Design and synthesis of porphyrin-containing catenanes and rotaxanes
Jonathan A.
Faiz
,
Valérie
Heitz
and
Jean-Pierre
Sauvage
*
Laboratoire de Chimie Organo-Minérale, LC 3, UMR 7177 du CNRS, Université Louis Pasteur, Faculté de Chimie, 4 rue Blaise Pascal, 67070 Strasbourg Cedex, France. E-mail: sauvage@chimie.u-strasbg.fr; Fax: +33 (0)3 90 24 13 68; Tel: +33 (0)3 90 24 13 61
First published on 24th November 2008
Abstract
Catenanes and rotaxanes containing porphyrin subunits have become popular synthetic targets because of the large variety of available synthetic strategies including the coordination chemistry of metallated porphyrins, coupled with the many attractive physical properties of porphyrins. This tutorial review outlines various synthetic approaches and templating strategies that have been used to prepare a range of mechanically interlocked architectures that incorporate porphyrins as fundamental subunits either grafted onto macrocycles or as stoppers. These species are of interest in relation to recreating natural processes such as the photosynthetic apparatus or enzyme binding sites.
 Jonathan Faiz | Jonathan Faiz obtained an MChem degree from the University of Oxford. He carried out his PhD and postdoctoral work at the University of Birmingham where he studied self-assembled systems based on photoactive cyclodextrin receptors. He is now an EU Marie Curie Fellow in the laboratory of Jean-Pierre Sauvage where his research interests include the synthesis of multiply interlocked species based on porphyrins. |
 Valérie Heitz | Valérie Heitz has been assistant professor in the group of Jean-Pierre Sauvage since 1993. She has worked for many years on the synthesis of interlocked multiporphyrinic systems as functional models of the photosynthetic reaction centre. Her interests also include non-covalent systems bearing porphyrins for their dynamic and electronic properties and the design of molecular machines based on catenanes and rotaxanes. |
 Jean-Pierre Sauvage | Jean-Pierre Sauvage is a CNRS director of research. His current interests span from models of the photosynthetic reaction centre, using transition metal complexes and porphyrins, to topology (catenanes and knots), molecular machines and motors and two dimensional interlocking arrays. He has given numerous lectures and has published more than 400 articles. He is a member of the French Academy of Sciences. |
1. Introduction
In the early days of simple catenanes and rotaxanes, the synthesis of these compounds used to represent a real challenge in itself.1 In the course of the past twenty five years, the introduction of synthesis strategies based on various templates (transition metal complexes2 or purely organic complexes3–5) has completely changed the scene, making these compounds accessible and, in parallel, allowing the introduction of various functional groups in their backbones or attached at their periphery. The main field of research derived from catenanes and rotaxanes is clearly that of controlled dynamic systems, i.e.molecular machines.6–8 This is very natural since interlocking rings or rings threaded by molecular strings are a priori ideally suited to large amplitude motions such as the pirouetting of a ring around the axle on which it is threaded, or translation of the ring along the thread, without any constraint. This flourishing field of research has indeed produced spectacular examples of such molecular machines, also including non-interlocking molecular systems.9,10Rotaxanes and, to a lesser extent, catenanes, have also been used in conjunction with electron or energy transfer processes. A particularly attractive feature of such species is that their constitutive elements are disconnected from one another in terms of classical chemical connection, the topology11 of the molecule being responsible for the fact that the ring(s) and thread(s) are held together, within the same molecular assembly, although they are not interlinked by classical bonds. In addition to the dynamic features associated with the topological properties, novel electron or energy transfer characteristics can thus be expected. If some of the components of the rotaxane or the catenane bear donor (D, electron or energy donor) or acceptor (A, electron or energy acceptor) groups, D and A being attached to chemically non-connected elements (ring or axis), transfer between D and A can be considered as “through space” since no chemical bond pathway between D and A can be invoked.
Porphyrins are particularly important compounds in relation to electron and energy transfer processes. In particular, they are the key elements of a myriad of synthetic multicomponent species aimed at mimicking the functions of the photosynthetic reaction centres (RC).12 The incorporation of porphyrins in catenanes and rotaxanes has led to new families of compounds, either in relation to the above-mentioned photosynthetic models or to other topics such as molecular machines. Porphyrin-containing catenanes and rotaxanes combine the electronic properties of porphyrins and metalloporphyrins in their ground states or photoexcited states (mostly singlet states, which have a strong propensity to act as powerful electron donors, when the central metal is zinc(II)) with the high flexibility and mobility of mechanically linked systems. This unique combination of properties allows control of the photochemical behaviour of the compounds by dictating the shape and mobility of the catenanes or rotaxanes in view of performing photoinduced electron transfer. It also paves the way to new molecular machine prototypes whose intramolecular motions can be driven by chemical or photonic processes. By combining the ability of catenanes and rotaxanes to experience complete overturn under the action of a certain signal and the capacity of porphyrins to undergo electron or energy transfer, new dynamic systems have been elaborated which allow testing of electron and energy transfer theories. They also permit control of the rate of the photoinduced transfer processes by using a first stimulus which triggers the interconversion between two different forms of the same molecule, before sending the photonic signal which gives rise to the transfer. In this way, the same organic backbone can be used to investigate the electron or energy transfer processes of markedly different molecules derived from the same compounds.
The aim of the present review article is to discuss the various templated synthetic approaches which have been proposed in the past fifteen years for constructing catenanes and rotaxanes, either with porphyrin nuclei as integral components of their backbone or as attached peripheral fragments. Some of the electronic or dynamic properties of the compounds will be very briefly mentioned but the focus of this review article will clearly be that of synthesis.
