volumetric storage targets is feasible. Unfortunately, the relatively heavy metals make high gravimetric capacity difficult.One way to achieve higher gravimetric capacity is by including lighter main-group elements. These complex metal hydrides, such as alanates, amide, and borohydride compounds, have been evaluated as reversible hydrogen storage materials.21 One of the early examples is titanium-doped sodium alanate (NaAlH4), which can reversibly release hydrogen to give a maximum material gravimetric capacity of 5.5 wt% (although 3–4 wt% is typically obtained). Chen et al. found that Li3N can add roughly two equivalents of hydrogen to form lithium amide (LiNH2) and two equivalents of lithium hydride at elevated temperatures (200–250 °C) to give a 9.3 wt% uptake of hydrogen (Scheme 1).22 Under vacuum and at temperatures below 200 °C, 6.3 wt% of hydrogen desorbs. The remaining 3 wt% could be removed by heating above 320 °C.
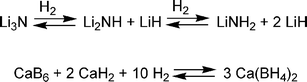 | ||
| Scheme 1 Reactions of some complex hydrides. | ||
Calcium borohydride can be heated to 400 °C to release 9.6 wt% hydrogen (Scheme 1).23 Upon addition of catalytic amounts of the dopants, TiCl3 and Pd, the ‘spent fuel’ can be rehydrogenated at 700 bar and 400–440 °C in 60% yield.
Recent effort has concentrated on mixtures of complex metal hydrides as potential hydrogen storage systems. Yang and Sudik found that ternary mixtures of MgH2, LiNH2 and LiBH4 have increased rates and extent of hydrogen release compared to binary mixtures of these components.24 At elevated temperatures (∼350 °C), 8–11 wt% hydrogen is released depending on the amount of MgH2 in the mixture. At lower temperatures, hydrogen release is reversible (2.8 wt% at 140 °C). Soloveichik and co-workers reported the decomposition of a compound that contains borohydride and amines, Mg(BH4)·2NH3.25 This compound has a maximum storage capacity of 16.0 wt%. Though hydrogen loss is observed at 150 °C, even heating to 400 °C results in a net loss of only 13.1 wt% hydrogen. Rehydrogenation has not yet been realised, but loss of hydrogen is reported to be endothermic, indicating that direct rehydrogenation may be possible.
Chemical hydrides
The so-called chemical hydrides typically use lighter elements than metal hydrides, which result in much higher gravimetric storage capacity. The necessary cleavage of covalent element–hydrogen bonds, however, puts more stringent requirements on reversibility. A reversible system will have a Gibb’s free energy (ΔG) of hydrogen release at or near 0 kcal mol−1. These materials will have a positive entropic term (ΔS) as hydrogen gas is being released. Thus, a slightly endothermic (ΔH > 0) dehydrogenation reaction is required to achieve a system that is reversible under reasonable/practical conditions.Most potential chemical hydrogen storage materials suffer from reaction enthalpies that are too endothermic or exothermic for reversible hydrogen release. Reaction kinetics can also be a significant problem. Indeed, for many years, the only known examples of hydrogen activation under mild conditions by non-metals involved complexes only isolable in matrices.26 However, there have been several recent examples of isolable non-metal compounds that add hydrogen under mild conditions. Power and co-workers demonstrated addition of two hydrogen molecules to a digermyne compound (Scheme 2).27 This was followed by the discovery of facile hydrogen addition across two diaryltin molecules.27 The reverse reaction, the conversion of two E–H bonds to hydrogen and an E–E bond, has been demonstrated by Himmel and co-workers for a B–H species.28 Bertrand, Schoeller, and co-workers designed N-heterocyclic carbene analogues, with an N-aryl group substituted with a carbon group, which add hydrogen under mild heating.29 Stephan and co-workers reported the reversible heterolysis of hydrogen over a phosphinoborane species [1,4-(R2P)C6F4(BR2)] to make a phosphonium-borate zwitterionic compound.30 Finally, mixtures of Lewis acids (BR3) and Lewis bases (PR′3,31N-heterocyclic carbenes,31,32 and NR′333) that are typically too sterically encumbered to form a dative bond (frustrated Lewis pairs) are capable of adding hydrogen under mild conditions to form borate salts. The ease of hydrogen addition depends on the sterics and electronics of the frustrated Lewis pairs. All of these systems have very low gravimetric capacity, but studying the basic reactions of hydrogen addition to non-metal systems can garner insight into the design of reversible systems with higher storage capacity.
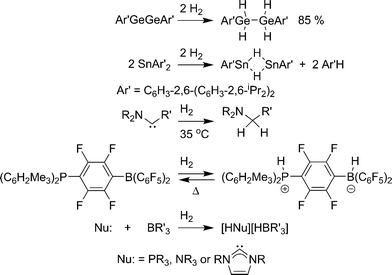 | ||
| Scheme 2 Non-metal compounds that add hydrogen under mild conditions. | ||
Cyclic organic molecules have also been evaluated as potential hydrogen storage materials.34 Dehydrogenation of organic molecules is typically too endothermic for hydrogen release at moderate temperatures. However, incorporation of fused rings and heteroatoms (O and especially N) can lower the heat requirements for hydrogen release.35 Due to the large kinetic barriers, a catalyst (typically noble metal-based) is also used to aid dehydrogenation and rehydrogenation. For instance, in a recent patent, Pez and co-workers reported that N-ethylcarbazole can be hydrogenated over Ru/LiAlO2 at 1000 psi of hydrogen and 160 °C. The hydrogen can then be released over Pd/LiAlO2 at 199 °C to give 5.6 wt% hydrogen (Scheme 3). More importantly, the system can be cycled 5 times with no detectable degradation of the N-ethylcarbazole.
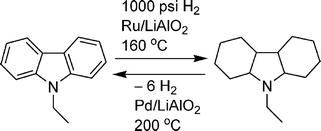 | ||
| Scheme 3 Reversible dehydrogenation of N-ethylcarbazole. | ||
There are several systems that rely on hydrolysis of active metals36 or chemical hydrides to produce hydrogen. Thorn and co-workers reported that a C–H in the 2-position of benzimidazoles is sufficiently hydridic to be protonated by a variety of acids including water over a Pd catalyst to produce hydrogen.37 Hydrolysis of another chemical hydride, sodium borohydride, has also been investigated. Although it can be stabilised in basic aqueous solutions, the addition of catalysts or acids can be used to initiate a controlled release of hydrogen by hydrolysis.38 Unfortunately, efficiently regenerating the BO2− to BH4− is difficult due to the stability of B–O bonds.
Under the current technology, chemical hydrides with high storage capacity will require off-board regeneration, which adds to the complexity of the hydrogen storage system. However, these compounds could offer advantages for the fuel distribution system. Current fuel distribution is based on transporting liquid hydrocarbons; converting this system to transport compressed or liquid hydrogen will be an expensive endeavour (potentially costing trillions of dollars).39 A stable chemical hydride could circumvent this problem, allowing for transportation of hydrogen using the existing infrastructure.
B–N compounds
Many of the previously mentioned hydrogen storage materials have gravimetric material capacities that may be too low for on-board hydrogen storage requirements. However, there are several B–N compounds with the potential to meet these requirements. B–N compounds are well suited for hydrogen storage because both boron and nitrogen are lightweight elements capable of bearing multiple hydrogens. Also, B–H and N–H bonds tend to be hydridic and protic, respectively, resulting in normally facile hydrogen release.40 A successful B–N hydrogen storage material must contain multiple hydrogen equivalents per main-group element, have a good match between the number of hydridic B–H bonds and protic N–H bonds, and have the stability requirements necessary for safe storage of hydrogen. Several classes of B–N materials that may be suitable for hydrogen storage applications will be mentioned, and then the simplest B–N compounds, amine boranes, will be covered in more detail.Hydrazine borane compounds
Much of the early interest in hydrazine borane compounds was centred around their use as propellants.41 Both hydrazine borane and -bis(borane) have been synthesised by various methods, the most common of which is addition of hydrazine salts to borohydride (Scheme 4). Hydrazine borane [-bis(borane)] has a potential hydrogen storage capacity of 13.1 [13.4] wt% (assuming loss of 3[4] eq. of H2). Unfortunately, hydrazine bis(borane) is a shock-sensitive explosive,42 explodes when heated in air,43 and is therefore poorly suited for hydrogen storage applications unless it can be stabilised.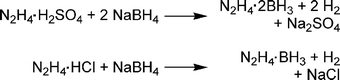 | ||
| Scheme 4 Synthesis of hydrazine bis(borane) and hydrazine monoborane. | ||
Amine triborane compounds
The first high yield synthesis of ammonia-triborane (H3N–B3H7) was reported by Kodama and co-workers in 1959.44 Tetraborane (B4H10) is treated with tetrahydropyran to yield tetrahydropyran-triborane and a half equivalent of diborane. Tetrahydropyran can be readily displaced by ammonia to yield the product. Unfortunately, tetraborane and diborane are toxic gasses causing large-scale synthesis by this route to be impractical. Yoon and Sneddon reported an alternate synthesis of ammonia-triborane in which Bu4NB3H845 salt (Scheme 5) is treated with 0.5 eq. I2 in glyme to yield glyme–B3H7, Bu4NI and 0.5 eq. of H2.46 Similar to the previous method, glyme is displaced by ammonia to yield the product. The structure of ammonia-triborane was established by X-ray methods and has a trigonal arrangement of B atoms with one bonded to ammonia.47 Ammonia-triborane has a storage potential of 10.6 wt% (assuming 3 eq. H2 are released), but direct dehydrogenation methods have yet to be reported. Kodama noted that upon treatment with sodium in ammonia, one equivalent of hydrogen is released giving a mixture of products including NaBH4. Sneddon found that H3N–B3H7 undergoes rapid hydrolysis after treatment with 1 M HCl (7.85 eq. of H2 after 120 min) or various metal catalysts, the best being Rh/Al2O3 (approx. 7.5 eq. after 25 min at 21 °C with 7 mol% catalysts).46 Similar to borohydride hydrolysis, products with B–O bonds are produced which will be energetically costly to regenerate.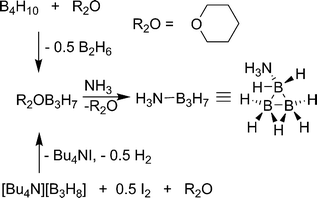
Ammonium hydrotriborate ([NH4][B3H8]) has also been synthesised by treatment of pentaborane with ammonium hydroxide.48 Surprisingly, this is a stable, colourless, crystalline solid in pure form, and no decomposition was evident on heating the solid at 60 °C for 70 hours. It is apparently stable in water and alcohols and slowly decomposes to form H3N–B3H7 and H2 when treated with benzene or ether. This is in stark contrast to [NH4][BH4]49 which decomposes at temperatures above −40 °C. Ammonium hydrotriborate has a potential hydrogen storage capacity of 13.9 wt% (assuming loss of 4 eq. H2). Hydrogen loss from alkylammonium hydrotriborate salts has been realised by addition of metallic Si or Al (Scheme 6).50 When [NH3Me][B3H8] is treated with Si, 5 eq.51 of hydrogen gas are released giving 9.9 wt% H2.
