B-Site cation diffusivity of Mn and Cr in perovskite-type LaMnO3 with cation-deficit nonstoichiometry
Shogo
Miyoshi†
* and
Manfred
Martin
Institute of Physical Chemistry, RWTH Aachen, Landoltweg 2, D-52056, Aachen, Germany. E-mail: miyoshi@alto.material.t.u-tokyo.ac.jp
First published on 13th March 2009
Abstract
The Mn tracer diffusivity and Cr impurity diffusivity in LaMnO3+δ were studied for the regime of cation-deficient nonstoichiometry with radio-isotope techniques. The effective Mn diffusion coefficient in polycrystalline LaMnO3+δ was successfully measured in the temperature range 1423 ≤T/K ≤ 1523 and oxygen partial pressure range 0.008 ≤PO2/bar ≤ 1. The estimated bulk diffusion coefficient of Mn in LaMnO3+δ is quite high compared with the other perovskite oxides reported in literature, which is regarded to reflect the high cation vacancy concentration of LaMnO3+δ. The Mn diffusivity shows a weak temperature dependence with an apparent activation energy of 0.6 eV and a strong PO2 dependence. Both dependencies may indicate a Mn diffusion mechanism by means of B-site vacancies which is, however, facilitated by the presence of A-site vacancies; that is, the Mn diffusivity depends on the vacancy concentrations not only at the B-sites but also at the A-sites. The Cr impurity diffusivity is significantly smaller than the Mn tracer diffusivity.
1. Introduction
The perovskite-type oxides of the series La1−xSrxMnO3 are technologically important materials that are applied as air electrodes of solid oxide fuel cells (SOFCs)1 and magneto-resistive read heads of electronic storage devices.2 From an aspect of fundamental science, the oxide is known to interestingly display “apparent” oxygen-excess nonstoichiometry under oxidizing conditions.3–7 That is, given amounts of La1−xSrxMnO3 can incorporate “apparent” excess oxygen from a gaseous phase, resulting in the nominal composition of La1−xSrxMnO3+δ (δ > 0). Such kind of nonstoichiometry is rarely observed among perovskite-type oxides, ABO3.The major ionic defects in the “apparent” oxygen-excess La1−xSrxMnO3+δ are believed to be vacancies at cationic sites which are charge-compensated by electron holes. Many diffraction studies involving refinement of the detailed crystal structure suggested cationic occupation numbers well lower than unity.8–11 The numerous reports of defect chemical analysis also claim the predominance of cation vacancies on the basis of atomistic and thermodynamic calculations which somewhat succeeded in reproducing the experimental results on the nominal oxygen content, 3+δ, as a function of temperature T and oxygen partial pressure PO2.3–7
In a preceding study, the predominance of cation vacancies was directly evidenced12 by studying the creation of extra unit cells, which is inevitable for the uptake of excess oxygenvia the cation vacancy mechanism. That is, if cation vacancies are the major ionic defects, they are formed upon the incorporation of the excess oxygen. From the observation of the morphological evolution and the overall volume change of the specimens, it was clearly demonstrated that the excess oxygen is accommodated by the creation of new lattice sites, and thus the defect structure is not determined by oxygen interstitials but by cation vacancies as denoted by the following reaction:
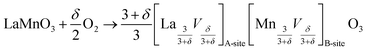 | (1) |
Due to the high population of cation vacancies, cations in La1−xSrxMnO3+δ are expected to be significantly more mobile than in other perovskite-type oxides. This may induce long-term stability problems of La1−xSrxMnO3+δ-based SOFC cathodes. SOFC cathodes in operation always involve a gradient of the oxygen potential in the vicinity of the interface between the electrode and electrolyte, so that the cathode reaction can progress against the reaction resistance. Such a gradient of oxygen potential in metal oxides induces gradients of the cation chemical-potentials in the opposite direction, which is derived from the Gibbs–Duhem relationship. The cation potential-gradients, as well as an electric field, are the driving forces of cation transport, which may cause several stability problems of SOFC cathodes.
Firstly, migration of the cations constituting the framework of the crystal must result in the evolution of morphology, which is one of the most important factors determining the electrode performance. In reality, the morphological change of SOFC cathodes after operation has been reported in the literature,13,14 although the mechanism has not been well recognized. Secondly, the cation transport driven by such potential gradients may induce kinetic demixing or decomposition in multi-component oxides.15 That is, when the cations diffuse to the high PO2 side, cations with high and low diffusivities may concentrate or precipitate at the high and low PO2 sides, respectively, which may deteriorate the electrode performance likewise.