Although, in the course of the past fifteen years, several research teams have produced novel and interesting systems combining catenanes, rotaxanes and porphyrins, our group has been involved in the elaboration and study of such systems from a very early stage.13 By analogy with the synthesis of many nonporphyrinic catenanes and rotaxanes, the construction of the compounds relies on the “gathering and threading” effect of a transition metal centre such as copper(I). As shown in Fig. 1, if a molecular “string” is threaded through a preliminarily synthesised ring, we are in a good situation to subsequently prepare a rotaxane by attaching two terminal functions (which are preferably bulky enough for preventing the system from unthreading once the metal template has been removed) at both ends of the threaded fragment. As will be discussed later, tetraaryl porphyrins and even etio porphyrins are sufficiently voluminous to fulfil this function while displaying a range of interesting electronic and photochemical properties. The strategy of Fig. 1 has been much exploited by our group and, as a consequence, it does not need much comment. The most important points of this threading reaction is that (i) it is performed under thermodynamic control and (ii) it relies on the strong preference of copper(I) for two-bidentate chelate complexes, a situation which corresponds to the threaded product of Fig. 1. Such a stable tetrahedral coordination could not be reached for all the copper(I) centres of the solution without threading the axle through the ring since two rings can not form a two-chelate complex with a copper(I) atom for obvious reasons.
 | ||
| Fig. 1 The formation of a pseudorotaxane is quantitative provided the stoichiometry of the reaction is strictly controlled (copper to macrocyclic ligand to complexing thread: 1 : 1 : 1) and the equilibrium has been reached. Formation of Cu(I) complexes with one chelate only is thermodynamically strongly disfavoured, which is the main driving force for the threading reaction to proceed efficiently. The chelating units are represented by U-shaped symbols. | ||
2. Examples from the Strasbourg group
2.1 Porphyrin-stoppered rotaxanes
 | ||
| Fig. 2 The macrocycle (A) incorporating a coordinating fragment (thick line) interacts with a metal centre (black circle) and an asymmetrical open chain chelate (B) bearing one porphyrin and a precursor function X which is small enough to pass through the ring; after the threaded intermediate C is formed, the additional porphyrin ring is constructed, affording the transition metal-complexed rotaxanes D and E. Demetalation leads to the free-ligand rotaxanes F and G from D and E, respectively. | ||
The macrocycle A is the dpp-incorporating 30-membered ring (dpp: 2,9-diphenyl-1,10-phenanthroline) used previously in our group to make various catenanes and rotaxanes. As shown from CPK molecular models, it is sufficiently small to prevent release of the phenanthroline-bridged bisporphyrin dumbbell from E. B is a non-symmetrical dpp derivative attached to a gold(III) porphyrin at one end and to an aromatic aldehyde at the other end. Fig. 2 also shows the two possible rotaxanes which can be formed (a [2]- or a [3]rotaxane) from a common threaded precursor C. The chemical structure corresponding to compounds A to G of Fig. 2 and the reactions leading to the transition metal-complexed [2]- and [3]rotaxanes 82+2+ and 104+4+, respectively, are indicated in Fig. 3.14
![Synthesis of the various [2]- and [3]rotaxanes: (i) CH2Cl2–CH3CN, room temperature; (ii) di-tert-butyl-3,5-benzaldehyde 5, (diethyl-3,3′-dimethyl-4,4′-dipyrryl-2,2′)methane 4, CF3COOH, CH2Cl2, room temperature, then chloranil, CH2C12, reflux.](/image/article/2009/CS/b710908n/b710908n-f3.gif) | ||
| Fig. 3 Synthesis of the various [2]- and [3]rotaxanes: (i) CH2Cl2–CH3CN, room temperature; (ii) di-tert-butyl-3,5-benzaldehyde 5, (diethyl-3,3′-dimethyl-4,4′-dipyrryl-2,2′)methane 4, CF3COOH, CH2Cl2, room temperature, then chloranil, CH2C12, reflux. | ||
In compound 1++, the porphyrin incorporates a trivalent gold metal centre which was selected for two reasons: (i) it forms very stable porphyrin complexes and will thus not be lost during the synthesis of the second porphyrin nucleus,14 (ii) its strong electropositive character confers a remarkable electron-accepting ability to the aromatic porphyrin ring to which it is complexed, resulting in a very accessible reduction potential.15
Following the synthetic strategy outlined above, prerotaxane 32+2+ was first formed by complexing macrocycle 2 with Cu(I) by reaction with [Cu(CH3CN)4]BF4, followed by addition of the gold porphyrin 1-(BF4). It is noteworthy that although Cu(dpp)2+-type complexes are notoriously highly coloured because of the presence of the gold(III) porphyrin, no colour change was observed in the course of the formation reaction of 32+2+. Thin-layer chromatography and NMR spectroscopy showed that complex 32+2+ had formed quantitatively and was thus used in the next step, without further purification. Importantly, the synthesis of the second porphyrin had to be compatible with the presence of the threaded prerotaxane intermediate C of Fig. 2. A mild reaction had thus to be selected, for which nodemetalation of the copper(I) complex 32+2+ was expected to take place. Since the Adler synthesis provides too harsh a set of conditions (boiling propionic acid) for copper(I) bischelate complexes to survive the reaction quantitatively, we investigated a milder procedure developed by Lindsey and co-workers. The condensation reaction was performed by mixing [3](BF4)2, di-tert-butyl-3,5-benzaldehyde 5 and (diethyl-3,3′-dimethyl-4,4′-dipyrryl-2,2′)methane 4 in molar ratio 1 : 4 : 40. Subsequently, a large excess of chloranil was added in order to oxidize the intermediate porphyrinogen. After work up, anion exchange (PF6−) and chromatographic separations, three porphyrins were isolated: etioporphyrin 6, the desired copper(I) [2]rotaxane 82+2+, and the biscopper(I) [3]rotaxane 104+4+. The latter product may be considered as what we could call a “compartmental” [3]rotaxane since the two macrocycles are separated by an inner porphyrin blocking group. The rotaxanes 82+2+ and 104+4+ were isolated as their PF6− salts in 25% and 32% yields, respectively. All three compounds contained a free-base etioporphyrin and could be readily metalated with Zn(OAc)2·2H2O to afford 7, 92+2+, and 114+4+, respectively. The rotaxanes that were synthesized in this way, are copper(I)-complexed rotaxanes. The two constituent parts, the ring(s) (macrocycle 2) and the dumbbell (the phenanthroline-bridged bisporphyrin), are linked together by coordination to copper(I).
Copper(I)-free [2]rotaxane 12-(PF6) was obtained by treatment of [9](PF6)2 with excess KCN (Fig. 4). The fact that demetalation of Cu+-complexed rotaxane 92+2+ afforded a single product 12++ and the absence of release of the bisporphyrin from macrocycle 2 is proof that the compound synthesized is a true rotaxane, at least under normal conditions (room temperature). Linking is provided only by steric crowding of the porphyrin moieties which prevent unthreading of the bisporphyrin from macrocycle 2.