 | ||
| Scheme 6 Synthesis of ammonium hydrotriborate and dehydrogenation of alkylammonium salts. | ||
Guanidinium hydrotriborate, [C(NH2)3][B3H8], has been prepared by treating di(guanidinium) sulfate with sodium hydrotriborate.52 At ∼100 °C, this compound violently decomposes to form 6.2 eq. H2 (by MS), which provides 12.3 material wt%. It was noted that some decomposition was evident if the compound was stored at 20 °C for ∼2 months. Also, triaminoguanidinium hydrotriborate, [C(NHNH2)3][B3H8] was synthesised by treatment of pentaborane with triaminoguanidine.53 A decomposition point was not reported, but a melting point of 72–78 °C was determined. The potential hydrogen storage capacity is 11.0 wt% (assuming loss of 8 eq. H2).
It is important to note that these polyborane compounds are likely to be explosive, as many related compounds have been used as rocket propellants.
Amine compounds of higher-order polyboranes
Borane has a very rich chemistry, and there are many polyborohydride compounds known. When ammonia is bubbled through a solution of decaborane (B10H14), the tris(ammoniate) of decaborane (TAD) is formed.54 This compound likely has a [NH4][B10H13(NH3)2] formulation, and one of the NH3 groups is only loosely bound. This formulation is thus similar to the compound [NH2Et2][B10H13(NHEt2)].55 Although TAD is stable under ambient conditions, upon heating to 75 °C, one equivalent of hydrogen and one equivalent of ammonia are released. Complete conversion to BN compounds was achievable by addition of hydrazine to TAD, and heating to 800 °C under an ammonia stream.56 It is noteworthy that decaborane reacts vigorously with hydrazine to induce a fire in air.In a theoretical study, Nguyen, Matus, and Dixon investigated the heats of formation of several ammonium salts of polyboranes.57 They found that formation of [B12H12][NH4]2 from H2 (10 eq.), BN (2 eq.), and B (10 eq.) has a heat of formation of only 10 to 12 kcal mol−1 (the reverse reaction would result in loss of 8.8 wt% of H2 from [B12H12][NH4]2). In a recent DOE progress report, Hawthorne and co-workers investigated a series of anionic polyborane compounds for hydrogen storage applications.58 Ammonium salts of (B11H14)−, (B12H12)2−, and (B10H10)2− were all synthesised and hydrolyzed in the presence of a metal catalyst to yield hydrogen, ammonium borate, and boric acid (Scheme 7).
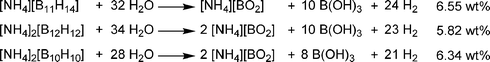 | ||
| Scheme 7 Hydrolysis of ammonium polyborane salts. | ||
Amine borane compounds
Amine boranes are the most thoroughly studied B–N hydrogen storage materials. By varying the substituents on B and N, a variety of properties can be altered such as melting and decomposition points as well as dehydrogenation enthalpy and nature of the reaction products. Nöth and Beyer investigated the physical properties of a variety of alkylamine boranes obtained by addition of the alkylammonium salt to lithium borohydride (Table 1).59 An alternative way to synthesise these compounds is by substitution of H3B·L (L = Me2S, THF, Me3N) with the amine.60 The physical properties are difficult to predict. For instance, H2EtNBH3 is one of the least stable of the group of monoalkylamine boranes, whereas HEt2NBH3 is one of the most stable dialkylamine boranes. Linear H2nBuNBH3 decomposes above 10 °C, whereas the branched isomer, H2tBuNBH3, is stable up to 120 °C.| Alkylamine borane | Melting point/°C | Decomp. point/°C |
|---|---|---|
| a Table adapted from ref. 59. | ||
| H3NBH3 | 104 | ∼100 |
| H2MeNBH3 | 56 | 70 |
| H2EtNBH3 | 19 | 30–40 |
| H2nPrNBH3 | 45 | 50–70 |
| H2iPrNBH3 | 65 | 90–100 |
| H2nBuNBH3 | −48 | 10–15 |
| H2tBuNBH3 | 96 | 120–140 |
| HMe2NBH3 | 37 | 150 |
| HEt2NBH3 | −18 | 200 |
| HnPr2NBH3 | 30 | 140 |
| HiPr2NBH3 | 23 | 250 |
| HnBu2NBH3 | 15 | 120 |
| HiBu2NBH3 | 19 | 150 |
Hawthorne reported that B-substituted amine boranes can be prepared by reduction of alkylboroxines [(BOR)3] using lithium aluminium hydride in the presence of trimethylamine.61 The majority of the Me3NBRH2 (R = nPr, iPr, nBu, 2-Bu, iBu, tBu, n-pentyl, and n-hexyl) compounds are liquids at room temperature with the exception of R = cyclohexyl (mp = 40–41 °C) and benzyl (mp = 58–60 °C). Treatment of these compounds with excess ammonia in the presence of catalytic ammonium chloride at 100–150 °C affords B-substituted borazines, [HNBR]3, in moderate to good yield (65–91%).62
In a theoretical study, Manners, Harvey and co-workers investigated the effect of B- and N-substituents on the ΔG and ΔH of dehydrogenation of HR2NBR′2H.63 Loss of hydrogen from amine boranes normally is too exergonic for reversibility (see the section: Thermal dehydrogenation of amine boranes). By altering the substituents (R and R′) on HR2NBR′2H, however, the ΔG of dehydrogenation can be made more neutral. A strong dative N–B σ-bond in the reactant (HR2NBR′2H) and a weak dative N–B π-bond in the product (R2NBR′2) results in a less exergonic dehydrogenation. In general, the σ-bond plays a more important role in determining the overall energetics, so HR2NBR′2H compounds with electron donating groups on nitrogen (resulting in a more Lewis-basic amine) and electron withdrawing groups on boron (resulting in a more Lewis-acidic borane) are best suited for reversible dehydrogenation. Evaluation of a series of cyclic amines indicates that four- and five-membered rings exhibit very similar dehydrogenation enthalpies, whereas mechanisms involving six-membered rings are more endothermic. This appears to be an effect of added ring strain going from an sp3- to an sp2-hybridised nitrogen centre.