In spite of their considerable importance, there have been only few reports of cation diffusion experiments for La1−xSrxMnO3+δ, while we can find several precedents for measuring the cation diffusivities for the other perovskite-type oxides such as LaCrO3, doped LaGaO3, LaFeO3, LaCoO3, and BaTiO3.16–24 With respect to La1−xSrxMnO3+δ, creep deformation has been studied in relation to the cation transport and defect chemistry.25 As a quantitative study on the cation diffusivity among the system of La1−xSrxMnO3+δ, the Pr impurity diffusivity in LaMnO3+δ measured with a SIMS technique was reported, which is rather higher than those in LaFeO3 and LaCoO3.26 In addition, the kinetics of the solid state reaction between La2O3 and Mn3O4 was studied in relation to the cation diffusion behaviour of LaMnO3+δ.27 The results suggest that the diffusivity of Mn is higher than that of La, and also higher than A-site and B-site diffusivities of other perovskite-type oxides such as LaCrO3 and LaGaO3. However, the solid state reaction technique can provide only the diffusion coefficient of the faster cationic species in principle, and is not capable of distinguishing bulk and grain boundary diffusivities; that is, the measurements may be affected by the cation transport along grain boundaries as a fast diffusion path. It is thus necessary to provide further, more detailed knowledge on cation diffusivity in La1−xSrxMnO3+δ.
In this paper, we report the tracer diffusivity of Mn in LaMnO3+δ in the region of cation-deficient (apparently oxygen-excess) nonstoichiometry. Since there are no stable isotopes of Mn, we employed a radio-tracer of Mn. The measured Mn tracer diffusivity will be presented as a function of temperature and PO2, and the experimental results will be discussed in terms of the defect structure and diffusion mechanism in LaMnO3+δ.
2. Experimental
2.1 Sample preparation
The powder of undoped lanthanum manganite was synthesized by a citrate process. The starting materials were La2O3 (Alfa Aesar, 99.99%) and MnCO3·xH2O (Alfa Aesar, 99.985% (metal basis)), whose net contents were assayed with TG/DTA analysis. They were dissolved in diluted aqueous HNO3 at an appropriate ratio, followed by addition of citric acid of three times the total molar number of cations. After well stirring, the mixed solution was heated up to ca. 700 K in a furnace, yielding an ash-like product which was ground in a mortar and then calcined at 1073 K in air for 1 h. XRD analysis confirmed that the calcined powder consisted of a single phase of perovskite-type structure. After ball-milling, the powder was uni-axially compacted into a cylindrical shape, and subsequently hydrostatic pressure of 100 MPa was applied. The cylindrical green bodies were sintered at 1723 K in air for 10 h and then sliced to ca. 1 mm thickness. One of the surfaces was polished using diamond abrasives. The relative densities of the samples were ca. 98%. SEM observation showed that the average grain size of as-sintered samples is over 10 μm.2.2 Radio-isotope diffusion experiments
The sintered samples were cut into planar shapes with the appropriate dimensions (typically 6 mm diameter and 1 mm thickness), and the surface was precisely polished down to 1 μm roughness. After that, the substrate was equilibrated for ca. twice as long as the diffusion annealing at the same temperature and PO2 as used later for the diffusion annealing. The tracer layer of the radio-active isotope 54Mn (and in some cases also 51Cr, both of which are γ-emitters) was fabricated by applying the aqueous solution including the isotope(s) (54MnCl2 aq., Perkin Elmer, 99.9% radionuclidic purity and 51CrCl3 aq., Perkin Elmer, 99.9% radionuclidic purity) to the substrate surface and subsequent drying with an infrared lamp. This specimen was annealed under a given condition to establish the radiotracer diffusion profile(s). The PO2 for the diffusion annealing was controlled by a flow of a mixture of O2 and N2 gases, and monitored with a yttria-stabilized zirconia-based oxygen concentration cell which was placed close to the specimen.After the diffusion annealing, the edges of the specimen were removed to minimize both the influence of surface diffusion of the radiotracer and its recondensation on the specimen surface after evaporation at elevated temperatures. Then, the diffusion profiles were obtained via two distinct methods in order to ensure the reliability.28 First, a thin layer with a thickness of several μm was ground off the surface of the specimen with abrasive papers. All of the resulting powder was collected, and the spectrum of the γ-rays emitted from the powder was measured (tracer sectioning method). Subsequently, the γ-ray spectrum of the remaining bulk specimen was also obtained (residual activity method). After that, a new thin layer was ground off and γ-ray spectra were similarly collected for the powder and the “new” remaining specimen. By repeating this process, two γ-ray activity profiles, a sectioning profile and a residual activity profile, were obtained for each specimen from which the diffusion coefficients were extracted as described in the next section.