![Decomplexation reaction of the Cu(i)-complexed [2]rotaxane [9](PF6)2 leading to the free-ligand [2]rotaxane 12-(PF6): (i) KCN, CH3CN–CH2C12–H2O.](/image/article/2009/CS/b710908n/b710908n-f4.gif) | ||
| Fig. 4 Decomplexation reaction of the Cu(I)-complexed [2]rotaxane [9](PF6)2 leading to the free-ligand [2]rotaxane 12-(PF6): (i) KCN, CH3CN–CH2C12–H2O. | ||
The couple (PZn, PAu+) turned out to be an excellent choice for studying electron transfer between porphyrins in relation to artificial photosynthesis. PZn has a singlet excited state which is a good electron donor and PAu+ is a powerful electron acceptor in its ground sate. In addition, the Au(III) porphyrin has a very high singlet excited state which avoids any energy transfer from the excited Zn(II) porphyrin. In the Cu+-complexed rotaxane 92+2+, very fast photoinduced electron transfer takes place from the excited zinc(II) porphyrin to the gold(III) porphyrin with a rate of 1.7 ps, close to the one measured in photosynthetic purple bacteria (Fig. 5).16 This very fast process is attributed to the presence of the central Cu(I) bisphenanthroline complex which provides efficient electronic coupling between the porphyrins.17
![(a) A fragment of the photosynthetic RC with the three important components: SP (special pair of bacteriochlorophylls), BCh (bacteriochlorophyll) and BPh (bacteriopheophytin). (b) Chemical structure of the gold(iii)/zinc(ii) Cu(i)-complexed [2]rotaxane 92+2+.](/image/article/2009/CS/b710908n/b710908n-f5.gif) | ||
| Fig. 5 (a) A fragment of the photosynthetic RC with the three important components: SP (special pair of bacteriochlorophylls), BCh (bacteriochlorophyll) and BPh (bacteriopheophytin). (b) Chemical structure of the gold(III)/zinc(II) Cu(I)-complexed [2]rotaxane 92+2+. | ||
![Synthetic strategy for making porphyrin-stoppered multirotaxanes. The threaded precursor is made of a two-coordination site molecular string ended with aldehyde groups passing through two coordinating rings by binding to two identical metals (dot). The porphyrin-forming reaction (i) requires a precise stoichiometry of reagents and can, in principle, be directed mostly towards the construction of porphyrins stoppers or of both terminal and bridging porphyrins. The Cu(i)-complexed [3]- and [5]rotaxanes obtained following this synthetic strategy are represented.](/image/article/2009/CS/b710908n/b710908n-f6.gif) | ||
| Fig. 6 Synthetic strategy for making porphyrin-stoppered multirotaxanes. The threaded precursor is made of a two-coordination site molecular string ended with aldehyde groups passing through two coordinating rings by binding to two identical metals (dot). The porphyrin-forming reaction (i) requires a precise stoichiometry of reagents and can, in principle, be directed mostly towards the construction of porphyrins stoppers or of both terminal and bridging porphyrins. The Cu(I)-complexed [3]- and [5]rotaxanes obtained following this synthetic strategy are represented. | ||
As previously shown with similar compounds, the threading steps were quantitative. Applying experimental conditions analogous to those originally described by Lindsey et al. to the threaded precursor, three porphyrin-containing compounds could be isolated (Fig. 6): 5,15-bis(3,5-di-tert-butylphenyl)-2,8,12,18-tetrahexyl-3,7,13,17-tetramethylporphyrin (not represented) in 32% yield, (Cu2)-[3]rotaxane 132+2+ in 34% yield, and (Cu4)-[5]rotaxane 144+4+ in 8% yield, the copper complexes being isolated as PF6− salts.18,19 The high yield of the (Cu2)-[3]rotaxane 132+2+ is noteworthy. Since two porphyrins are created in the same reaction, the yield of individual porphyrin formation is approximately 60%. (Cu4)-[5]rotaxane 144+4+ deserves some comment. First of all, it is a molecule with four threaded macrocycles, that is, it has a [5]rotaxane structure; secondly, it results from the simultaneous formation of three porphyrins, the central one arising from the condensation of two prerotaxane-like units. The yield of individual porphyrin formation is approximately 45% in this case. This copper(I)-complexed [5]rotaxane may be considered, like 104+4+ as a “compartmental” [5]rotaxane since two groups of two macrocycles each, are separated by an inner porphyrin blocking group. As these rotaxanes bear several coordination sites, a variety of coordination chemistry could be performed on both the porphyrin and the phenanthroline units.18
![Principle of transition metal-templated synthesis of a [2]rotaxane whose ring incorporates a porphyrin. The thick line represents a dpp chelate, the black dot represents a metal cation, a hatched diamond symbolizes a Au(iii) porphyrin and an empty diamond stands for a Zn(ii) porphyrin. The transition metal controls the threading of Au(iii) porphyrin-incorporated macrocycle A onto chelate B, to form prerotaxane C. Construction of the porphyrin stoppers at the X functions leads to the metal complex [2]rotaxane D. Removal of the template cation forms the free rotaxane E.](/image/article/2009/CS/b710908n/b710908n-f7.gif) | ||
| Fig. 7 Principle of transition metal-templated synthesis of a [2]rotaxane whose ring incorporates a porphyrin. The thick line represents a dpp chelate, the black dot represents a metal cation, a hatched diamond symbolizes a Au(III) porphyrin and an empty diamond stands for a Zn(II) porphyrin. The transition metal controls the threading of Au(III) porphyrin-incorporated macrocycle A onto chelate B, to form prerotaxane C. Construction of the porphyrin stoppers at the X functions leads to the metal complex [2]rotaxane D. Removal of the template cation forms the free rotaxane E. | ||
This construction approach is not restricted to the [2]rotaxane described below and can also be applied to other three-porphyrin incorporating [2]rotaxanes. For example, [2]rotaxanes in which a gold(III) porphyrin is appended to the macrocycle were synthesized.20
Cu(I)-complexed [2]rotaxane 182+2+ was prepared using the classical template approach to assemble the gold(III) porphyrin containing macrocycle 15++ and a precursor to the dumbbell, the open chelate 2,9-bis[p-(formylphenyl)]-1,10-phenanthroline 16 (Fig. 8).21,22 The prerotaxane 172+2+ was obtained quantitatively and used directly in the next step. Porphyrin stoppers were constructed from the protruding aldehyde functions as follows: a mixture of prerotaxane 172+2+ (1 equiv.), 3,5-di-tert-butylbenzaldehyde (8 equiv.), dipyrrylmethane derivative (10 equiv.) and a few drops of trifluoroacetic acid in CH2Cl2 was stirred at room temperature overnight. The tetrapyrrole assembly was fixed by controlled oxidation of the porphyrinogens with chloranil. Cu(I)-complexed [2]rotaxane 182+2+ was isolated in 13% yield, after chromatographic purification and Zn(II) insertion into the porphyrin stoppers.