Hydrogen loss from amine boranes is typically effected by solvolysis (including acid- and metal-catalysed variants) or thermolysis, in which the product distribution depends on the reaction conditions and presence of additives or catalysts. The easiest way to evaluate the extent of hydrogen loss is by measuring the amount of hydrogen gas generated by either Toepler pump or GC/MS methods. Gas burette and thermogravimetric analysis (TGA) may also be used, but other gaseous products may also contribute to the volume measured without a method for direct identification or quantification. Temperature-programmed desorption (TPD) provides a means to analyze the volatile products by MS. If soluble products are formed, NMR spectroscopy (especially 1,2H, 11B, and 14,15N)64 can also give a good indication of the extent of dehydrogenation by both the chemical shift and multiplicity of peaks formed (Fig. 2). The wide chemical shift range for 15N NMR is particularly useful, with sp2-hybridised N in substituted borazines from −230 to −280 ppm and sp3-hybridised N in alkylamine boranes from −340 to −375 ppm.65 Many of these nuclei are quadrupolar and observed line widths will depend on the electric field gradient at the nucleus and the nuclear correlation time. Large molecules with low symmetry (around the quadrupolar nucleus) in viscous solvents will thus give rise to broad resonances. The increased molecular motion and decreased viscosity at higher temperatures can often be used to reduce linewidths in solution NMR experiments. Conversely, cooling the sample effectively ‘decouples’ the quadrupolar interaction to adjacent nuclei thus reducing line widths of, for example, 1H and 13C NMR signals in alkylboranes. Finally, double rotation experiments or use of high magnetic fields allows for useful information to be obtained for quadrupolar nuclei in the solid state as well.66
Thermal solvolysis of amine boranes
One of the conceptually easiest ways to release hydrogen from molecules with B–H bonds is by solvolysis (see borohydride and polyborane hydrolysis). Reactions of amine boranes with alcohol or water are thermodynamically downhill. However, high temperatures (above the boiling point of water) are necessary to induce hydrolysis under neutral or basic conditions.67 In a recent article, thermally-induced solvolysis was used in two different ways.68 The first capitalised on an exothermic hydrogen release to induce a self-sustained reaction, and the second relied on pressurising water to increase its boiling point. A gelled mixture of AB, Al, and H2O in a 2 : 3 : 3 ratio can be ignited. The excess heat generated causes the reaction to be self-sustaining, releasing 7.7 wt% of hydrogen in the process. Also, a mixture of water and AB can be heated to 135 °C (under Ar pressure) to release 3 equivalents of hydrogen. The reaction was very clean by 11B-NMR and MS; no borazine formation was detected.Acid-catalysed solvolysis
Acid-catalysed hydrolysis is the oldest known process for releasing hydrogen from amine boranes (Scheme 8).69 It is likely that the acid functions by protonating the amine, which releases BH3 for subsequent hydrolysis. The nature of the amine has a profound influence on the reaction rate. For instance, AB is hydrolysed 600 times faster than H2MeNBH3 and 4.8 × 104 times faster than HMe2NBH3. More interesting from an engineering point of view is successful use of immobilised acids such as ion exchange resins or various zeolites to activate ammonia borane.70 Even CO2 (which generates carbonic acid in situ) proved to be a catalyst for the dehydrogenation (albeit with a slow rate). | ||
| Scheme 8 Acid catalysed hydrolysis of ammonia borane. | ||
Metal-catalysed solvolysis
Many metals and metal complexes have been found to catalyse amine borane solvolysis (Table 2).71–80 The recent focus in catalyst development has been on the use of non-precious metals. In two recent heterogeneous systems, catalyst morphology greatly affected catalytic activity. Hollow spheres of nickel metal were shown to exhibit substantial catalytic activity versus nickel powder. By doping the nickel hollow spheres with Pt, the rate could be increased such that three equivalents of hydrogen were released within 30 minutes.79 In a recent study, iron nanoparticles were also found to catalyse solvolysis. Borohydride reduction of Fe(SO4) results in nanoparticles that slowly catalyse hydrolysis (160 min at RT with 12% cat. loading). However, if the nanoparticles are generated in the presence of ammonia borane, a very active catalyst forms (8 min, RT, 12% cat. loading), which can be recycled without loss of activity, and was similarly effective in air.80 It was found that Fe(SO4) reduction forms crystalline material in the absence of AB, but forms amorphous nanoparticles in the presence of AB, which may account for the difference in activity.| Amine borane | Catalyst | Solvent | Eq. of H2 released | Temp/°C | Time | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | H2tBuNBH3 | 10% Pd/C (50%wet) | MeOH | Approx 3 | 30 | 100 min | 71, 72 |
| Me3NBH3 | 20 h | ||||||
| 2 | Various | 10% Pd/C (50%wet) | H2O, various alcohols | High efficiency | 20 | 5 min (MeOH) to 190 min (tBuOH) | 73 |
| Raney Ni (5 mol%) | |||||||
| 3 | H3NBH3 | Pt (20% on C) (2 mol%) | H2O | Approx 3 | 20 | 2 min | 74 |
| [Rh(1,5-cod)(μ-Cl)]2 (2 mol%) | Approx 2.7 | 20 min | |||||
| Pd (2 mol%) | Approx 2.5 | 250 min | |||||
| 4 | H3NBH3 | Dowex (12 wt%) | H2O | Approx 2.8 | 20 | 8 min | 70 |
| CO2 | No AB left | 7 days | |||||
| 5 | H3NBH3 | Co (10% on C) (2 mol%) | H2O | Approx 2.9 | 20 | 60 min | 75 |
| Ni (10% on γ-Al2O3) (2 mol%) | Approx 2.9 | 60 min | |||||
| 6 | H3NBH3 | Ni0.88Pt0.12 hollow sphere (2 mol%) | H2O | Approx 3 | 20 | 30 min | 79 |
| 7 | H3NBH3 | Rh colloids (1 mol%) | H2O | Approx 2.8 | 20 | 40 s | 76 |
| Ir colloids (1 mol%) | Approx 3 | 105 min | |||||
| Co colloids (1 mol%) | Approx 3 | 60 min | |||||
| 8 | H3NBH3 | RuCl3 (0.5 mol%) | MeOH | Approx 3 | 20 | 5 min | 77 |
| 9 | H3NBH3 | Amorphous Fe nanoparticles | H2O | Approx 3 | 20 | 8 min | 80 |
| 10 | H3NBH3 | Various Co, Ni, Cu nanoparticles | H2O | Approx 3 | 20 | 20–300 min | 78 |
Thermal dehydrogenation of amine boranes
Although amine boranes are isoelectronic to alkanes, the energetics and process of dehydrogenation are much different. Dehydrogenation of ethane to ethylene, for example, is endothermic (ΔH = 32.6 kcal mol−1) as cleavage of two strong C–H bonds is not totally compensated for by formation of H2 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π-bond. In contrast, dehydrogenation of ammonia borane to aminoborane (H2NBH2) is exothermic (ΔH = −5.09 kcal mol−1)81 as the dative B–N bond is converted into a stronger covalent one. Upon evaluating the kinetics of intramolecular hydrogen loss from ammonia borane in the gas phase, Dixon and co-workers found that the intramolecular activation barrier (32–33 kcal mol−1)82 is actually larger than the B–N bond dissociation energy (25.9 kcal mol−1).83 According to this result, AB should dissociate into NH3 and BH3 before H2 loss. However, the newly formed BH3 can catalyse AB dehydrogenation through a six-membered transition state (Scheme 9) at a barrier only 6.1 kcal mol−1 higher in energy than separated AB and BH3.
C π-bond. In contrast, dehydrogenation of ammonia borane to aminoborane (H2NBH2) is exothermic (ΔH = −5.09 kcal mol−1)81 as the dative B–N bond is converted into a stronger covalent one. Upon evaluating the kinetics of intramolecular hydrogen loss from ammonia borane in the gas phase, Dixon and co-workers found that the intramolecular activation barrier (32–33 kcal mol−1)82 is actually larger than the B–N bond dissociation energy (25.9 kcal mol−1).83 According to this result, AB should dissociate into NH3 and BH3 before H2 loss. However, the newly formed BH3 can catalyse AB dehydrogenation through a six-membered transition state (Scheme 9) at a barrier only 6.1 kcal mol−1 higher in energy than separated AB and BH3.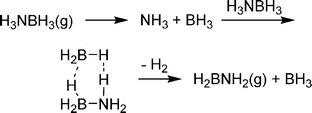 | ||
| Scheme 9 Mechanism of BH3 catalysed AB dehydrogenation. | ||
In the condensed phase, thermolysis of amine boranes such as AB and methylamine borane (H2MeNBH3, MeAB) have been shown to proceed by an intermolecular mechanism that involves initial formation of a diaminoboronium borohydride salt (Scheme 10). Further reaction of this salt with additional amine borane molecules builds up aminoborane chains with formation of a new B–N bond for each hydrogen molecule released. Computational investigations of presumed linear polyaminoborane products, H3N(BH2NH2)nBH3, showed that low energy coiled and helical conformations are favoured that feature B–H⋯H–N dihydrogen bonding.84
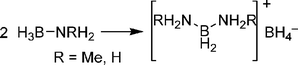 | ||
| Scheme 10 Formation of the diaminoboronium borohydride salt. | ||
Dehydrogenation of amine boranes typically yields a variety of oligomeric products depending on conditions and methods unless they are sterically blocked (Fig. 2, for AB). Dixon and co-workers calculated the thermodynamics of the formation of smaller oligomers [BxNxHy (x = 2, 3)] in both the gas and condensed phase.85 Larger oligomeric products were evaluated in the condensed phase by Miranda and Ceder.86 These products result from both a polymeric ammonia borane cycle (AB to PAB to PIB; see Fig. 2 for structures) and a cyclic oligomeric pathway (AB to CTB, borazine or 1,4-polyborazylene). While the overall reaction enthalpies depend on the products formed, all reactions in the study are estimated to be mildly exothermic [−1.6 to −20 kcal mol−1 AB]. If one considers that the entropic term contributes ∼8 kcal mol−1 H2, it is clear that direct rehydrogenation will not be possible under practical conditions and that amine boranes will need to be regenerated in a chemical process. A few products, such as borazine, are volatile. Loss of these products leads to contamination of the hydrogen stream (potentially poisoning the fuel cell), and material loss (limiting regeneration efficiency).
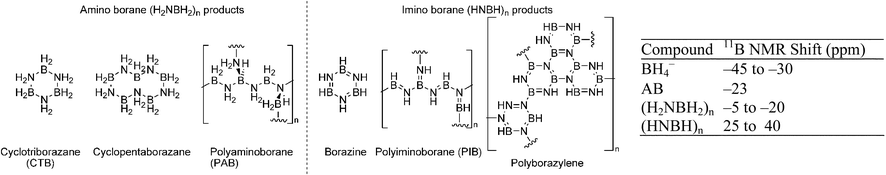 | ||
| Fig. 2 Some products of dehydrogenation from AB, and 11B NMR shifts. | ||
Solid state thermolysis
Initial studies of alkylamine borane (H2RNBH3) thermolysis (at 90–120 °C) afforded mixtures of cyclic amino- and iminoborane oligomers as well as undefined products.87 Framery and Vaultier found that heating N- and B-substituted amine boranes (H2RNBH3 or H3NBRH2) to 200 °C gave the corresponding borazine compounds in good yield.65 Detailed studies on the thermolysis of MeAB revealed that hydrogen is released in two stages, one at ∼100 °C, and the second at 190 °C. For the latter, a competing pathway to borazine formation was identified as dehydrogenative cross-linking of (HMeNBH2)3 to give an insoluble polymer.88The parent, ammonia borane, decomposes in three distinct steps (Scheme 11).89 The first commences at ∼100 °C and peaks between 107 and 117 °C with an initial weight loss of ∼1.1 eq. of dihydrogen (∼7.2 wt%). The second equivalent is lost over a much broader temperature range, with a maximum rate at ∼150 °C, and the rest released at much higher temperatures.90 Since all steps are exothermic, the high temperature requirement is a reflection of the significant kinetic barriers. The decomposition temperatures and products of dehydrogenation are dependent on the rate that the temperature is elevated. Lower temperature ramping rates result in a higher decomposition temperature. Analysis (IR and MS) of the volatile thermolysis products for the first dehydrogenation step revealed traces of B2H6, H2NBH291 and borazine accompanying the evolved hydrogen.92 Subsequent detailed 11B solid state NMR studies at high field showed that formation of a new AB mobile phase preceded formation of the diammoniate of diborane (DADB, [BH2(NH3)2][BH4]). DADB, formed from two AB molecules by a hydride transfer, actually initiates hydrogen loss and concomitant B–N bond formation.93 A similar intermediate was proposed for the thermal decomposition of MeAB.88 The second hydrogen-releasing step produces cyclic iminoborane oligomers (including borazine and B–N linked borazines, polyborazylene) whose proposed graphitic structure is reminiscent of hexagonal and rhombohedral phases of boron nitride.94
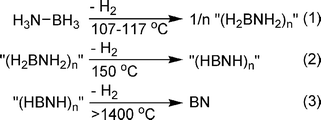 | ||
| Scheme 11 Thermolysis of ammonia borane. | ||
The dehydrogenation rate can be increased by the inclusion of additives or by intercalation of AB in a solid scaffold. Benedetto and co-workers found that AB samples doped or milled with Pt (ca. 1%) had a greater extent of H2 release at low temperatures (23% increase in H2 release at 140 °C).95 Autrey and co-workers found that a nanocomposite of mesoporous silica and AB (1 : 1 by weight) releases hydrogen at 50 °C with a half-reaction time of 85 min compared to a half-reaction time of 290 min at 80 °C for neat ammonia borane.96 The peak dehydrogenation temperature was lowered from ∼110 °C to ∼98 °C when a heating rate of 1 °C min−1 was used. Encapsulation of AB in a 24 wt% carbon cryogel97 decreased the peak dehydrogenation temperature to ∼90 °C, and there was no further decomposition at higher temperatures. Volumetric measurements indicated a 9 wt% loss of hydrogen, and no borazine formation was detected (MS).