3. Results and discussion
3.1 Evaluation of the diffusion coefficients
Fig. 1 shows typical γ-ray activity profiles of 54Mn in LaMnO3+δ obtained by the tracer sectioning method and the residual activity method, respectively. Because the depth profiling in these methods relies on mechanical grinding, the attainable depth resolution is a few μm at best; therefore, the diffusion annealing was performed for a sufficient time to obtain a long diffusion profile ranging over, for example, 100 μm as observed in Fig. 1. In contrast, the thickness of the tracer layer is thinner than 10 nm (as estimated from the amount of the radio-isotope solution applied), which is negligibly small compared with the typical diffusion distance in Fig. 1. This condition allows us to apply the infinite thin film solution29 to describe the diffusion behaviour.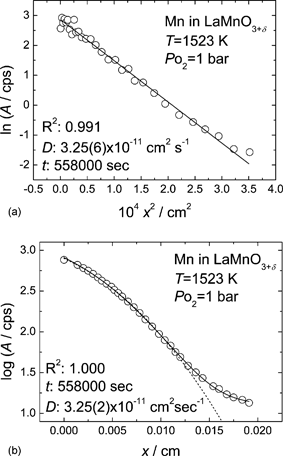 | ||
| Fig. 1 Diffusion profile of 54Mn in LaMnO3+δ obtained with the sectioning method (a) and the residual activity method (b). The diffusion annealing was performed at 1523 K and PO2 = 1 bar for 155 h. The solid line in (a) represents the result of fitting with eqn (2). The dotted and solid lines in (b) represent the results of fitting with eqn (3) before and after correcting the bottom-side activity, respectively. | ||
In the tracer sectioning method, the γ-ray activity measured for each section AS(x) is regarded as directly proportional to the isotope concentration c(x), and thereby AS(x) is given by:
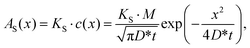 | (2) |
In the residual activity method, all radiotracer atoms in the remaining specimen contribute to the measured residual activity (= integral over the activity profile). However, we must consider that γ-rays from the inner part of the substrate undergo self-absorption by the specimen itself and, in addition, that their intensity is weakened due to the increasing distance to the detector. These effects can be corrected by means of the self-absorption factor α and the geometrical factor β, both of which were determined in independent experiments. The residual activity, AR(x), in this case is formulated as
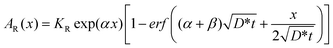 | (3) |
Table 1 summarizes the tracer diffusion coefficients of 54Mn in LaMnO3+δ under various conditions of temperature and PO2. All of the diffusion profiles which give the diffusivity values in Table 1 are fitted to the theoretical curves as well as those shown in Fig. 1. The diffusivity values extracted from the two methods agree well with each other, indicating the reliability of both methods. With respect to the error window, the fluctuations in measuring the penetration depth x, which relies on a digital micrometer, and the statistic error in measuring the γ-ray activity are included in the fitting error. The error originating from the absolute accuracy of the micrometer is 1% at most in consideration of the thickness of the specimen and specification of the micrometer, which is comparable or lower than the fitting error. The width of the error bar in each diffusivity value is thus estimated to be too small to be indicated in the following figures showing D*.
| Exp. # | T/K | log (PO2/bar) | t/h | D eff/cm2 s−1 | Harrison’s classification | |
|---|---|---|---|---|---|---|
| Sectioning method | Residual activity method | |||||
| a Extracted from the shallow part in sectioning profile of the Harrison B-type kinetics; can be approximated as bulk diffusion coefficient. | ||||||
| 54Mn in LaMnO3+δ | ||||||
| 1 | 1423 | −0.678 | 264 | 1.03(±0.02) × 10−11 | 1.03(±0.01) × 10−11 | A-type |
| 2 | 1473 | −0.678 | 260 | 1.26(±0.03) × 10−11 | 1.28(±0.01) × 10−11 | A-type |
| 3 | 1523 | 0 | 155 | 3.25(±0.06) × 10−11 | 3.25(±0.02) × 10−11 | A-type |
| 4 | 1523 | −0.678 | 162 | 1.47(±0.02) × 10−11 | 1.45(±0.01) × 10−11 | A-type |
| 5 | 1523 | −1.38 | 246 | 3.86(±0.08) × 10−12 | 3.70(±0.03) × 10−12 | A-type |
| 6 | 1523 | −2.08 | 305 | 2.9(±0.8) × 10−13a | — | B-type |
| 51Cr in LaMnO3+δ | ||||||
| 7 | 1523 | 0 | 155 | 5.4(±0.2) × 10−12 | 5.3(±0.1) × 10−12 | A-type |
| 8 | 1523 | −1.38 | 246 | 6.8(±0.9) × 10−13a | — | B-type |
Since the LaMnO3+δ substrates used in this study are polycrystalline, it is necessary to consider the possible contributions of both bulk diffusion and grain boundary diffusion. Diffusion in polycrystalline materials can be classified into Harrison’s three types of kinetics.30 When the penetration depth due to volume diffusion is large enough, 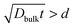 , where d denotes the grain diameter and Dbulk the bulk diffusion coefficient, the diffusion profile appears to follow Fick’s law as for a homogeneous system, which is termed A-type kinetics. In the opposite case,
, where d denotes the grain diameter and Dbulk the bulk diffusion coefficient, the diffusion profile appears to follow Fick’s law as for a homogeneous system, which is termed A-type kinetics. In the opposite case, 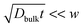 where w denotes the grain boundary width, diffusional transport takes place only along grain boundaries (C-type kinetics). B-Type kinetics is the intermediate case, for which the typical criterion is
where w denotes the grain boundary width, diffusional transport takes place only along grain boundaries (C-type kinetics). B-Type kinetics is the intermediate case, for which the typical criterion is 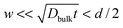 . In this case, the diffusion profile consists of two regions; that is, the profile in the shallow region follows the Gaussian form similarly to the A-type kinetics, while it has a long tailing in the deep region due to grain boundary diffusion.