![Synthesis of the Cu(i)-complexed [2]rotaxane 182+2+.](/image/article/2009/CS/b710908n/b710908n-f8.gif) | ||
| Fig. 8 Synthesis of the Cu(I)-complexed [2]rotaxane 182+2+. | ||
The metal template (Cu(I)) was selectively removed by reacting the [2]rotaxane complex 182+2+ with KCN (50 equiv.). This decomplexation reaction liberated the free [2]rotaxane 19++ quantitatively. As studied by 1H NMR spectroscopy, and depicted in Fig. 9, the template imprint (a bisdpp, tetrahedral coordination sphere) completely vanishes by rearrangement of the threaded macrocycle around its axle. Recomplexation of 19++ with Ag+ or Li+ by reaction with AgBF4 or LiBF4 restored the template imprint and afforded the Ag+- and Li+-[2]rotaxane complexes 202+2+ and 212+2+ quantitatively.
![Illustration of the demetalation/remetalation reactions carried out on Cu(i)-complexed [2]rotaxane 182+2+ to afford free [2]rotaxane 19++ and metallo-[2]rotaxanes 202+2+ and 212+2+ and showing the pirouetting motion of the Au(iii)-incorporating macrocycle upon removal of the central metal.](/image/article/2009/CS/b710908n/b710908n-f9.gif) | ||
| Fig. 9 Illustration of the demetalation/remetalation reactions carried out on Cu(I)-complexed [2]rotaxane 182+2+ to afford free [2]rotaxane 19++ and metallo-[2]rotaxanes 202+2+ and 212+2+ and showing the pirouetting motion of the Au(III)-incorporating macrocycle upon removal of the central metal. | ||
The rotaxanes described in this paragraph constitute representative examples showing that complexing or decomplexing the appropriate metal in a coordination site can either bring into close proximity, or spread over a long distance, the porphyrin components of the system, which can be exploited in relation to molecular machines.6–8 In the complex, the Au porphyrin is remote from the two Zn porphyrins. After removal of the central metal, weak forces may favour an attractive interaction between the Au(III) porphyrin and the Zn(II) nuclei leading to a situation in which the Au(III) porphyrin is pinched between the two Zn(II) porphyrin units. The interconversion between both situations implies a half-turn rotation of the threaded fragment within the ring. It also leads to dramatic electron transfer property differences between the two situations.22 In the case of the copper-complexed [2]rotaxane 182+2+, photoinduced electron transfer leading to a Zn(II) porphyrin cation radical and an Au(III) porphyrin neutral radical occurs via a multistep pathway involving the Cu(I) centre. Once the copper(I) is removed, the same charge separated state is formed in the major conformation of 19++, (see Fig. 9), with a rate constant faster than 50 × 109 s−1.
![Copper(i)-templated strategy and synthetic steps for the preparation of a linear Cu(i)complexed [2]rotaxane 252+2+.](/image/article/2009/CS/b710908n/b710908n-f10.gif) | ||
| Fig. 10 Copper(I)-templated strategy and synthetic steps for the preparation of a linear Cu(I)complexed [2]rotaxane 252+2+. | ||
Fig. 10 shows the strategy for rotaxane construction and the synthetic route that was followed to prepare [2]rotaxane 252+2+.23 Effective copper(I)-templated threading to form prerotaxane 23 was observed upon reaction of the dicarboxylate of 22·HCl, (generated by deprotonation by Et3N), with the copper complex of the macrocycle [15·Cu](PF6)2. The stoppering reaction was performed by coupling prerotaxane 23 with zinc aminoporphyrin 24, preparing the activated esters in situ. 1-Hydroxybenzotriazole (HOBt) and 1-ethyl-3-(3-dimethylamino)propylcarbodiimide (EDC) were used as mild coupling reagents for the formation of the activated ester affording copper(I)-complexed [2]rotaxane [25](PF6)2 in 34% yield.
2.2 Porphyrin-containing catenanes
[2]Catenanes are topologically non-trivial molecules (non-planar molecular graph) in which two rings are interlocked but not linked.1 Therefore, differentiating the rings with Zn and Au porphyrins enables the study of photoinduced electron transfer in mechanically bonded systems and can also give information on the conformation of the system.The target Cu(I)-complexed [2]catenane contains two different macrocycles. Therefore, two routes can be envisaged for its construction by the transition metal templated strategy. They are shown in Fig. 11. Both involve the preparation of an intermediate precatenane species, C or F, in which either Zn(II) or Au(III) porphyrin-containing macrocycle A or E is threaded onto chelate B, by copper(I) coordination. Formation of the second interlocking macrocycle is achieved in the next step by reaction of the precatenane with the appropriate porphyrin, D in the case of C, G in the case of F, to produce the desired Cu(I)-complexed [2]catenate H. Finally, removal of the metal template affords the free catenane species I.