The mechanism(s) by which the solid scaffolds increase the rate may be similar for both cases. It is possible that the observed effect may be a function of increasing the surface area of AB, as it is intercalated in nano-scale pores, which is known to lower the phase transition temperature and thereby presumably the dehydrogenation temperature. Another possibility is that dehydrogenation of AB is catalysed by the exposed functional groups (SiOH for silica and carboxylic acid for carbon cryogel) on the nanocomposite. Acid catalysed dehydrogenation of AB is well known (see below). Both mechanisms are consistent with the observation that smaller pore sizes reduce the decomposition temperature.
Solution thermolysis of ammonia borane
Thermal decomposition of AB in a variety of aprotic, polar solvents is slow and results in a mixture of cyclic amino- and iminoborane oligomeric dehydrogenation products.98 Sneddon and co-workers found that addition of ionic liquids can greatly increase both the rate and extent of dehydrogenation.99 While heating pure ammonia borane at 85 °C gave a negligible amount of dihydrogen after 3 h, heating in certain ionic liquids resulted in immediate release of dihydrogen (0.95 equivalents after 3 h at 85 °C and even 1.5 eq. after 3 h at 95 °C, compared to 0.8 eq. at 95 °C after 3 h for AB). Monitoring these reactions in situ using 11B NMR provided evidence for rapid formation and stabilisation of DADB in the ionic liquid. 11B NMR analysis of pyridine extracts of the colorless non-volatile residue indicated linear and branched acyclic aminoborane chains, such as H3N(BH2NH2)nBH3 and H3NBH(NH2BH3)2, in addition to DADB.Acid-catalysed dehydrogenation of ammonia borane
Another way to effectively dehydrogenate AB is by the addition of Lewis or Brønsted acids. Treatment of AB with the strong Lewis acid B(C6F5)3 at 25 °C in ether affords the boronium cation salt [BH2(NH3)(OEt2)][BH(C6F5)3] by hydride abstraction. Strong Brønsted acids, such as trifluoromethane sulfonic acid (HOTf), protonate a B–H bond in AB yielding hydrogen and the analogous boronium triflate. These boronium cations are more reactive versions of that found in DADB and can, as a result, initiate hydrogen release from AB even at 25 °C. Computational studies showed that the cation interacts initially with a B–H bond of AB, drawing a protic N–H in proximity to a hydridic B–H, resulting in loss of hydrogen. Further molecules of AB then interact similarly with the resultant cationic complex to build the aminoborane chains stepwise (Scheme 12). The relative concentration of acid needs to be kept low (0.5 mol%) to avoid chain termination to aminodiborane, B2H5(μ-NH2), and concentrated solutions afford high yields of borazine at 60 °C in 4 h.100![A Lewis-acidic [H2BNH3]+ molecule interacts with ammonia borane to lose hydrogen and form a new compound that is capable of attack at two positions.](/image/article/2009/CS/b800312m/b800312m-s12.gif) | ||
| Scheme 12 A Lewis-acidic [H2BNH3]+ molecule interacts with ammonia borane to lose hydrogen and form a new compound that is capable of attack at two positions. | ||
Anionic dehydropolymerisation of ammonia borane
In a recent DOE progress report, Sneddon and co-workers reported that generation of catalytic amounts of H2NBH3− increases the rate of hydrogen release.101 Metal complexes of this anion have also been investigated as potential hydrogen storage materials (see the Metal amidoboranes section). The anion can be generated in situ by addition of LiNH2, LiH, or proton sponge [1,8-bis(dimethylamino)naphthalene]. The use of proton sponge eliminates the formation of LiBH4 and NH3 side products identified when LiNH2 or LiH was used as the base. Although, the mechanism is currently unknown, it has been suggested that the increased hydricity of the B–H bond in H2NBH3− leads to facile H2 loss from its reaction with AB.102Metal-catalysed dehydrogenation of amine boranes
Metal-catalysed dehydrogenation of amine boranes offers the potential for additional control over both extent and rate of hydrogen production.103–113 In a 1989 patent, Laine and Blum claimed the dehydrogenation of several amine boranes using Ru3(CO)12 at 60 °C (Table 3; 8–10).103 Using a heterogeneous Pd/C catalyst, Roberts and co-workers reported conversion of HMetBuNBH3 to the corresponding aminoborane at 120 °C.104 Despite the wide range of catalytic systems developed subsequently (Table 3), there has yet to be a catalyst capable of both a high rate and large extent of hydrogen release.105–113 Moreover, if these systems are to be practical, base metals need to be used at low catalyst loadings. Finally, engineering controlled hydrogen release has heretofore only been demonstrated for solid catalysts so effective heterogenisation strategies will be required.| Catalyst (mol%) | Substrate | Conditions | Products | Eq. of H2 | Ref. | |
|---|---|---|---|---|---|---|
| a Inferred from reported TOF values.b CTB is cyclotriborazane.c NR is “not reported.”d Yield of products not quantified; other products possible.e Isolated yields, actual yield will be higher but other products detected.f κ3-2,6-[OP(t-Bu)2]2C6H3.g Enders’ carbene: (1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene).h IDipp is 1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene. | ||||||
| 1 | Cp2TiMe2 (0.5%) | HMe2NBH3 | 16 h, 25 °C | No reaction | 0 | 106 |
| 2 | Cp2Ti (2%) | HMe2NBH3 | 4 h, 20 °C | (Me2NBH2)2 | 1 | 110 |
| 3 | Cp2Ti (2%) | H(i-Pr)2NBH3 | 1 h, 20 °C | iPr2NBH2 | 1 | 110 |
| 4 | {[Cp(SiMe3)2]2Ti}N2 (2%) | HMe2NBH3 | 7 min, 23 °Ca | (Me2NBH2)2 | 1 | 111 |
| 5 | {[Cp(SiMe3)2]2Ti}N2 (2%) | H3NBH3 | 161 h, 65 °Ca | CTBb, borazine | NRc | 111 |
| 6 | [indenyl-(SiMe3)2]2Zr | HMe2NBH3 | 147 h, 65 °Ca | (Me2NBH2)2 | 1 | 111 |
| 7 | [P(iPr)3]2Br2(CH3CN)(NO)Re (1%) | HMe2NBH3 | 4 h, 85 °C | (Me2NBH2)2 | 1 | 105 |
| 8 | Ru3(CO)12 (0.2%) | H3NBH3 | 85 h, 60 °C | BN1.13H4.7 (elemental analysis) | NR | 103 |
| 9 | Ru3(CO)12 (0.1%) | Me3NBH3, PrNH2 | 32 h, 60 °C | [–N(Pr)B(H)–]3 (57%) | NR | 103 |
| 10 | Ru3(CO)12 (0.1%) | Me3NBH3, MeNH2 | 9.5 h, 60 °C | [–B(NMeH)N(Me)–]3, B(NHMe)3d | NR | 103 |
| 11 | trans-RuMe2(PMe3)4 (0.5%) | HMe2NBH3 | 16 h, 25 °C | (Me2NBH2)2 | 1 | 106 |
| 12 | FeH(PMe2CH2)(PMe3)3 (9%) | H3NBH3 | 96 h, 25 °C | CTB, borazine, polyborazylened | NR | 108 |
| 13 | [Rh(1,5-cod)(μ-Cl)]2 (0.5%) | HMe2NBH3 | 8 h, 25 °C | (Me2NBH2)2 | 1 | 106 |
| 14 | [Rh(1,5-cod)(μ-Cl)]2 (5%) | HMe2NBH3 | <2 h, 25 °C | (Me2NBH2)2 | 1 | 106 |
| 15 | RhCl3 (0.5%) | HMe2NBH3 | 22.5 h, 25 °C | (Me2NBH2)2 (90%) | 0.9 | 106 |
| 16 | HRh(CO)(PPh3)3 (0.5%) | HMe2NBH3 | 160 h, 25 °C | (Me2NBH2)2 (5%) | 0.05 | 106 |
| 17 | [Cp*Rh(μ-Cl)Cl]2 (0.5%) | HMe2NBH3 | 112 h, 25 °C | (Me2NBH2)2 | 1 | 106 |
| 18 | [Rh(1,5-cod)(μ-Cl)]2 (0.5%) | H(1,4-C4H8)NBH3 | 24 h, 25 °C | [(1,4-C4H8)NBH2]2 (73%)e | NR | 106 |
| 19 | [Rh(1,5-cod)(μ-Cl)]2 (0.5%) | HMe(PhCH2)NBH3 | 24 h, 25 °C | [Me(PhCH2)NBH2]2 (79%)e | NR | 106 |
| 20 | [Rh(1,5-cod)(μ-Cl)]2 (1%) | H2MeNBH3 | ∼60 h, 45 °C | (MeNBH)3 (40%)e | NR | 106 |
| 21 | [Rh(1,5-cod)(μ-Cl)]2 (0.6%) | H2PhNBH3 | 16 h, 45 °C | (PhNBH)3 (56%)e | NR | 106 |
| 22 | [Rh(1,5-cod)(μ-Cl)]2 (0.6%) | H3NBH3 | ∼60 h, 45 °C | Borazine (10%),e PIB, polyborazylene | NR | 106 |
| 23 | [Rh(1,5-cod)(μ-Cl)]2 (1%) | HiPr2NBH3 | 24 h, 25 °C | (iPr)2NBH2 (49%)e | NR | 106 |