. In this case, the diffusion profile consists of two regions; that is, the profile in the shallow region follows the Gaussian form similarly to the A-type kinetics, while it has a long tailing in the deep region due to grain boundary diffusion.
Most of the diffusion profiles obtained in this study are regarded as of the A-type kinetics for two reasons: the typical penetration depths of these profiles are larger than the grain size (over 10 μm in this study), and in each case the complete profile is well reproduced by the thin film solution in eqn (2) corresponding to A-type kinetics. Here it should be noted that the tracer diffusion coefficient thus obtained is an effective tracer diffusion coefficient consisting of the bulk tracer diffusion coefficient, D*bulk, and the grain boundary tracer diffusion coefficient, D*g.b.:
| D*eff = (1 −r)D*bulk + rD*g.b. | (4) |
| r≈ 3w/d | (5) |
As a clue to estimate the contribution of the bulk diffusion coefficients to the observed effective diffusion coefficients, we mention a particular diffusion profile which seems to be of the B-type kinetics. Fig. 2(a) and (b) show the sectioning profiles of 54Mn in LaMnO3+δ obtained in experiment #6 (T = 1523 K, PO2 = 0.008 bar, t = 305 h) with the activities plotted against x2 and x6/5, respectively. In Fig. 2(a), we can not observe linearity between ln AS and x2; therefore, the A-type kinetics is not applicable to this diffusion profile. In Fig. 2(b) the activity is plotted versus x6/5 which is the penetration depth dependence obtained for the B-type kinetics at large penetration depth (grain boundary tail) according to the Le Claire solution,31
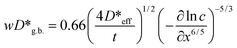 | (6) |
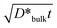 , the measure of Harrison’s classification, is calculated to be ∼6 μm, which indicates B-type kinetics in agreement with the above-mentioned linearity between ln AS and x6/5, because the average grain size of the LaMnO3+δ substrates is over 10 μm. On the other hand, D*g.b./D*bulk is ∼4 × 104 with the grain boundary width assumed to be ∼1 nm.
, the measure of Harrison’s classification, is calculated to be ∼6 μm, which indicates B-type kinetics in agreement with the above-mentioned linearity between ln AS and x6/5, because the average grain size of the LaMnO3+δ substrates is over 10 μm. On the other hand, D*g.b./D*bulk is ∼4 × 104 with the grain boundary width assumed to be ∼1 nm.
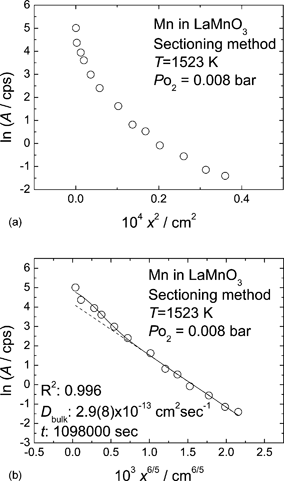 | ||
| Fig. 2 Diffusion profile of 54Mn in LaMnO3+δ obtained with the sectioning method: (a) plotted against x2; (b) plotted against x6/5. The solid and broken lines represent the results of fitting eqn (6) to the obtained profile with and without considering bulk diffusion represented by eqn (2). The diffusion annealing was performed at 1523 K and PO2 = 0.008 bar for 305 h. | ||
If the value of D*g.b./D*bulk obtained in experiment #6 holds also for the other diffusion conditions which gave the A-type kinetics, the contribution of the bulk diffusion coefficient to the effective diffusivity is estimated from eqn (4) and (5) to be D*bulk/D*eff∼ 0.07 with w and d assumed to be 1 nm and 10 μm, respectively. Taking experiment #5 (T = 1523 K, PO2 = 0.04 bar, t = 246 h) as an example of the A-type kinetics, D*bulk and 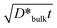 are consequently calculated as 3(±1) × 10−13 cm2 s−1 and 5 μm, respectively. The value of 5 μm for
are consequently calculated as 3(±1) × 10−13 cm2 s−1 and 5 μm, respectively. The value of 5 μm for 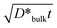 corresponds to B-type kinetics as well as experiment #6, while we obtained in experiment #5 a diffusion profile indicating A-type kinetics (cf.Table 1). This discrepancy indicates that D*bulk/D*eff in experiment #5 is appreciably higher than in experiment #6, where its value was 0.07. The difference in D*bulk/D*eff between experiments #5 and #6 can be attributed to the positive dependence of D*bulk on PO2 (cf. section 3.3) corresponding to an increase in the vacancy concentration at the corresponding sites, and a weaker PO2 dependence of D*g.b.., which may be less sensitive to the bulk defect concentration than D*bulk. Therefore, it is deduced that the contribution of the bulk diffusion coefficient to the effective diffusion coefficient is no less than that in experiment #6, 0.07 also for the other diffusion profiles of the A-type kinetics, all of which were obtained at higher PO2 than experiment #6.