![Two strategies for the transition metal-templated synthesis of a [2]catenane made with Zn and Au porphyrin-incorporating interlocked macrocycles. The thick lines represent chelating fragments, the black disk symbolises copper(i), the empty diamonds are Zn(ii) porphyrins, and the hatched diamonds are Au(iii) porphyrins.](/image/article/2009/CS/b710908n/b710908n-f11.gif) | ||
| Fig. 11 Two strategies for the transition metal-templated synthesis of a [2]catenane made with Zn and Au porphyrin-incorporating interlocked macrocycles. The thick lines represent chelating fragments, the black disk symbolises copper(I), the empty diamonds are Zn(II) porphyrins, and the hatched diamonds are Au(III) porphyrins. | ||
The two steps of the most efficient route leading to the Cu(I)-complexed [2]catenane are shown in Fig. 12.24Mixing of equimolar solutions of [Cu(CH3CN)4]PF6 and Zn porphyrin-containing macrocycle 26, followed by addition of phenanthroline derivative 27, afforded precatenate 28++ in quantitative yield. Subsequently, this complex was combined with a stoichiometric amount of gold(III) 5,10-di(p-hydroxyphenyl)-15,20-di(3,5-di-tert-butylphenyl)porphyrinate [29]PF6 in DMF. The resulting solution was treated with Cs2CO3 in DMF. This procedure allows to overcome the relative instability of the present precatenate in basic medium. In these conditions, the Cu(I)-complexed [2]catenate [30](PF6)2 was isolated in 12% yield after chromatography. The alternative route, which involves Au porphyrin-containing macrocycle 15++ and a Zn(II) 5,10-di(p-hydroxyphenyl)-15,20-di(3,5-di-tert-butylphenyl)porphyrinate as reactants afforded the same copper catenate in 5% yield. Demetalation leading to the free [2]catenane species 31++ was carried out by treating the Cu(I) complex with KCN. [2]Catenane 31++ was obtained in 83% yield after purification by column chromatography. The 1H NMR spectrum of the free [2]catenane 31++ is dramatically different from that of its parent Cu(I) complex 302+2+. Dipolar correlations show that upon demetalation the catenane goes from an extended conformation to a more compact one in which the two porphyrins are closer together on average but have no significant interactions, as represented in Fig. 12. Photophysical studies were particularly useful to determine the different conformers of the free catenane 31++ in acetonitrile. In the compact conformer represented in Fig. 12, which accounts for 60% of the total, a very rapid charge separated state (>50 × 109 s−1) is formed upon excitation of the Zn(II) porphyrin followed by electron transfer to the Au(III) porphyrin. In the extended conformers (40% of the total), the charge-separated state is formed in 1.3 × 109 s−1 with a rate similar to the one in the Cu(I)-complexed [2]catenane 302+2+.25
![Three-step preparation of gold(iii)/zinc(ii) [2]catenane 31++.](/image/article/2009/CS/b710908n/b710908n-f12.gif) | ||
| Fig. 12 Three-step preparation of gold(III)/zinc(II) [2]catenane 31++. | ||
These results, as well as the one obtained in the rotaxanes of section 2.1.c, clearly show that fast electron transfer can occur between Zn(II) porphyrin and Au(III) porphyrin despite the lack of a covalent link between them, which is important to relate with natural photosynthesis. Using interlocked systems as catenanes and rotaxanes we have also shown that we can control the dynamics of the systems by complexation or decomplexation reactions. This is a first important step towards molecular machines based on these molecules.
3. Catenanes and rotaxanes reported by other groups
3.1 Rotaxanes with covalently attached porphyrin stoppers
The first examples of a porphyrin-containing rotaxane were reported by us13 (see section 2.1.a) and at the same time by Stoddart et al.26 This latter example consists of a thread containing either one or two hydroquinol rings as molecular recognition sites and a porphyrin at each end. The rotaxanes 32 and 33 are formed by a ‘clipping’ technique, where the π–π stacking interactions between the hydroquinol rings of the thread and the bipyridinium units of the ring act as a template, as well as the formation of hydrogen bonds between particular H atoms of the pyridinium nuclei (α to the nitrogen atoms) and oxygen atoms of the –OCH2CH2O– fragments. Typical yields are in the range of 9%. It was demonstrated by variable temperature NMR experiments that shuttling occurs between the two hydroquinol stations in the rotaxane 33 with an activation energy of 13.6 kcal mol−1.26 The compounds are represented in Fig. 13.
A rotaxane bearing porphyrin stoppers has been reported by the group of Hunter (Fig. 14).27 In contrast to the previous example, the macrocycle consists of two zinc porphyrin-pyridine units that dimerise in non-coordinating solvents to form a rectangle, with a dimerisation constant of 2 × 108 M−1. In the presence of a thread that contains hydrogen-bonding recognition sites the dimer can self-assemble to form pseudorotaxane or rotaxane architectures. NMR studies showed that a new set of signals appears in a mixture of H434 and Zn35, corresponding to a 2 : 1 complex 36 with the constitution [H434·(Zn35)2]. 36 is formed with an association constant of 1.8 ± 0.4 × 104 M−1. Variable-temperature NMR experiments have highlighted a fundamental difference between 36 and a pseudorotaxane with the same backbone as 36 but without the porphyrin stoppers. Although the structure and stability constants for the two species are similar, the energy barrier for the exchange of bound and free species is significantly higher for the stoppered rotaxane than the pseudorotaxane. This is interpreted by the occurrence of a slipping mechanism for the pseudorotaxane but a ‘de-clipping’ process (where the self-assembled macrocycle opens prior to dethreading) for the stoppered rotaxane.27
 | ||
| Fig. 14 Self-assembly of rotaxane 36 from individual components. | ||
3.2 Rotaxanes stoppered by axial coordination of metallated porphyrins
As well as systems containing covalently bound porphyrin stoppers, rotaxanes using axially coordinated metallated porphyrins as stoppers have also been investigated. These species use two varieties of non-covalent interactions for their assembly—those between the thread and the macrocycle and those between the thread (which typically bears pyridine units at each end) and the stoppers—so that the whole rotaxane self-assembles from the three different components. The first example of this class of rotaxanes was reported by Branda and co-workers.28 The various threads, rings and stoppers used by this group are represented in Fig. 15a. The principle of the rotaxane-forming reaction is also indicated in a schematic fashion. Mixing equal amounts of 37 and dibenzo-24-crown-8 (DB24C8) led to the formation of the corresponding pseudorotaxane. NMR titrations showed the association constant to be between 3 × 103 and 1 × 105 M−1, and revealed the presence of π–π interactions and hydrogen bonds between the bispyridinium cation and the crown ether. The value of the association constant was found to be dependent on the concentration of the samples, which is attributed to residual water in the NMR solvents influencing the precision of the titrations. Addition of two equivalents of 38 to the pseudorotaxane solution resulted in significant shifts in the proton NMR spectrum, which were assigned to arise from rotaxane formation. This was confirmed by an X-ray crystal structure. Addition of competitive coordinating solvents was found to remove the stoppers to leave the starting pseudorotaxane.28 | ||