| 24 | [Ir(1,5-cod)(μ-Cl)]2 (0.5%) | HMe2NBH3 | 136 h, 25 °C | (Me2NBH2)2 (95%) | 0.95 | 106 |
| 25 | (POCOPf)Ir(H)2 (0.5%) | H3NBH3 | 14 min, 20 °C | Cyclopentaborazane | 1 | 113 |
| 26 | Ni(1,5-cod)2, 2 NHCg (9%) | H3NBH3 | 3 h, 60 °C | Polyborazylened | 2.8 | 108 |
| 27 | Pd/C | HMetBuNBH3 | 1 h, 120 °C | [MetBuNBH2]2 | 1 | 104 |
| 28 | Pd/C (0.5%) | HMe2NBH3 | 68 h, 25 °C | (Me2NBH2)2 (95%) | 0.95 | 106 |
| 29 | (IDipp)hCuCl (12.5%) | HMe2NBH3 | 24 h, 20 °C | (Me2NBH2)2d | NR | 108 |
Under the heavily reducing conditions of amine borane dehydrogenation, the metal complex catalyst precursor will often undergo changes. Frequently, the active species is much different than the precatalyst. Manners and co-workers found that [Rh(1,5-cod)(μ-Cl)]2 catalyses the dehydrogenation of a variety of amine boranes at room temperature or with mild heating (Table 3; 13, 14, 18–23).106 The analogous Ir precursor or Rh precursors with different supporting ligands exhibited much lower activity for HMe2NBH3 (DMAB) dehydrogenation (Table 3; 15–17, 24). The dehydrogenation of DMAB using [Rh(1,5-cod)(μ-Cl)]2 exhibits an induction period, during which a black, opaque suspension forms. TEM analysis indicated Rh aggregation; the UV-Vis spectrum was similar to the spectrum of Rh colloids; addition of Hg (either at the onset, or during the reaction progress) resulted in complete loss of activity; and although the dark powder isolated from catalysis still had activity, the solution after filtration had almost no activity. These observations all point to Rh(0) colloids as the catalytically-active species. However, subsequent in situ EXAFS (extended X-ray absorption fine structure) analysis found that the same catalytic precursor, under different conditions, may form soluble Rh clusters that also catalyse this reaction.107 These Rh4–6 clusters, observed in solution during the reaction, eventually precipitate, likely due to a ligand exchange process with the formed products. The clusters can be redissolved by treatment with DMAB. The process of forming colloids or clusters can be complicated, and minor variations in conditions can have a profound influence on metal species development. However, it is clear in both cases that the precatalyst is drastically altered during the reaction.
Minor alterations to supporting ligands can also greatly influence the catalytic activity. Baker and co-workers measured the relative catalytic activity of N-heterocyclic carbene (NHC) complexes of Ni.108 The Enders’ carbene Ni complex was found to be 11.5 times faster than the IDipp complex and 8.8 times faster than the IMes complex.109 Also, the Enders’ Ni complex was 4.1 times faster than a Rh(NHC) complex and 1.9 times faster than a Ru(NHC) complex. The Enders’ carbene Ni complex has the largest extent of dehydrogenation yet seen (>2.5 eq.) but requires mild heating. Initially, a minor amount of borazine is formed. However, borazine reacts further by crosslinking reactions to form polyborazylene. Using borazine as the substrate under similar catalytic conditions to AB results in almost no activity, indicating that AB either activates the catalyst or is involved in the crosslinking reactions. During the course of the reaction a dark, homogeneous maroon solution forms. Hg addition results in no loss of activity, which is indicative that metal colloids are not the active catalyst. A kinetic isotope effect (KIE) is observed for D3NBH3, H3NBD3, and D3NBD3 (2.3, 1.7, and 3.0, respectively) indicating that both N–H and B–H bond cleavage are involved in the turnover limiting step or that N–H and B–H bond cleavage steps have similar rates.
Solvent and substrate can also play a significant role. An example of this was noted in the titanocene-based dehydrogenation of amine boranes initially reported by Manners and co-workers (Table 3; 2, 3)110 and extended by Chirik and co-workers to include other Cp-based ligands and Zr (Table 3; 4–6).111 Chirik and co-workers noted that {[η5-C5H3(SiMe3)2]2Ti}N2 has a TOF of >420 h−1 in benzene-d6 and 0.29 h−1 in THF for the dehydrogenation of DMAB. Also, AB was found to have a much slower rate than DMAB (Table 3; 4 and 5).
Finally, the catalyst identity controls the extent of dehydrogenation. Heinekey, Goldberg and co-workers reported that (POCOP)IrH2112 is an extremely active catalyst for AB dehydrogenation (Table 3; 25).113 Unfortunately, only a single equivalent of hydrogen is released per equivalent of AB. During the course of the reaction, an insoluble colorless precipitate is formed. The X-ray powder diffraction and IR data agree closely with that previously reported for cyclopentaborazane, [H2NBH2]5.114 However, Manners et al. found that alkylamine boranes, and mixtures of alkylamine boranes and AB under similar conditions gave soluble aminoborane polymers.115 The measured wide angle X-ray scattering pattern and the IR for the white precipitate from the (POCOP)IrH2 catalysed dehydrogenation of AB were different from that reported for [H2NBH2]5. They suggested that the white precipitate may be polymeric in nature similar to the products formed from alkylamine borane dehydrogenation. Although the actual nature of the precipitate has not yet been completely established, it is clear from the solid state 11B NMR data (−18 ppm) and the measured amount of hydrogen released that an [H2NBH2]n product is formed.
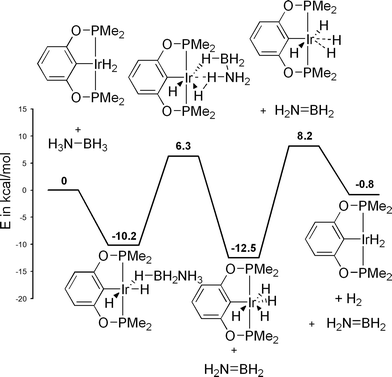
The mechanisms of metal-catalysed dehydrogenation have been investigated by computational methods for the titanocene system reported by Manners, the (POCOP)IrH2 system reported by Goldberg and Heinekey, and the Ni(NHC)2 system reported by Baker. Although the relative energies of intermediates are different, the overall reaction pathways are similar. In the initial step, B–H coordinates to the metal complex. In the titanocene case, the N–H is activated followed by hydride transfer from B–H (Scheme 13).116 Two mechanisms were evaluated in the (POCOP)IrH2 case. The first follows generation of monovalent (POCOP)Ir with a concerted B–H and N–H bond activation at the IrI centre. The second one involves a concerted B–H and N–H bond activation at trivalent (POCOP)IrH2 (Scheme 14). In the second mechanism, a hydride ligand acts as a proton acceptor yielding pentavalent (POCOP)IrH4.117 In the Ni(NHC)2 case, N–H deprotonation is proposed to occur at the Ni-bound NHC carbon, forming a coordinated imidazolium-type ligand (Scheme 15).118 The imidazolium subsequently protonates the Ni centre and hydrogen formation follows. The proposed mechanism is not consistent with the observed KIE, so further investigation is warranted.
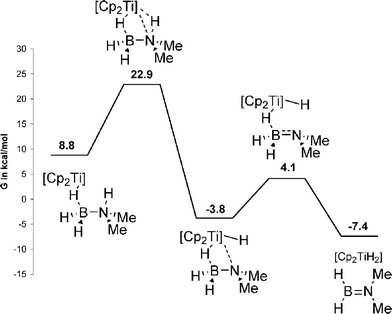 | ||
| Scheme 13 Mechanism of TiCp2 catalysed dehydrogenation of Me2HNBH3 as proposed by Luo and Ohno.116 Gibbs free energies corrected for toluene (CPCM). | ||
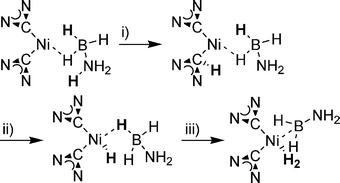 | ||
| Scheme 15 Simplified partial mechanism for NHC2Ni catalysed dehydrogenation of AB. (i) Protonation of an NHC ligand. (ii) Transfer of a proton to metal centre. (iii) Formation of hydrogen. | ||
All mechanisms proposed to date are focused on the initial loss of hydrogen from amine boranes. Establishing the role of the metal complex in subsequent oligomerisation and loss of the second equivalent of hydrogen will also be very important.