corresponds to B-type kinetics as well as experiment #6, while we obtained in experiment #5 a diffusion profile indicating A-type kinetics (cf.Table 1). This discrepancy indicates that D*bulk/D*eff in experiment #5 is appreciably higher than in experiment #6, where its value was 0.07. The difference in D*bulk/D*eff between experiments #5 and #6 can be attributed to the positive dependence of D*bulk on PO2 (cf. section 3.3) corresponding to an increase in the vacancy concentration at the corresponding sites, and a weaker PO2 dependence of D*g.b.., which may be less sensitive to the bulk defect concentration than D*bulk. Therefore, it is deduced that the contribution of the bulk diffusion coefficient to the effective diffusion coefficient is no less than that in experiment #6, 0.07 also for the other diffusion profiles of the A-type kinetics, all of which were obtained at higher PO2 than experiment #6.
3.2 Temperature dependence of the Mn diffusion coefficients and comparison with cation diffusivities of the other perovskites
Fig. 3 shows the Arrhenius plot of the extracted effective tracer diffusion coefficients of Mn in LaMnO3+δ in air together with previously reported cation diffusion coefficients in the other perovskite systems, which were also measured in air. The closed circles denote the effective Mn tracer diffusion coefficients D*Mn,eff obtained in this study. The downward error bars correspond to the range of the bulk tracer diffusion coefficient of Mn, D*Mn,bulk, discussed above. The Mn diffusivity in LaMnO3+δ is quite high compared with the diffusivity of B-site cations in the other perovskites, and also no smaller than the other A-site diffusivities, which have been regarded as being generally higher than B-site diffusivities. The magnitude of the Mn diffusivity obtained from the kinetics of the solid state reaction between La2O3 and Mn3O427 is in agreement with the present study.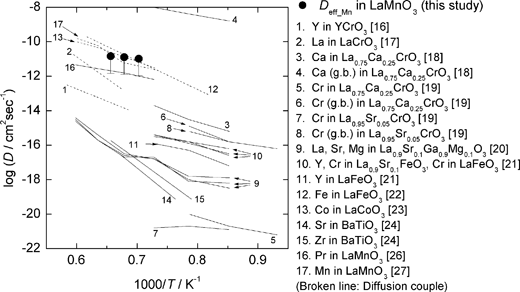 | ||
| Fig. 3 Arrhenius plot of the effective tracer diffusion coefficients of Mn in LaMnO3+δ. The downward error bars represent the possible ranges of the tracer bulk diffusion coefficients of Mn. The solid and broken lines denote previously reported cation diffusion coefficients in other perovskite systems obtained via SIMS depth profiling and solid-state reactions, respectively. Both the present and literature data were measured in air. | ||
The significantly high Mn diffusivity in LaMnO3+δ implies a rather high population of B-site vacancies in its cation-deficient defect regime, since it is expected that the cation diffusion in perovskite systems takes place via a vacancy-based mechanism. Here, it is necessary to pay attention to the preferential formation of vacancies at the A-sites. Provided that the formation of cation vacancies involves only creation of extra perovskite-type cells and no precipitation of second phases, the following defect models may be proposed. One of them (i) is that vacancies appear at both A- and B-sites with the same concentration; therefore, the chemical formula can be expressed as [La3/(3+δ)Vδ/(3+δ)]A[Mn3/(3+δ)Vδ/(3+δ)]BO3, where V denotes a vacancy. This simple model has been assumed in the majority of the investigations on the nonstoichiometry.3,4,6,8,9 On the other hand (ii), one may claim that vacancies preferentially exist at A-sites, because the B-site of the perovskite-type structure is more stable due to its 6-fold octahedral coordination with oxide ions compared to the A-site having 12-fold coordination. If this is the case, part of La will enter the B-sites to keep them fully occupied and to avoid the formation of secondary phases, resulting in the chemical formula of [La(3−δ)/(3+δ)V2δ/(3+δ)]A[Mn3/(3+δ)Laδ/(3+δ)]BO3. It should be noted that the above-mentioned cation vacancy/lattice creation mechanism still holds in this model (ii). While it may be difficult for La having a large ionic radius to occupy the B-site that is normally occupied by small cations, there are also some researchers who suggested this mechanism for the formation of A-site vacancies7,10,11. Recently, the bulk diffusivity of Pr3+ as the A-site tracer in LaMnO3+δ has been reported,26 which is slightly lower than the Mn diffusivity. The comparable A-site and B-site diffusivities may suggest the low probability of strong preferential formation of A-site vacancies over B-site vacancies.