| Fig. 15 Threads, stoppers and macrocycles used for the construction of rotaxanes stoppered by axial coordination to a metallated porphyrin. The principle of the rotaxane construction is represented in the bottom line. | ||
Rotaxanes stoppered at one end only by the axial coordination method have been prepared by the group of Shimizu (Fig. 15b).29 In this study, rotaxane formation between two threads (39 and 40) bearing a tertiary butyl stopper and a central amine unit have been examined in different combinations with three porphyrins 41a–c and DB24C8 in acidic medium to protonate the amine group and bind the crown ether. A range of NMR techniques showed that rotaxanes constructed from the long thread have the same configuration of the porphyrin relative to the macrocycle. Those containing the short thread, however, change their conformation with the identity of the porphyrin stopper. Thus the tertiary butyl groups of 41c (TBPP) make the benzene rings of the crown lie between those of the porphyrin, whereas 41b (TPP) adopts the conformation where the benzene rings on the porphyrin and crown eclipse each other due to the CH–π interactions of the two rings.29
Gunter and Sanders have also reported rotaxanes of this genre using a thread 42 and macrocycle 43 (Fig. 15c).30 In a systematic study the central metal of the porphyrin core 44 was varied as it was expected that the association constants would increase in the order Zn < Ru(II)CO < Rh(III)I allowing the temperature dependence and timescale of exchange to be fully explored. Mixing the components in the stoichiometry required for rotaxane formation led to the appearance of new peaks in the proton NMR spectrum that corresponded to the rotaxanes. These peaks showed NOE (nuclear Overhauser effect) connectivities between the diimide and macrocyclic protons, confirming rotaxane formation. Variation in concentration and temperature was found to influence the proportion of rotaxane in the mixture, implying a dynamic equilibrium. This was further probed by variable temperature NMR experiments, the equilibrium in peak intensities for bound and unbound species was reached at 0 °C for RhI, −20 °C for RuCO and −65 °C for Zn, corroborating the hypothesis of the respective stability constants. It was also shown that the RuCO and RhI rotaxanes could be chromatographed intact at low temperatures.30
An extension of the class of dynamic three-component systems has been recently reported in the form of bis zinc porphyrin tweezers.31 The example shown in Fig. 16 uses axial coordination of a zinc porphyrin for the stoppering reaction, but in this example the two stoppers are linked by a rigid hydrogen-bonded foldamer 45 that keeps them separated and parallel. The porphyrins were grafted onto the foldamer (itself prepared in four steps from hydroquinone) by peptide coupling. The association constants for the formation of the macrocyclic structures were calculated from UV/Vis titration experiments to be (5.7 ± 0.7 × 106) M−1 for 45·46 and (7.9 ± 0.9 × 104) M−1 for 45·47, the latter being smaller due to the electron-withdrawing effect of the two pyridinium units. Mixing equal amounts of 45, macrocycle 48 and thread 46 or 47 led to rotaxane formation in 75% and 62% yields for 45·46·48 and 45·47·48, respectively, determined by the integrated intensity of the signals corresponding to catenane and unbound species in the proton NMR spectra at −13 °C. Catenane formation was confirmed by two-dimensional ordered NMR (DOSY) spectra which revealed that 45·46·48 exists as one single structure. Furthermore, addition of DABCO to 45·46·48 led to the disappearance of characteristic catenane peaks in the NMR spectrum due to dethreading of the crown ether, implying formation and subsequent deconstruction of the desired catenane.31
 | ||
| Fig. 16 Catenanes formed from a bis zinc porphyrin tweezer with a conformation defined by a hydrogen-bonded foldamer spacer. | ||
3.3 Porphyrin catenanes and rotaxanes incorporating fullerene units
Rotaxanes bearing porphyrin stoppers and a macrocycle containing a fullerene have been synthesised to study the efficiency of the forward and back electron transfer between the zincated porphyrins (ZnP) and the fullerene. Incorporating these two centres, which are known to form a long-lived charge separated state, into rotaxane architectures was expected to mirror the intricate organisation of the subunits of the photosynthetic reaction centre and as such mimic its function. An initial example of a rotaxane of this nature was reported by Takata et al., and was formed by stoppering a porphyrin-containing pseudorotaxane by a Diels–Alder reaction with C60.32 A further example of a pseudorotaxane with one fullerene stopper (but bearing TTF units on the macrocycle) has also been reported, in which the threading occurs by interaction of a dibenzylammonium salt with the crown ether.33 The first porphyrin/fullerene rotaxane that was subjected to a photophysical investigation was 49 (Fig. 17), which used hydrogen bonding interactions to thread the axle through the macrocycle.34Rotaxane formation was achieved in a one-pot reaction between the macrocycle (containing a sulfolene group), the thread bearing acid chlorides and the amine-functionalised porphyrin stoppers, with the high yield (55%) of 49c being particularly noteworthy. Subsequent deprotection of the sulfolene group revealed a diene that was reacted with the fullerene by a Diels–Alder reaction in 69–74% yield to give the target rotaxane. | ||
| Fig. 17 Synthesis of rotaxanes bearing porphyrin stoppers and a fullerene-containing macrocycleviahydrogen-bond templating. | ||
Comprehensive photophysical studies have shown that formation of the charge-separated state (i.e.ZnP C60→ ZnP* C60→ ZnP˙+ C60˙−) occurs in all three rotaxanes. As the length of the thread increases the rate of formation of the charge-separated state decreases (kCS = 1.1 × 1010 s−1, 9.0 × 108 s−1, 4.2 × 108 s−1 for a, b and c, respectively, in benzonitrile), however, the lifetime of the charge-separated species increases (i.e. the rate of recombination ZnP˙+ C60˙−→ ZnP C60 decreases) (kb 5.5 × 106 s−1, 4.3 × 106 s−1, 1.6 × 106 s−1 for a, b and c in benzonitrile). It was also observed that the mechanism for electron transfer switches between the excited singlet or triplet state of the porphyrin depending on the identity of the thread and the temperature of measurement.34
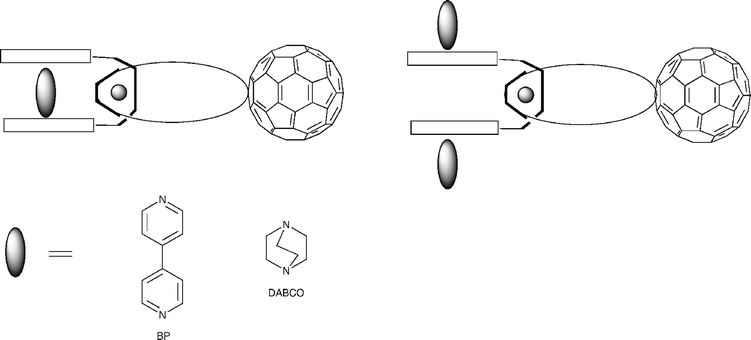