Regeneration of spent fuel
Spent fuel from solvolysis methods
The advantage of solvolysis methods is their high efficiency, chemical robustness, and the fact that inexpensive base-metal catalysts have been developed. A significant drawback of these systems is that B–H bonds are converted to much stronger B–O bonds, resulting in a more exothermic reaction than dehydrogenation. Regeneration of spent fuel from rehydrogenation methods requires strong reducing agents. Ramachandran and co-workers demonstrated a system based on transition metal catalysed solvolysis of AB to yield [NH4][B(OMe)4], which could be converted back to AB by treatment with NH4Cl and lithium aluminium hydride (Scheme 16).77 It will likely be energetically costly to convert the oxidation product, Al(OMe)3, back to the complex hydride.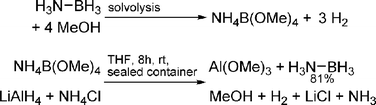 | ||
| Scheme 16 Solvolysis and subsequent regeneration of ammonia borane. | ||
Spent fuel from dehydrogenation methods
The strategies presented to date for regeneration of BNHx-spent fuel involve two important steps, digestion and reduction. Although a multitude of products can result from AB dehydrogenation, they all contain newly formed B–N linkages that must be broken. Addition of an acid (HX) can protonate these linkages, releasing amine and making B–X bonds (digestion). The B–X bonds can then be reduced by chemical reductants to form B–H bonds. A practical system must be as energetically efficient as possible, so no reaction step can be too exo- or endothermic.The method mentioned above by Ramachandran and co-workers could potentially be used to regenerate ammonia borane. However, the generation of strong B–O bonds in the reaction pathway will likely limit overall efficiency. Alternatively, Sneddon and co-workers102 and Mertens and co-workers119 have independently developed reduction schemes that form B–Br and B–Cl bonds, respectively.
Sneddon and co-workers reported that spent BNHx fuel was successfully digested using HBr/AlBr3 (super-acid) in CS2 to form H2, NH4Br, BBr3 as well as [H2NBBr2]3. The relative ratio of BBr3 to [H2NBBr2]3 depends on the material being digested. Mertens and co-workers treat a THF solution of spent fuel with an ether solution of HCl to generate BCl3, NH4Cl and H2. Both aminoborane [(BH2NH2)x] and iminoborane [(BHNH)x] materials were successfully digested. Unfortunately, the yields of BCl3 were low due to decomposition in THF. Switching to a similar super-acid solution that Sneddon and co-workers used increases the yield of BCl3 to >60%. BCl3 is difficult to directly hydrodechlorinate and reduction by hydrogen requires high temperatures (600–700 °C) to yield the partially reduced product, BHCl2 (which must be removed from the side-product HCl and can subsequently disproportionate into BCl3 and B2H6). However, addition of a Lewis-base such as NMe3 reduces the hydrodechlorination temperature to 200 °C (but still requires high pressures of 2000 atm).120 Unfortunately, these conditions are energetically costly, and the yield is poor (25%). However, a similar concept can be used for reduction of BX3 compounds by chemical hydrides. Another advantage offered by a Lewis-base is that it eliminates formation of B2H6, a hazardous material. Thus, Sneddon and co-workers treat BBr3 with N,N′-diethylaniline, which yields a compound easily reduced by triethylsilane under mild conditions. Mertens and co-workers found that stronger bases such as NEt3, inhibit the reduction of BCl3 by triethylsilane and MgH2. Addition of a weaker base, NPh3, allows for the complete reduction of Ph3NBCl3 by MgH2 after 20 minutes at 80 °C. The final step is displacement of R3N with ammonia, to yield H3NBH3.
In a DOE progress report regarding Los Alamos National Laboratory’s effort toward BNHx-spent fuel processing, effective digestion of polyborazylene with 1,2-benzenedithiol was demonstrated to form ammonia adducts of dithioboron compounds.121 These compounds contain relatively weak B–S bonds that are readily reduced by Sn–H. Optimisation of reaction conditions and energy efficiency are still being developed, but this is an intriguing result.
Horizons
Despite the vast amount of progress made on understanding and controlling the properties of B–N compounds, more work needs to be accomplished for these materials to be practical hydrogen storage materials. From an engineering point of view, fuels and spent fuels that are liquids are preferable, especially for materials that require off-board regeneration. A tailored mixture of B–N compounds, such as substituted amine boranes, could possibly be mixed with AB to afford such a liquid fuel that could still maintain acceptable gravimetric storage capacity. Combinations of this fuel mix with solid catalysts that maintain acceptable extent and rate of hydrogen release would still need to generate a pure hydrogen stream for the fuel cell and a spent fuel that can be regenerated in high yield with minimum energy input. Given the energy required just to transport the spent fuel for chemical processing, research is underway to discover new B–N hybrid compounds that are capable of on-board reversible hydrogen storage. Two promising examples are metal amidoboranes and C–B–N compounds.Metal amidoboranes
Just as complex hydrides use main-group elements to increase the gravimetric capacity of metal hydrides, metal amidoboranes use metals to potentially decrease the enthalpy of hydrogen loss, eliminate formation of volatile coproducts (increasing purity of hydrogen stream), and help control the rate of H2 release. AB is readily deprotonated by a variety of metal hydrides to form a new family of metal amidoboranes. MI(NH2BH3) (MI = Li, Na) were prepared by ball-milling solid AB with sodium- or lithium hydride.122 Thermal decomposition of these materials was monitored using TPD, coupled with an MS analyser. Under the conditions of measurement, the Li and Na compounds released hydrogen at considerably lower temperatures (89 and 92 °C, respectively) than AB (108 °C). Formation of borazine, observed at 154 °C for AB, was not observed in the case of the alkali-metal amidoboranes. Both compounds have shorter B–N bond distances than that in AB, which may be an indication of a stronger dative bond.123 The different dative bond may be responsible for the different decomposition temperature, but this is still a matter of speculation.Burrell and co-workers synthesised Ca(NH2BH3)2 by addition of AB to CaH2 in THF.124 A THF adduct is formed, but the THF can be removed in vacuo. While this compound is reported to lose hydrogen at 90 °C, only <0.3 equivalents of H2 are released. Slowly ramping the temperature to 170 °C results in 3.6 equivalents of H2 released (corresponding to 7.2 material wt%) with <0.1% ammonia and borazine released.
In addition to expanding the range of metals used in metal amidoborane complexes, the role of dopants or catalysts in controlling dehydrogenation, and possibly rehydrogenation, needs to be explored.
C–B–N compounds
Another strategy to obtain reversible chemical hydrogen storage materials is through C–B–N compounds. Since dehydrogenation of C–C bonds is significantly endothermic, new compounds containing both C–C and B–N bonds may be tailored to undergo dehydrogenation with ΔG≈ 0. Indeed, computational results from Dixon and co-workers show that replacement of 2 methylene groups of cyclohexane with an H2NBH2 group gives a material with 7.1 wt% hydrogen for loss of 3 eq. of H2 and ΔH = 23.5 kcal mol−1 H2 (Scheme 17).125 Similar compounds have been reported recently and identification of suitable dehydrogenation catalysts is in progress.126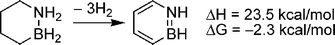 | ||
| Scheme 17 Calculated energy of dehydrogenation for 1,2-azaboracyclohexane. | ||
Conclusions
The high gravimetric capacity of B–N materials makes them particularly appealing for hydrogen storage applications. It is important to note that basic research in this area also impacts several other fields such as transfer hydrogenation,33,127 ceramic precursor production, and B–N polymer synthesis.115 A wide range of materials have been synthesised, and our understanding of hydrogen release mechanisms is rapidly increasing. As the next generation of hybrid B–N materials are designed to enable energy efficient regeneration or even reversible storage, these compounds continue to show promise for practical hydrogen storage.References
- Intergovernmental panel on climate change, Climate change 2007: Synthesis Report, http://www.ipcc.ch/ipccreports/ar4-syr.htm.
- Annual Energy Outlook 2008 (Revised Early Release) http://www.eia.doe.gov/oiaf/aeo/index.html.
- Eurostat: Energy, transport and environment indicators http://epp.eurostat.ec.europa.eu/.
- F. X. Han, J. S. Lindner and C. Wang, Naturwissenschaften, 2007, 94, 170 CrossRef CAS; A. Yamasaki, J. Chem. Eng. Jpn., 2003, 36, 361 CrossRef CAS.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246 CrossRef CAS.
- A. Boudghene Stambouli and E. Traversa, Renewable Sustainable Energy Rev., 2002, 6, 297 CAS.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613 RSC; T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS; H. W. Langmi and G. S. McGrady, Coord. Chem. Rev., 2007, 251, 925 CrossRef CAS.
- http://www.gm.com/explore/technology/news/2007/fuel_cells/.
- http://automobiles.honda.com/fcx-clarity/.
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 668 RSC.
- G. G. Tibbetts, G. P. Meisner and C. H. Olk, Carbon, 2001, 39, 2291 CrossRef CAS.
- J. Dong, X. Wang, H. Xu, Q. Zhao and J. Li, Int. J. Hydrogen Energy, 2007, 32, 4998 CrossRef CAS.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673 CrossRef CAS.
- P. M. Budd, A. Butler, J. Selbie, K. Mahmood, N. B. McKeown, B. Ghanem, K. Msayib, D. Book and A. Walton, Phys. Chem. Chem. Phys., 2007, 9, 1802 RSC; N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- J.-Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670 RSC.
- Two recent reviews: J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 Search PubMed; D. J. Collins and H.-C. Zhou, J. Mater. Chem., 2007, 17, 3154 CrossRef CAS.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110 CrossRef CAS.
- Y. Li and R. T. Yang, J. Phys. Chem. B, 2006, 110, 17175 CrossRef CAS.
- Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 726 CrossRef CAS.
- A. D. Lueking and R. T. Yang, Appl. Catal., A, 2004, 265, 259 CrossRef CAS.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302 CrossRef CAS.
- E. Rönnebro and E. H. Majzoub, J. Phys. Chem. B, 2007, 111, 12045 CrossRef.