The apparent activation energy of the Mn diffusivity in LaMnO3+δ is calculated as 0.6 ± 0.1 eV. In the literature, the activation energies of cation diffusion in perovskites as well as the diffusion coefficients themselves are rather scattered depending on the system. As an example of a high activation energy, a value of 2.8 eV was reported for the Y diffusivity in YCrO3 which was determined from the kinetics of the solid state reaction between Y2O3 and Cr2O3.16 On the other hand, the activation energy of Cr impurity diffusion in La0.9Sr0.1FeO3 is reported as 1.55 eV,21 which is a sort of low activation energy for a cation bulk diffusion coefficient in perovskites reported so far. The value of the activation energy of Mn diffusion from this study is again lower than the latter one.
For a vacancy diffusion mechanism, the self-diffusion coefficient D is proportional to the vacancy fraction in the concerned sublattice, [V], and the vacancy diffusion coefficient DV,
| D≈DV·[V] | (7) |
| Ea = Ed + Em | (8) |
With respect to the migration energy Em, De Souza et al. studied the energetics of cation migration in LaMnO3+δ with an atomistic calculation.32 After examination of several migration paths of B-site cations, the energetically most favourable one was found to be the “curved path” between adjacent Mn sites along the pseudo cubic 〈100〉 direction. This migration path lies in the (110) plane, and the saddle point locates between an oxide ion and a La ion in the (001) plane. However, the migration energy for the curved path still takes a rather high value around 8 eV. Interestingly, this migration energy reduces to 3.5 eV after removing the La ion near the saddle point. This drastic decrease is ascribed to the electrostatic repulsion between the migrating Mn ion and the neighbouring La ion which is missing when the La ion is removed. Thus the A-site vacancy facilitates the diffusion of Mn.
If we assume at first a simple vacancy mechanism of Mn diffusion, a combination of the above-mentioned Ed_B-site of −0.36 eV and Em of 8 eV32 yields Ea of ca. 7.6 eV from eqn (8), which is far larger than the value obtained in this study. On the other hand, the migration process that is mediated by a vacancy on an A-site gives Em of 3.5 eV, and the Mn diffusivity depends also on the vacancy fraction at A-sites, [V‴La], and not only on the B-site vacancy fraction [V‴Mn]. This assumption gives the following expression,
| DMn≈DV_B-site·[V‴La]·[V‴Mn] | (9) |
| Ea = Ed_A-site + Ed_B-site + Em | (10) |
A further decrease of Ea would result if both vacancies, VA and VB, would form a bound vacancy pair with a sufficiently negative binding energy, Epair. Then eqn (10) would change to
| Ea = Ed_A-site + Ed_B-site + Epair + Em | (11) |
3.3 P O2 dependence of the Mn diffusivity
Fig. 4 shows the Mn tracer diffusion coefficients in LaMnO3+δ as a function of PO2. Similarly to Fig. 3, the symbols with downward error bars denote the effective tracer diffusion coefficients determined from the diffusion profiles of the Harrison A-type kinetics, and the downward error bars indicate the possible range of the tracer bulk diffusion coefficients. The symbols with both-side error bars denote the tracer bulk diffusion coefficients extracted from the diffusion profile of the Harrison B-type kinetics. The PO2 dependence of the Mn diffusivity is rather large, and it does not seem to be linear (in the double logarithmic plot in Fig. 4). At high PO2 the slope is about 0.6 while at low PO2 it is nearly 1.5.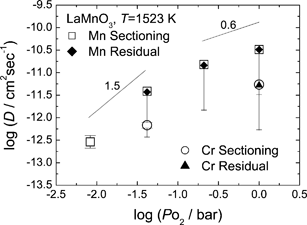 | ||
| Fig. 4 P O2 dependence of the effective tracer diffusion coefficients of Mn and Cr in LaMnO3+δ at 1523 K. The downward error bars represent the possible range of the tracer bulk diffusion coefficient. The symbols with both-side error bars denote effective diffusion coefficients extracted from the shallow part in sectioning profile of the Harrison B-type kinetics, and can be approximated as bulk diffusion coefficient. | ||
Assuming equal fractions of vacancies at A- and B-sites, the defect species that should be taken into consideration for the region of cation-deficient nonstoichiometry are V‴La, V‴Mn, and h˙. The charge neutrality is described as
| h˙ = 3[V‴La] + 3[V‴Mn] = 6[V‴Mn]. | (12) |
| 3/2O2(g) = V‴La + V‴Mn + 6h˙ + 3OO×. | (13) |
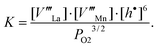 | (14) |
| [V‴Mn] = [V‴La] ∝PO23/16. | (15) |
If we assume a simple vacancy mechanism and a negligible dependence of the vacancy diffusivity on the defect concentration, the PO2 exponent of the diffusion coefficient is identical to that of the vacancy concentration as described by eqn (7). Then, both PO2 exponents of the Mn diffusivity expected from the above two defect models are much smaller than the present experimental results. If we take on the other hand the above-mentioned hypothesis of Mn diffusion that is mediated by A-site vacancies, DMn can be described by eqn (9). The PO2 dependence of DMn is then derived on the assumption of equal concentrations of A- and B-site vacancy as follows,
| DMn∝PO22n, | (16) |
However, the discrepancy between the predicted and experimentally-obtained PO2 dependence is still rather large. While the estimated Mn bulk diffusivities involve large error bars, the discrepancy may imply a diffusion mechanism which yields a stronger dependence on the cation vacancy concentrations. Such a suggestion can be also raised by the small temperature dependence mentioned above; that is, relying on the migration energy Em = 3.5 eV suggested by De Souza et al.,32 the contribution of the defect formation energy, Ed_B-site + Ed_A-site = −0.7 eV, is too small to explain the observed activation energy Ea of 0.6 eV.
3.4 Comparison of Mn diffusivity and Cr diffusivity
At some conditions, the radio-isotope 51Cr was also applied to observe the impurity diffusion behaviour. Similarly to the 54Mn tracer, diffusion profiles of both the Harrison A-type and B-type were observed, and successfully analyzed to extract the effective Cr tracer diffusion coefficients (and bulk diffusion coefficient in the diffusion profile of the B-type kinetics). As found in Fig. 4, the Cr diffusivity is ca. one order of magnitude lower than that of Mn, with the PO2 dependence being similar to the Mn diffusivity. The difference can be attributed to unequal jump frequencies of Mn and Cr ions, ω0 and ω2, respectively. The resulting impurity diffusion coefficient, of Cr is then obtained from D, in eqn (7) by multiplying with the factor ω2/ω0 (neglecting correlation effects).On the other hand, the Cr diffusivity in LaMnO3+δ is still significantly larger than cation diffusivities in other perovskite oxides summarized in Fig. 3. The fact that both of the isotope and impurity diffusion coefficients are significantly high among perovskites verifies that the fast cation diffusion in LaMnO3+δ originates from high vacancy population at cationic sites, not from jump frequency of cations.
4. Conclusions
The effective Mn tracer diffusion coefficient in polycrystalline LaMnO3+δ was successfully measured in the temperature range of 1423 K and 1523 K and PO2 range of 0.008 bar and 1 bar. The contribution of the bulk diffusion coefficient to the effective diffusion coefficient is estimated to be no less than 10%. The estimated bulk diffusion coefficient of Mn in LaMnO3+δ is quite high compared with other perovskite oxides, which is regarded to reflect the high cation vacancy concentration of LaMnO3+δ. Since the Mn diffusivity is comparable to the reported A-site diffusivity in LaMnO3+δ, it may not be plausible to assume a significant preference in the formation of A-site vacancies. The Mn diffusivity shows a small apparent activation energy of 0.6 eV and a large PO2 exponent m in DMn∝PO2n of 0.6–1.5. The weak temperature dependence and strong PO2 dependence are qualitatively in agreement with a Mn diffusion mechanism that is mediated by A-site vacancies. The Cr impurity diffusivity is significantly smaller than the Mn tracer diffusivity, which verifies the necessity of employing the isotope in order to properly evaluate the B-site diffusivity in LaMnO3+δ.Since the peculiar cation-deficient nonstoichiometry of LaMnO3+δ facilitates the fast cation diffusion in LaMnO3+δ, it is essential to pay attention to the cation nonstoichiometry in application to SOFC cathodes.
Acknowledgements
One of the authors (SM) acknowledges the financial support from The Canon Foundation in Europe for the research stay at the Institute of Physical Chemistry of RWTH Aachen University. The authors are also grateful to Dr Michael Schroeder and Ms Christa Fuhs for help in the radio−isotope experiments.References
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CAS.
- A. Urushibara, Y. Moritomo, T. Arima, A. Asamitsu, G. Kido and Y. Tokura, Phys. Rev. B, 1995, 51, 14103–14109 CrossRef CAS.
- N. Kamegashira, Y. Miyazaki and H. Yamamoto, Mater. Chem. Phys., 1984, 11, 187–194 CrossRef CAS.
- J. H. Kuo, H. U. Anderson and D. M. Sparlin, J. Solid State Chem., 1989, 83, 52–60 CrossRef CAS.
- J. Mizusaki, H. Tagawa, Y. Yonemura, H. Minamiue and H. Nambu, Proceedings of the 2nd International Symposium on Ionic and Mixed Conducting Ceramics, The Electrochem. Soc. Proceeding Series, Pennington, NJ, 1994, PV 94-12, 402-411 Search PubMed.
- J. A. M. van Roosmalen and E. H. P. Cordfunke, J. Solid State Chem., 1994, 110, 109–112 CrossRef CAS.
- J. Mizusaki, N. Mori, H. Takai, Y. Yonemura, H. Minamiue, H. Tagawa, M. Dokiya, H. Inaba, K. Naraya, T. Sasamoto and T. Hashimoto, Solid State Ionics, 2000, 129, 163–177 CrossRef CAS.
- J. A. M. van Roosmalen, E. H. P. Cordfunke and R. B. Helmholdt, J. Solid State Chem., 1994, 110, 100–105 CrossRef CAS.
- Q. Huang, A. Santoro, J. W. Lynn, R. W. Erwin, J. A. Borchers, J. L. Peng and R. L. Greene, Phys. Rev. B, 1997, 55, 14987–14999 CrossRef CAS.
- B. C. Tofield and W. R. Scott, J. Solid State Chem., 1974, 10, 183–194 CrossRef CAS.
- J. F. Mitchell, D. N. Argyriou, C. D. Potter, D. G. Hinks, J. D. Jorgensen and S. D. Bader, Phys. Rev. B, 1996, 54, 6172–6183 CrossRef CAS.
- S. Miyoshi, J.-O. Hong, K. Yashiro, A. Kaimai, Y. Nigara, K. Kawamura, T. Kawada and J. Mizusaki, Solid State Ionics, 2002, 154–155, 257–263 CrossRef CAS.
- S. Linderoth and M. Mogensen, Proc. 4th European Solid Oxide Fuel Cell Forum, European Fuel Cell Forum, Oberrohrdorf, 2000, vol. 1, 141 Search PubMed.
- M. Kuznecov, P. Otschik, P. Obenaus, K. Eichler and W. Schaffrath, Solid State Ionics, 2003, 157, 371–378 CrossRef CAS.
- M. Martin, Pure Appl. Chem., 2003, 75, 889–903 CrossRef CAS.
- K. Kawamura, A. Saiki, T. Maruyama and K. Nagata, J. Electrochem. Soc., 1995, 142, 3073–3077 CrossRef CAS.
- T. Akashi, M. Nanko, T. Maruyama, Y. Shiraishi and J. Tanabe, J. Electrochem. Soc., 1998, 145, 2090–2094 CAS.
- T. Horita, M. Ishikawa, K. Yamaji, N. Sakai, H. Yokokawa and M. Dokiya, Solid State Ionics, 1999, 124, 301–307 CrossRef CAS.
- N. Sakai, K. Yamaji, T. Horita, H. Negishi and H. Yokokawa, Solid State Ionics, 2000, 135, 469–474 CrossRef CAS.
- O. Schulz, M. Martin, C. Argirusis and G. Borchardt, Phys. Chem. Chem. Phys., 2003, 5, 2308–2313 RSC.
- I. Wærnhus, N. Sakai, H. Yokokawa, T. Grande, M.-A. Einarsrud and K. Wiik, Solid State Ionics, 2004, 175, 69–71 CrossRef CAS.
- J. B. Smith and T. Norby, Solid State Ionics, 2006, 177, 639–646 CrossRef CAS.
- M. Palcut, K. Wiik and T. Grande, J. Phys. Chem. B, 2007, 111, 2299–2308 CrossRef CAS.
- S. Koerfer, R. A. De Souza, H. I. Yoo and M. Martin, Solid State Sciences, 2008, 10, 725–734 CrossRef CAS.
- R. E. Cook, K. C. Goretta, J. Wolfenstine, P. Nash and J. L. Routbort, Acta Mater., 1999, 47, 2969–2980 CrossRef CAS.
- M. Palcut, J. S. Christensen, K. Wiik and T. Grande, Phys. Chem. Chem. Phys., 2008, 10, 6544–6552 RSC.
- M. Palcut, K. Wiik and T. Grande, J. Phys. Chem. C, 2007, 111, 813 CrossRef CAS.
- J.-H. Lee, M. Martin and H.-I. Yoo, J. Phys. Chem. Solids, 2000, 61, 1597–1605 CrossRef CAS.
- J. Crank, The Mathematics of Diffusion, Clarendon Press, Oxford, 1975 Search PubMed.
- I. Kaur and W. Gust, Fundamentals of Grain and Interphase Boundary Diffusion, Ziegler Press, Stuttgart, 1989 Search PubMed.
- A. D. Le Claire, Br. J. Appl. Phys., 1963, 14, 351 CrossRef.
- R. A. De Souza, M. S. Islam and E. Ivers-Tiffée, J. Mater. Chem., 1999, 9, 1621–1627 RSC.
Footnote |
| † Present address: Department of Materials Engineering, School of Engineering, The University of Tokyo, 113–8656 Tokyo, Japan |
| This journal is © the Owner Societies 2009 |