A similar system has been explored by Schuster and Guldi. As shown in Fig. 18, a Cu(I)-templated reaction around a phenanthroline core is used to direct formation of rotaxanes such as 50.35 The potential for catenane formation of these rotaxanes has also been exploited. Complexes of bidentate guests DABCO and BP that bind axially to the zinc porphyrins have been formed, either in a 1 : 1 or 2 : 1 ratio, as represented in Fig. 19.36
 | ||
| Fig. 18 Schematic showing the stepwise copper(I)-templated synthesis of the porphyrin-stoppered rotaxanes reported by Guldi and Schuster, and rotaxane 50. | ||
3.4 Porphyrin-containing catenanes and rotaxanes
Catenanes 52 formed from porphyrins ‘strapped’ with polyether units 51 have been pioneered by the research group of Gunter (Fig. 20).37 These receptor molecules are synthesised by condensation of a dialdehyde with a dipyrromethane giving the desired porphyrin 51 in 9–16% yield. When the longer straps are present two isomers of porphyrin are formed—the desired strapped product and a ‘twisted’ porphyrin that has the two ortho-substituted meso-aryl substituents in opposite configurations. These two isomers equilibrate in solution, and the ‘clipping’ procedure to form the strapped catenane 52 from an equilibrated solution was found to yield a single catenane product when carried out under high-pressure conditions. The catenanes were found to display a number of interesting properties. It was discovered that the protonation constant (pKa) of the porphyrins increased with strap length. This is interpreted by considering the coulombic repulsion that exists after protonation. In the unprotonated state the bipyridinium dication sits between the macrocycle and the porphyrin due to the favourable π–π interactions between the three units. After protonation the coulombic repulsion between the two positively charged centres outweighs the π–π interactions and the macrocycle attempts to turn through ninety degrees to relieve the repulsion. This rotation occurs more easily in the catenanes with longer straps, hence their higher affinity for protons. Photoinduced electron transfer has been shown to occur from the porphyrin to the macrocycle and back again. Forward electron transfer occurs rapidly (rate ≥ 5 × 1010 s−1), whilst back electron transfer occurs with a rate of 2.5 × 1010 s−1 for all catenanes. This observation is attributed to the occurrence of two electron transfer steps, initially between the porphyrin and the inner bipyridinium, followed by a subsequent transfer of charge from the inner to the outer bipyridinium before charge recombination. The behaviour of the catenanes has also been studied electrochemically. All rotaxanes in the series initially undergo two one-electron reduction processes, indicating that there are two inequivalent sites on the bipyridinium unit. It is postulated that the first reduction takes place at the outer whilst the second takes place at the inner bipyridinium unit, which is more difficult to reduce as it is sandwiched between two electron donating groups. Subsequent reductions depend on the structure of the catenane.38 A rotaxane system has been reported by Morinet al. in which two crown ether appended porphyrins are threaded through a central backbone that contains two binding sites. This more flexible example has been shown to act as a receptor for fullerenes.39 The final examples to be discussed in this section are architectures that have been directly inspired by specific natural architectures. Hamachi et al. have reported a triad composed of a Zn heme sensitiser, ruthenium(II) (2,2′-bipyridyl)3 as an electron donor, which bears a crown ether and a cyclic viologen as a catenated electron acceptor. The lifetimes of the charge-separated states were found to increase when the system was embedded in cytochrome b562 or myoglobin matrices.40 The rotaxane 54 that acts as an enzyme analogue was reported by Rowan and Nolte (Fig. 21).41 This rotaxane incorporates two porphyrin ‘clips’53 as macrocycles that function as the catalytic centres. The clips, which are synthesised by condensation of a diphenylglycouril core bearing four pendant aldehydes with pyrrole, have a high affinity for the bipyridinium ion. The rotaxane is built from a central polybutadiene unit with a pyridinium binding site on each side and 3,5-di-tert-butylphenyl stoppers in 43% yield. It has been demonstrated that the porphyrin clips act as catalytic centres for the epoxidation of the alkene units in the presence of an oxidant such as iodosylbenzene (PhIO), moreover the rotaxane 54 in the presence of 4-tert-butylpyridine (tbpy) has been shown to undergo the epoxidation process nearly twice as rapidly than the non-stoppered pseudorotaxane analogue. The tbpy ligand is required to activate the manganese catalyst and coordinates axially to the top face of the clip, implying that the catalytic process must take place inside the clip cavity rather than the thread ‘folding’ to reach the porphyrin on the exterior.40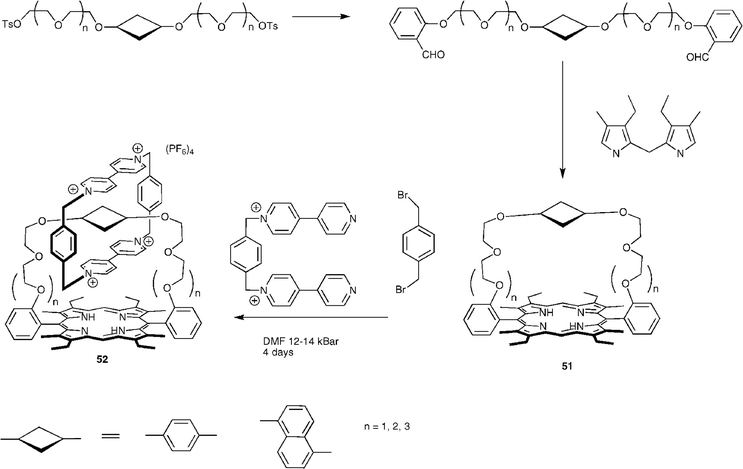 | ||
| Fig. 20 Synthesis of strapped porphyrins and their catenanes. | ||
 | ||
| Fig. 21 Rotaxane 54 incorporating manganese porphyrin ‘clips’ that act as catalytic centres for oxidation of olefins. | ||
4. Conclusions
Porphyrins and metallo-porphyrins display specific properties which make them attractive components of catenanes and rotaxanes, both as building blocks and as electro- or photo-active elements. The ability of zinc-complexed porphyrins to bind pyridinic groups has been exploited in a very ingenious way by several groups who could construct particularly novel interlocking species. In addition, the template principles which were used in the past for building non-porphyrinic catenanes and rotaxanes (transition metal-based entwining or threading of ligands, generation of threaded species viahydrogen bonding or formation of donor–acceptor complexes) could be extended to porphyrin-incorporating compounds of this wide family of molecules. Since free-base and metal-complexed porphyrins are groups susceptible to undergo energy and electron transfer par excellence, interesting intramolecular reactions of these types have been investigated, in relation to photosynthesis mimics. The catenane or rotaxane nature of the multiporphyrinic assemblies discussed in the present review article makes them prone to undergo large amplitude motions under the action of an external signal (metalation/demetalation of a coordination site, for instance) which allows the study of various photochemical processes within groups of molecules displaying the same organic backbone but that are markedly different as far as their geometrical properties are concerned. Here again, such processes can be related to natural photosynthesis which combines electron transfer steps with molecular motion. Finally, a recent example demonstrates that porphyrinic systems displaying catalytic properties combined with rotaxanes are particularly promising and may be utilised in the fabrication of polymers with high control over the properties of the product. Porphyrinic catenanes and rotaxanes thus have a bright future that encompasses the areas of photosynthesis, molecular machines and catalysis.References
- G. Schill, Catenanes, Rotaxanes and Knots, Academic Press, New York, 2001 Search PubMed.