- J. Yang, A. Sudik, D. J. Siegel, D. Halliday, A. Drews, R. O. Carter, III, C. Wolverton, G. J. Lewis, J. W. A. Sachtler, J. J. Low, S. A. Faheem, D. A. Lesch and V. Ozolinš, Angew. Chem., Int. Ed., 2008, 47, 882 CrossRef CAS; A. Sudik, J. Yang, D. Halliday and C. Wolverton, J. Phys. Chem. C, 2008, 112, 4384 CrossRef CAS.
- G. Soloveichik, J.-H. Her, P. W. Stephens, Y. Gao, J. Rijssenbeek, M. Andrus and J.-C. Zhao, Inorg. Chem., 2008, 47, 4290 CrossRef CAS.
- Some representative publications: Z. L. Xiao, R. H. Hauge and J. L. Margrave, Inorg. Chem., 1993, 32, 642 Search PubMed; H.-J. Himmel, L. Manceron, A. J. Downs and P. Pullumbi, Angew. Chem., Int. Ed., 2002, 41, 796 CrossRef CAS; H.-J. Himmel, L. Manceron, A. J. Downs and P. Pullumbi, J. Am. Chem. Soc., 2002, 124, 4448 CrossRef CAS; A. Köhn, H.-J. Himmel and B. Gaertner, Chem.–Eur. J., 2003, 9, 3909 CrossRef CAS.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS; Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268 CrossRef CAS.
- O. Ciobanu, P. Roquette, S. Leingang, H. Wadepohl, J. Mautz and H.-J. Himmel, Eur. J. Inorg. Chem., 2007, 4530 CrossRef CAS.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS; Y. Guo and S. Li, Inorg. Chem., 2008, 47, 6212 CrossRef CAS.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS; P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS; S. J. Geier, T. M. Gilbert and D. W. Stephan, J. Am. Chem. Soc., 2008, 130, 12632 CrossRef CAS; T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435 CrossRef CAS.
- D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CAS; V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskelä, T. Repo, P. Pyykkö and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117 CrossRef CAS.
- S. Hodoshima, H. Arai, S. Takaiwa and Y. Saito, Int. J. Hydrogen Energy, 2003, 28, 1255 CrossRef CAS.
- G. P. Pez, A. R. Scott, A. C. Cooper and H. Cheng, US Pat., 7 101 530, 2006; G. P. Pez, A. R. Scott, A. C. Cooper, H. Cheng, F. C. Wilhelm and A. H. Abdourazak, US Pat., 7 351 395, 2008; A. Moores, M. Poyatos, Y. Luo and R. H. Crabtree, New J. Chem., 2006, 30, 1675 Search PubMed; E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 2231 Search PubMed.
- M.-H. Grosjean, M. Zidoune, L. Roué and J.-Y. Huot, Int. J. Hydrogen Energy, 2006, 31, 109 CrossRef CAS.
- D. E. Schwarz, T. M. Cameron, P. J. Hay, B. L. Scott, W. Tumas and D. L. Thorn, Chem. Commun., 2005, 5919 RSC.
- S. C. Amendola, M. Binder, S. L. Sharp-Goldman, M. T. Kelly and P. J. Petillo, US Pat., 6 534 033, 2003.
- Estimated $380 million for 1.41 million vehicles in California, USA alone: J. M. Ogden, Int. J. Hydrogen Energy, 1999, 24, 709 Search PubMed.
- While one valence bond structure for ammonia borane would place a formal negative charge on B and a formal positive charge on N, in spite of charge transfer from N to B, the nitrogen actually carries a net negative charge due to the electronegativity difference between N and B. R. Hoffmann, J. Chem. Phys., 1964, 40, 2474 Search PubMed.
- L. R. Grant and J. E. Flanagan, US Pat., 4 381 206, 1983.
- F. C. Gunderloy, Jr, B. Spielvogel and R. W. Parry, Inorg. Synth., 1967, 9, 13.
- H. J. Emeleus and F. G. A. Stone, J. Chem. Soc., 1951, 840 Search PubMed.
- G. Kodama, R. W. Parry and J. C. Carter, J. Am. Chem. Soc., 1959, 81, 3534 CrossRef CAS.
- K. C. Nainan and G. E. Ryschkewitsch, Inorg. Nucl. Chem. Lett., 1970, 6, 765 CrossRef CAS; G. E. Ryschkewitsch, K. C. Nainan, S. R. Miller, L. J. Todd, W. J. Dewkett, M. Grace, H. Beall, M. F. Hawthorne and R. Leyden, Inorg. Synth., 1974, 15, 113 CAS.
- C. W. Yoon and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 13992 CrossRef CAS.
- C. E. Nordman and C. Reimann, J. Am. Chem. Soc., 1959, 81, 3538 CrossRef CAS.
- W. V. Hough and J. M. Makhlouf, US Pat., 3 313 603, 1967.
- R. W. Parry, D. R. Schultz and P. R. Girardot, J. Am. Chem. Soc., 1958, 80, 1 CrossRef CAS.
- J. E. Flanagan, US Pat., 4 166 843, 1979.
- In the original figure of this patent, (6 + n) H2 should be (6 −n) H2.
- L. V. Titov, M. D. Levicheva and G. N. Dubikhina, Izv. Akad. Nauk SSSR, Ser. Khim., 1976, 8, 1856.
- D. A. L. Carvalho and N. W. Shust, US Pat., 3 564 561, 1971.
- R. H. Toenikoetter, PhD Thesis, St. Louis Univ., Missouri, 1959; J. Williams, R. L. Williams and J. C. Wright, J. Chem. Soc., 1963, 5816 Search PubMed.
- M. F. Hawthorne, A. R. Pitochelli, R. D. Strahm and J. J. Miller, J. Am. Chem. Soc., 1960, 82, 1825 CrossRef CAS; B. M. Graybill, A. R. Pitochelli and M. F. Hawthorne, Inorg. Chem., 1962, 1, 622 CrossRef CAS.
- T. Yogo and S. Naka, J. Mater. Sci., 1990, 25, 374 CrossRef CAS.
- M. T. Nguyen, M. H. Matus and D. A. Dixon, Inorg. Chem., 2007, 46, 7561 CrossRef CAS.
- M. F. Hawthorne, S. S. Jalisatgi and A. Safronov, University of Missouri-Columbia’s Progress Towards Chemical Hydrogen Storage Using Polyhedral Borane Anion Salts, DoE Hydrogen Annual Progress Report, 2007 (http://www.hydrogen.energy.gov/pdfs/progress07/iv_b_5d_hawthorne.pdf).
- H. Nöth and H. Beyer, Chem. Ber., 1960, 93, 928 CAS.
- For a general review on amine borane synthesis and properties see: B. Carboni and L. Monnier, Tetrahedron, 1999, 55, 1197 Search PubMed.
- M. F. Hawthorne, J. Am. Chem. Soc., 1961, 83, 831 CrossRef CAS.
- M. F. Hawthorne, J. Am. Chem. Soc., 1961, 83, 833 CrossRef CAS.
- A. Staubitz, M. Besora, J. N. Harvey and I. Manners, Inorg. Chem., 2008, 47, 5910 CrossRef CAS.
- A. R. Siedle, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, London, 1988, vol. 20, ch. 2, pp. 205–306 Search PubMed; H. Nöth, in NMR: Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer-Verlag, Berlin, 1978, vol. 14 Search PubMed.
- E. Framery and M. Vaultier, Heteroat. Chem., 2000, 11, 218 CrossRef CAS.
- H. Beall and C. H. Bushweller, Chem. Rev., 1973, 73, 465 CrossRef CAS; A. Jerschow, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 63 CrossRef CAS.
- P. A. Storozhenko, R. A. Svitsyn, V. A. Ketsko, A. K. Buryak and A. V. Ul’yanov, Russ. J. Inorg. Chem. (Transl. of Zh. Neorg. Khim.), 2005, 50, 980.
- M. Diwan, V. Diakov, E. Shafirovich and A. Varma, Int. J. Hydrogen Energy, 2008, 33, 1135 CrossRef CAS.
- H. C. Kelly, F. R. Marchelli and M. B. Giutso, Inorg. Chem., 1964, 3, 431 CrossRef CAS; G. E. Ryschkewitsch, J. Am. Chem. Soc., 1960, 82, 3290 CrossRef CAS; G. E. Ryschkewitsch and E. R. Birnbaum, J. Phys. Chem., 1961, 65, 1087 CrossRef CAS; G. E. Ryschkewitsch and E. R. Birnbaum, Inorg. Chem., 1965, 4, 575 CrossRef CAS; H. C. Kelly and V. B. Marriott, Inorg. Chem., 1979, 18, 2875 CrossRef CAS; A. D’Ulivo, M. Onor and E. Pitzalis, Anal. Chem., 2004, 76, 6342 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 159, 855 CrossRef CAS.
- M. Couturier, B. M. Andresen, J. L. Tucker, P. Dubé, S. J. Brenek and J. T. Negri, Tetrahedron Lett., 2001, 42, 2763 CrossRef CAS.
- M. Couturier, J. L. Tucker, B. M. Andresen, P. Dubé, S. J. Brenek and J. T. Negri, Tetrahedron Lett., 2001, 42, 2285 CrossRef CAS.
- M. Couturier, J. L. Tucker, B. M. Andresen, P. Dubé and J. T. Negri, Org. Lett., 2001, 3, 465 CrossRef CAS; M. Couturier, B. M. Andresen, J. B. Jorgensen, J. L. Tucker, F. R. Busch, S. J. Brenek, P. Dubé, D. J. am Ende and J. T. Negri, Org. Process Res. Dev., 2002, 6, 42 Search PubMed.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190 CrossRef CAS.
- Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364 CrossRef CAS.
- T. J. Clark, G. R. Whittell and I. Manners, Inorg. Chem., 2007, 46, 7522 CrossRef CAS.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810 CrossRef CAS.
- S. B. Kalidindi, M. Indirani and B. R. Jagirdar, Inorg. Chem., 2008, 47, 7424 CrossRef CAS.
- F. Cheng, H. Ma, Y. Li and J. Chen, Inorg. Chem., 2007, 46, 788 CrossRef CAS.
- J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287 CrossRef CAS.
- D. A. Dixon and M. Gutowski, J. Phys. Chem. A, 2005, 109, 5129 CrossRef CAS.
- J. Zhang, S. Zhang and Q. S. Li, J. Mol. Struct. (THEOCHEM), 2005, 717, 33 CrossRef CAS; Q. S. Li, J. Zhang and S. Zhang, Chem. Phys. Lett., 2005, 404, 100 CrossRef CAS.
- M. T. Nguyen, V. S. Nguyen, M. H. Matus, G. Gopakumar and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 679 CrossRef CAS.
- J. Li, S. M. Kathmann, G. K. Schenter and M. Gutowski, J. Phys. Chem. C, 2007, 111, 3294 CrossRef CAS; D. Jacquemin, E. A. Perpète, V. Wathelet and J.-M. André, J. Phys. Chem. A, 2004, 108, 9616 CrossRef CAS.
- M. H. Matus, K. D. Anderson, D. M. Camaioni, S. T. Autrey and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 4411 CrossRef CAS.
- C. R. Miranda and G. Ceder, J. Chem. Phys., 2007, 126, 184703 CrossRef.
- M. P. Brown, R. W. Heseltine and L. H. Sutcliffe, J. Chem. Soc. A, 1968, 612 RSC.
- O. T. Beachley, Inorg. Chem., 1967, 6, 870 CrossRef CAS; M. E. Bowden, I. W. M. Brown, G. J. Gainsford and H. Wong, Inorg. Chim. Acta, 2008, 361, 2147 CrossRef CAS.
- M. G. Hu, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1978, 23, 249 CrossRef CAS; R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1985, 86, 375 CrossRef; V. Sit, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1987, 113, 379 CrossRef CAS; G. Wolf, J. Baumann, F. Baitalow and F. P. Hoffmann, Thermochim. Acta, 2000, 343, 19 CrossRef CAS; F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Rößler and G. Leitner, Thermochim. Acta, 2002, 391, 159 CrossRef CAS; J. Baumann, F. Baitalow and G. Wolf, Thermochim. Acta, 2005, 430, 9 CrossRef CAS.
- A good indication of whether pure boron nitride has been obtained is the IR spectrum: often, authors cite WAXS data as proof, where BN can indeed be identified, but some N–H bonds remain, which can be identified by a prominent N–H stretch band around 3600 cm−1.
- Both compounds were confirmed as volatile products in matrix isolation studies: J. D. Carpenter and B. S. Ault, Chem. Phys. Lett., 1992, 197, 171 Search PubMed.
- P. M. Kuznesof, D. F. Shriver and F. E. Stafford, J. Am. Chem. Soc., 1968, 90, 2557 CrossRef CAS.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831 RSC; M. Bowden, T. Autrey, I. Brown and M. Ryan, Curr. Appl. Phys., 2008, 8, 498 CrossRef.
- The thermal dehydrogenation of borazine to form boron nitride has elicited considerable interest due to the material properties of boron nitride. While this expands the chemical knowledge of formally the third dehydrogenation step of ammonia borane, this is of no interest for hydrogen storage purposes, as firstly, borazine is a fuel cell poison and secondly, formation of boron nitride as a thermodynamically extremely stable product makes recycling difficult. Readers who are interested in this aspect of ammonia borane chemistry are referred to, for example: A. W. Laubengayer, P. C. Moews, Jr and R. F. Porter, J. Am. Chem. Soc., 1961, 83, 1337 Search PubMed; P. J. Fazen, J. S. Beck, A. T. Lynch, E. E. Remsen and L. G. Sneddon, Chem. Mater., 1990, 2, 96 CrossRef CAS; P. J. Fazen, E. E. Remsen, J. S. Beck, P. J. Carroll, A. R. McGhie and L. G. Sneddon, Chem. Mater., 1995, 7, 1942 CrossRef CAS; D.-P. Kim, K.-T. Moon, J.-G. Kho, J. Economy, C. Gervais and F. Babonneau, Polym. Adv. Technol., 1999, 10, 702 CrossRef CAS.
- S. De Benedetto, M. Carewska, C. Cento, P. Gislon, M. Pasquali, S. Scaccia and P. P. Prosini, Thermochim. Acta, 2006, 441, 184 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578 CrossRef CAS.
- A. M. Feaver, S. Sepehri, P. J. Shamberger, A. C. Stowe, T. Autrey and G. Cao, J. Phys. Chem. B, 2007, 111, 7469 CrossRef CAS.
- J. S. Wang and R. A. Geanangel, Inorg. Chim. Acta, 1988, 148, 185 CrossRef CAS; W. J. Shaw, J. C. Linehan, N. K. Szymczak, D. J. Heldenbrandt, C. Yonker, D. M. Camaioni, R. T. Baker and T. Autrey, Angew. Chem., Int. Ed., 2008, 47, 7493 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS.
- F. H. Stephens, R. T. Baker, M. Hernandez-Matus, D. J. Grant and D. A. Dixon, Prepr. Pap. - Am. Chem. Soc., Div. Fuel Chem., 2006, 51, 573 Search PubMed.
- L. G. Sneddon, Amineborane Hydrogen Storage - New Methods for Promoting Amineborane Dehydrogenation/Regeneration Reactions, DoE Hydrogen Annual Progress Report, 2007 (http://www.hydrogen.energy.gov/pdfs/progress07/iv_b_5e_sneddon.pdf).
- L. G. Sneddon, Amineborane Based Chemical Hydrogen Storage, DoE Hydrogen Annual Merit Review, 2007 (http://www.hydrogen.energy.gov/pdfs/review07/st_27_sneddon.pdf).
- Y. D. Blum and R. M. Laine, US Pat., 4 801 439, 1989.
- I. G. Green, K. M. Johnson and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1989, 1963 RSC.
- Y. Jiang and H. Berke, Chem. Commun., 2007, 3571 RSC.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962 RSC; C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS; C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 9776 CrossRef CAS.
- Y. Chen, J. L. Fulton, J. C. Linehan and T. Autrey, J. Am. Chem. Soc., 2005, 127, 3254 CrossRef CAS; J. L. Fulton, J. C. Linehan, T. Autrey, M. Balasubramanian, Y. Chen and N. K. Szymczak, J. Am. Chem. Soc., 2007, 129, 11936 CrossRef CAS.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS.
- Enders’ carbene: 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene; IMes: 1,3-bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene; IDipp: 1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene.
- T. J. Clark, C. A. Russell and I. Manners, J. Am. Chem. Soc., 2006, 128, 9582 CrossRef CAS.
- D. Pun, E. Lobkovsky and P. J. Chirik, Chem. Commun., 2007, 3297 RSC.
- POCOP is κ3-2,6-[OP(t-Bu)2]2C6H3.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS.
- K. W. Böddeker, S. G. Shore and R. K. Bunting, J. Am. Chem. Soc., 1966, 88, 4396 CrossRef.
- A. Staubitz, A. P. Soto and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 6212 CrossRef CAS.
- Y. Luo and K. Ohno, Organometallics, 2007, 26, 3597 CrossRef CAS.
- A. Paul and C. B. Musgrave, Angew. Chem., Int. Ed., 2007, 46, 8153 CrossRef CAS.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2008, 130, 1798 CrossRef CAS.
- S. Hausdorf, F. Baitalow, G. Wolf and F. O. R. L. Mertens, Int. J. Hydrogen Energy, 2008, 33, 608 CrossRef CAS.
- F. M. Taylor and J. Dewing, US Pat., 3 103 417, 1963.
- K. C. Ott, R. T. Baker, A. K. Burrell, B. L. Davis, H. V. K. Diyabalanage, J. C. Gordon, C. W. Hamilton, M. Inbody, K. K. Jonietz, R. J. Keaton, V. Pons, T. A. Semelsberger, R. Shrestha, F. H. Stephens, D. L. Thorn and W. Tumas, Chemical Hydrogen Storage Research at Los Alamos National Laboratory, DoE Hydrogen Annual Progress Report, 2007 (http://www.hydrogen.energy.gov/pdfs/progress07/iv_b_5g_ott.pdf).
- Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS.
- A shorter bond distance on its own cannot per se serve as proof for a stronger bond. For a discussion see for example: F. Bessac and G. Frenking, Inorg. Chem., 2006, 45, 6956 Search PubMed.
- H. V. K. Diyabalanage, R. P. Shrestha, T. A. Semelsberger, B. L. Scott, M. E. Bowden, B. L. Davis and A. K. Burrell, Angew. Chem., Int. Ed., 2007, 46, 8995 CrossRef.
- A. J. Arduengo and D. A. Dixon, Main Group Element and Organic Chemistry for Hydrogen Storage and Activation, DoE Hydrogen Program Review, 2008 (http://www.hydrogen.energy.gov/pdfs/review08/st_9_dixon.pdf); K. Goldberg and M. Heinekey, Solutions for Chemical Hydrogen Storage: Dehydrogenation of B–N Bonds, DoE Hydrogen Program Review, 2008 (http://www.hydrogen.energy.gov/pdfs/review08/st_10_goldberg.pdf).
- E. R. Abbey, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2008, 130, 7250 CrossRef CAS; E. R. Abbey, J. T. Jenkins, L. N. Zakharov and S.-Y. Liu, Org. Lett., 2007, 9, 4905 CrossRef; M. Scheideman, G. Wang and E. Vedejs, J. Am. Chem. Soc., 2008, 130, 8669 CrossRef CAS.
- C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 2698 CrossRef CAS; D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130 RSC.
Footnotes |
| † Part of the renewable energy theme issue. |
| ‡ Dedicated to Prof. M. Frederick Hawthorne on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2009 |