- C. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kitzinger, Tetrahedron Lett., 1983, 24, 5095 CrossRef CAS.
- P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396 CrossRef.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS.
- F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619 CrossRef.
- Molecular Machines and Motors, Structure and Bonding, ed. J.-P. Sauvage, Springer, Berlin, Heidelberg, 2001, vol. 99 Search PubMed.
- V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2003 Search PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS.
- B. L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, 2001 Search PubMed.
- V. Amendola, L. Fabbrizzi, C. Mangano and P. Pallavicini, Acc. Chem. Res., 2001, 34, 488 CrossRef CAS.
- Strictly speaking, rotaxanes are topologically trivial species. Nevertheless, if the stoppers are bulky enough, unthreading is physically impossible. The thread can thus be assimilated to an infinitely long line, making under these conditions the rotaxane a topologically non-trivial species. Although this definition is not rigorous, it is convenient and allows one to consider that the topological properties of catenanes and rotaxanes are similar.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS.
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1992, 1131 RSC.
- J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1993, 115, 12378 CrossRef CAS.
- V. Heitz, S. Chardon-Noblat and J.-P. Sauvage, Tetrahedron Lett., 1991, 32, 197 CrossRef CAS.
- J.-C. Chambron, A. Harriman, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1993, 115, 6109 CrossRef.
- J.-C. Chambron, A. Harriman, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1993, 115, 7419 CrossRef CAS.
- N. Solladié, J.-C. Chambron and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 3684 CrossRef CAS.
- N. Solladié, J.-C. Chambron and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1996, 35, 906 CrossRef CAS.
- M. Linke, J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1997, 119, 11329 CrossRef CAS.
- M. Linke, J.-C. Chambron, V. Heitz, J.-P. Sauvage and V. Semetey, Chem. Commun., 1998, 2469 RSC.
- M. Linke, J.-C. Chambron, V. Heitz, J.-P. Sauvage, S. Ensinas, F. Barigelletti and L. Flamigni, J. Am. Chem. Soc., 2000, 122, 11834 CrossRef CAS.
- M.-J. Blanco, J.-C. Chambron, V. Heitz and J.-P. Sauvage, Org. Lett., 2000, 2, 3051 CrossRef CAS.
- M. Linke, N. Fujita, J.-C. Chambron, V. Heitz and J.-P. Sauvage, New J. Chem., 2001, 25, 790 RSC.
- L. Flamigni, A. M. Talarico, J.-C. Chambron, V. Heitz, M. Linke, N. Fujita and J.-P. Sauvage, Chem.–Eur. J., 2004, 10, 2689 CrossRef CAS.
- P. R. Ashton, M. R. Johnston, J. F. Stoddart, M. S. Tolley and J. W. Wheeler, J. Chem. Soc., Chem. Commun., 1992, 1128 RSC.
- C. A. Hunter, C. M. R. Low, M. J. Packer, S. E. Spey, J. G. Vinter, M. O. Vysotsky and C. Zonta, Angew. Chem., Int. Ed., 2001, 40, 2678 CrossRef CAS.
- K. Chichak, M. C. Walsh and N. R. Branda, Chem. Commun., 2000, 847 RSC.
- T. Ikeda, M. Asakawa and T. Shimizu, New J. Chem., 2004, 28, 870 RSC.
- M. J. Gunter, N. Bampos, K. D. Johnstone and J. K. M. Sanders, New J. Chem., 2001, 25, 166 RSC.
- J. Wu, F. Fang, W.-Y. Lu, J.-L. Hou, C. Li, Z.-Q. Wu, X.-K. Jiang, Z.-T. Li and Y.-H. Yu, J. Org. Chem., 2007, 72, 2897 CrossRef CAS.
- H. Sasabe, N. Kihara, Y. Furusho, K. Mizuno, A. Ogawa and T. Takata, Org. Lett., 2004, 6, 3957 CrossRef CAS.
- M. C. Diaz, B. M. Illescas, N. Martin, J. F. Stoddart, M. A. Canales, J. Jiménez-Barbero, G. Sarovo and D. M. Guldi, Tetrahedron, 2006, 62, 1998 CrossRef CAS.
- A. S. D. Sandanayaka, N. Watanabe, K.-I. Ikeshita, Y. Araki, N. Kihara, Y. Furusho, O. Ito and T. Takata, J. Phys. Chem. B, 2005, 109, 2516 CrossRef CAS.
- K. Li, D. I. Schuster, D. M. Guldi, M.-A. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 3388 CrossRef CAS.
- D. I. Schuster, K. Li, D. M. Guldi and J. Ramey, Org. Lett., 2004, 6, 1919 CrossRef CAS.
- M. J. Gunter, T. P. Jeynes and P. Turner, Eur. J. Org. Chem., 2004, 193 CrossRef CAS.
- L. Flamigni, A. M. Talarico, S. Serroni, F. Puntoriero, M. J. Gunter, M. R. Johnstone, T. P. Jeynes and P. Turner, Chem.–Eur. J., 2003, 9, 2649 CrossRef CAS.
- J.-S. Marois, K. Cantin, A. Desmarais and J.-F. Morin, Org. Lett., 2008, 10, 33 CrossRef CAS.
- Y.-Z. Hu, H. Takashima, S. Tsukiji, S. Shinkai, T. Nagamune, S. Oishi and I. Hamachi, Chem.–Eur. J., 2000, 6, 1907 CrossRef CAS.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |