Ligand-dependent blinking of zinc-blende CdSe/ZnS core/shell nanocrystals†
Yongwook
Kim
a,
Nam Woong
Song
*bc,
Hyunung
Yu
b,
Dae Won
Moon
b,
Sung Jun
Lim
a,
Wonjung
Kim
a,
Hye-Joo
Yoon
a and
Seung
Koo Shin
*a
aBionanotechnology Center, Department of Chemistry, Pohang University of Science and Technology, San31 Hyoja-dong Nam-gu Pohang, Kyungbuk, 790-784, Korea. E-mail: skshin@postech.ac.kr; Fax: +82 (0)54 279![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3399; Tel: +82 (0)54 2792123
3399; Tel: +82 (0)54 2792123
bNanobio Fusion Research Center, Korea Research Institute of Standards and Science, 1 Doryong-dongYuseong-gu, Daejeon, 305-340, Korea. E-mail: nwsong@kriss.re.kr; Fax: +82 (0)42 8685032; Tel: +82 (0)42 8685214
cCell Dynamics Research Center, Gwangju Institute of Science and Technology, 261 Oryong-dong Buk-gu, Gwangju, 500-712, Korea
First published on 3rd March 2009
Abstract
Blinking of zinc-blende CdSe/ZnS core/shell nanocrystals are studied as a function of surface passivating ligands. Organic-soluble CdSe/ZnS core/shell nanocrystals are prepared by colloidal synthesis free of trioctylphosphine oxide and converted into water-soluble ones by ligand exchange with three different hydrophilic thiols, 2-aminoethanethiol, 3-mercapto-1-propanol, and 3-mercaptopropionic acid. The zinc-blende lattice structure is confirmed by powder X-ray diffraction, the size and shape distributions are visualized by high-resolution transmission electron microscopy, and hydrodynamic size distributions of water-soluble nanocrystals are determined by dynamic light scattering. Ligand-dependent optical properties, such as the absorption and emission spectra as well as the photoluminescence lifetime, are obtained in both solution and glass substrate to characterize the effects of ligand on the bright state of nanocrystals. The time trace of blinking is recorded for single nanocrystals in polymer film. Both on- and off-time distributions are fit to a power law. The off-time exponents are clustered at 1.67 ± 0.05, whereas the on-time exponents are scattered in the range of 1.71–2.25. The thiolate conjugation on the surface zinc atom greatly reduces the on-time duration, suggesting that the rate of photoinduced charge separation from the bright (on) to the dark (off) state increases as the number of surface hole-trap states increases.
Introduction
Optical properties and blinking of semiconductor nanocrystals have attracted much interest1–4 as the various needs for replacing conventional organic dyes with this new class of fluorophores grow in biological imaging5,6 and flow cytometry.7 Fluorescent nanocrystals allow a wide range of excitation wavelengths to yield size-tunable emission with a narrow bandwidth and offer great photostability with high photoluminescence (PL) efficiency. Thus long-term and/or multi-color tracking of biomolecules in a flow system or live cell can be carried out with a single excitation source. However, for quantitative measurements at the single-molecule level, the fluorescence intermittency or blinking of single nanocrystals poses a real challenge.8–10Although many types of single fluorophores, such as organic dyes, fluorescent proteins and conjugated polymers, are known to emit light intermittently,3,4 semiconductor nanocrystals exhibit by far the most long-lasting blinking phenomena that follow a simple power law,11p(t) ∝t−α, for both on- and off-time probability distributions.11–13 The power-law statistics holds over a wide range of timescale from microseconds to seconds regardless of nanocrystal composition, shape, and size under various temperatures and excitation fluences.14–17 To explain seemingly universal power-law behaviors, a number of models have been proposed to date.14–21 A general consensus is that blinking is a random switching between the neutral (on) and charged (off) states.2–4 For instance, the photoinduced ejection of an electron from the core of nanocrystal to the surrounding can leave a hole behind, thus creating a charge-separated state that efficiently quenches emission. However, the predicted power-law exponents vary with blinking models.2–4,14–21 The key difference among proposed models lies in the mobility of the separated charges.3 Trap models assume tunneling of the photoexcited electron between the emitting and immobile trap states,14,15 whereas diffusion models assume random walk of the ejected electron around the ionized nanocrystal.16,17 A more elaborate diffusion-controlled electron transfer18–20 or Anderson localization model21 extends the nature and dynamics of the trap or localized state. To unravel the mechanism of power-law blinking, we need to characterize the charge-separated states under well-controlled conditions. Such an attempt has been recently made by Cichos and co-workers by varying the dielectric media surrounding CdSe/ZnS nanocrystals.15 They have found an interesting correlation of the off-time exponent with the dielectric constant and proposed self-trapped states of the ejected electron in dielectric media as a source of power-law blinking.
Of the semiconductor nanocrystals, the CdSe/ZnS core/shell nanocrystal is one of the most extensively studied systems in blinking. Most of them are based on the wurtzite (W) CdSe nanocrystal.11–14,16,20,22 However, blinking studies on the zinc-blende (ZB) CdSe/ZnS nanocrystal have rarely been reported. We have previously shown that ZB CdSe-based nanocrystals have an advantage over W nanocrystals in that the shell can be grown more uniformly over the core and water-soluble ZB nanocrystals are highly luminescent (∼50% PL efficiency) with the least amount of shell thickness.23 We have also characterized the ligand coordinated on the surface of ZB CdSe/ZnS nanocrystals by time-of-flight (TOF) secondary ion mass spectrometry (SIMS).24 TOF-SIMS imaging has shown that organic-soluble nanocrystals are covered with stearate, whereas water-soluble ones are coated with thiolate via zinc-thiolate linkage. Thus our well-characterized ZB CdSe/ZnS nanocrystals allow variations in density of hole-trap states and offer a new opportunity to study the effects of surface ligand on blinking of single nanocrystals. Herein, we present evidence of hole-trap states mediating the on-time probability distribution.
We employ both organic- and water-soluble ZB CdSe/ZnS nanocrystals, obtain the absorption and emission spectra as well as PL lifetimes to characterize the effects of surface ligand on the bright emitting state of nanocrystals in solution and glass substrate, and monitor the blinking of single nanocrystals in polymer film.
Experimental
Preparation of zinc-blende CdSe/ZnS nanocrystals
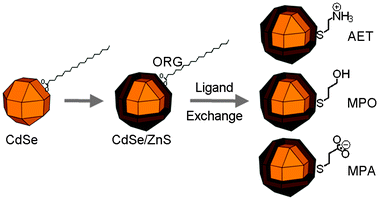 | ||
| Scheme 1 Preparation of organic-soluble (ORG) CdSe/ZnS nanocrystals and water-soluble CdSe/ZnS nanocrystals by ligand exchange with 2-aminoethanethiol (AET), 3-mercaptopropanol (MPO), and 3-mercaptopropinic acid (MPA). | ||
Characterization of nanocrystals
The output from a 543.5 nm HeNe laser was coupled to a single-mode fiber, filtered at the end, and directed to a Carl Zeiss Fluar 100× oil-immersion objective. At the sample spot, a Gaussian laser beam was ∼300 nm in diameter, and the laser fluence of 2.8 and 28 kW cm−2 was used for blinking and spectrum acquisition, respectively. The emitted light was collected with the same objective and focused onto a detector. A Dongwoo Optron 150-mm spectrograph (300 gr mm−1) equipped with a Princeton Instruments 1340 × 100 TE/CCD was used to take the PL spectra. A PerkinElmer APD photon counting module SPCM-AQR-13-FC was used to take the image and blinking. A 100 × 100 pixel image of single nanocrystals was obtained over a 20 × 20 μm area by rastering a sample on a Physik Instrumente P-611.3S nanopositioning system with 10 ms duration at each pixel. A time trace of blinking was monitored for 10–20 s with 1 ms bin by positioning the laser beam over a bright spot. About 50–100 single spots were tested from 4–5 different areas.
Results and discussion
The lattice structure and size distributions of nanocrystals
The XRD patterns of the CdSe core and CdSe/ZnS core/shell nanocrystals are shown in Fig. 1. An HRTEM image of a single nanocrystal is also presented in the inset. Both the core and core/shell nanocrystals show the ZB patterns, indicating the epitaxial growth of the ZnS shell. The size and shape of nanocrystals are displayed in Fig. S1 of the ESI.† CdSe nanocrystals appear monodisperse in size and nearly spherical in shape, while CdSe/ZnS nanocrystals are bigger in size. The average size is 3.8 ± 0.5 and 5.7 ± 0.6 nm for CdSe and CdSe/ZnS nanocrystals, respectively.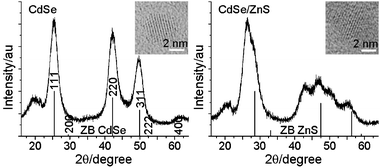 | ||
| Fig. 1 Powder X-ray diffraction patterns of CdSe and CdSe/ZnS nanocrystals. The inset shows an HRTEM image of a single nanocrystal. Vertical lines represent the diffraction patterns for bulk zinc-blende (ZB) compounds. | ||
The hydrodynamic size distributions of WS-NCs are shown in Fig. S2 of the ESI.† The MPA-capped CdSe nanocrystal is also compared with CdSe/ZnS WS-NCs. The average hydrodynamic size is 10.0 ± 2.0, 10.2 ± 2.4, 12.7 ± 3.0 nm for AET-, MPO-, and MPA-capped CdSe/ZnS, respectively, and 9.4 ± 1.6 nm for MPA-capped CdSe. Yamamoto and co-workers have reported a broad size distribution around 40 nm for AET-capped CdSe/ZnS synthesized in trioctylphosphine oxide (TOPO).26 By contrast, our TOPO-free nanocrystals yield a narrow size distribution with no indication of clusters greater than 20 nm. In TOPO-based nanocrystals, the impurities present in TOPO, such as phosphinic acids, may coordinate strongly onto the surface, survive the ligand exchange step and facilitate the cluster formation via electrostatic interactions with the terminal ammonium groups on adjacent nanocrystals. MPA-capped nanocrystals appear to be larger than others. A difference of 3.3 nm between the two MPA-capped CdSe/ZnS and CdSe nanocrystals is greater than the thickness of the ZnS shell (6 × 3.1 Å).
Ligand-dependent absorption and emission spectra
Because all WS-NCs showed narrow hydrodynamic size distributions without forming clusters at 2 μM concentration, we took the optical spectra in solution at 1 μM concentration to represent the ensemble average of single nanocrystals under cluster-free conditions.Both the absorption and emission spectra are presented in Fig. 2. The 1S3/21Se absorption band appears at 596 and 606 nm for CdSe and CdSe/ZnS ORG-NCs in chloroform, respectively, and 600, 596 and 600 nm for AET-, MPO- and MPA-capped WS-NCs, respectively. The ZnS shell passivation induces a 34-meV redshift in chloroform, while the zinc-thiolate linkage on the ZnS shell leads to a 20–34-meV blueshift in water. The solvatochromatic effect is supposed to induce a redshift in absorption rather than a blueshift as the solvent dielectric constant (ε) increases from 4.8 (chloroform) to 78 (water).27 The observed spectral shifts indicate the perturbation of the CdSe core electronic structure by surface passivation. TOF-SIMS imaging has shown that ORG-NCs are covered with weakly-coordinating stearates, while WS-NCs are covered with covalently-coordinating thiolate ligands.24 The surface dangling bonds on the ZnS shell are different in the two types of nanocrystals. Stearate-covered ORG-NCs face the full extent of surface reconstruction due to the dangling bonds, which could lead to the lattice distortion of the CdSe core that induces a redshift in absorption.23 On the other hand, thiolate-covered WS-NCs face such a distortion to a lesser degree because the zinc-thiolate linkage effectively uses up the dangling bonds of the surface zinc atoms, which could turn the spectral shift back to normal.
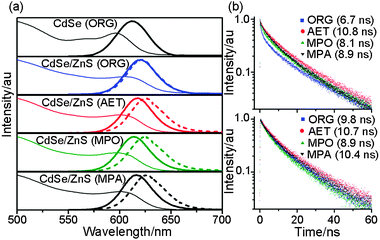 | ||
| Fig. 2 (a) Absorption (thin line) and emission (thick line) spectra of CdSe and CdSe/ZnS nanocrystals soluble in chloroform and of AET-, MPO-, and MPA-capped CdSe/ZnS nanocrystals soluble in water. The dashed line denotes the emission spectra of nanocrystal aggregates on a glass substrate; (b) (top) PL decay of organic-soluble (ORG) CdSe/ZnS nanocrystals in chloroform and AET-, MPO-, and MPA-capped CdSe/ZnS nanocrystals in water; (bottom) PL decay of organic- and water-soluble CdSe/ZnS nanocrystal aggregates on a glass substrate. | ||
The emission spectra of nanocrystals in solution (Fig. 2) show the variation of the Stokes shift between the absorption peak and the 1Se1S3/2 emission maximum depending on the type of surface ligands. The PL maximum appears at 611 and 621 nm for CdSe and CdSe/ZnS ORG-NCs in chloroform, respectively, but with almost identical Stokes shift of 49–51 meV. WS-NCs yield the PL maximum at 615–618 nm with Stokes shift of 57–61 meV. Values of full width at half maximum are listed in Table 1 along with other optical properties. The Stokes shift of WS-NCs is 8–12 meV greater than that of ORG-NCs.
| Nanocrystals in solutiona | Aggregates on glass substrateb | Single nanocrystals in polymer filmc | ||||
|---|---|---|---|---|---|---|
| Type of surface ligands | λ abs d/nm | λ em e/nm | τ eff f/ns | λ em e/nm | τ eff f/ns | 〈λem〉g/nm |
| a ORG-NCs were in chloroform and WS-NCs were in distilled water. b Nanocrystal aggregates were formed by drying a drop of nanocrystal solution on glass substrate. c ORG-NCs were in PMMA, while WS-NCs were in PVA. d The band edge absorption peak for the 1S3/21Se electronic transition. e The emission maximum (full width at half maximum in parenthesis). f The effective lifetime calculated from the multiple-component PL decay curve. g The average value of emission maximum from 50 different single NCs. | ||||||
| ORG | 606 | 621 (36) | 6.7 | 622 (36) | 9.8 | 614 |
| AET | 600 | 618 (34) | 10.8 | 627 (37) | 10.7 | 618 |
| MPO | 596 | 615 (35) | 8.1 | 626 (39) | 8.9 | 620 |
| MPA | 600 | 617 (35) | 8.9 | 628 (38) | 10.4 | 617 |
The emission spectra of nanocrystal aggregates on a glass substrate (dotted lines in Fig. 2) are red-shifted by different degrees depending on the surface ligand. Stearate-covered ORG-NCs show little shift in emission from 621 to 622 nm, whereas thiolate-covered WS-NCs yield 26–36 meV redshift from 615–618 to 626–628 nm. The two types of ligands differ in their alkyl chain lengths. Stearate has a C18-alkyl chain, but thiolates have C2–C3 alkyl chains. To check the effects of alkyl chain length on the spectral shift in aggregates, we replaced C3-alkyl MPO with C11-alkyl mercaptoundecanol (MUO). MUO-capped NCs result in only a small redshift in emission upon transition from solution to aggregates (data not shown). Thus, the observed redshift in emission from WS-NCs covered with short-chain thiolates is very likely due to the Förster resonance energy transfer (FRET) between different size nanocrystals within the aggregates. Because the FRET efficiency is inversely proportional to the sixth power of the distance between the donor and acceptor fluorophores, the amount of spectral shift should decrease with increasing chain length of the surface ligand. Provided that nanocrystals form close-packed but amorphous aggregates on a glass substrate as the solvent evaporates slowly,28 the inter-particle distance in WS-NC aggregates might be 3–4 nm shorter than the distance in ORG-NC aggregates, which could lead to the redshift in emission.
Ligand-dependent photoluminescence lifetimes
The temporal variation of PL decay is presented in Fig. 2b. In solution (Fig. 2b top), the PL lifetimes are 6.7 ns for ORG-NCs, 8.1 and 8.9 ns for MPO- and MPA-capped WS-NCs, respectively, and 10.8 ns for AET-capped WS-NCs. These lifetime variations correlate well with a depletion of the initial 0.17–0.29 ns decay component and a concomitant rise of the 12–15 ns decay component as shown in Fig. S3 of the ESI.† In aggregates (Fig. 2b bottom), the PL lifetime increases to 9.8 ns for ORG-NCs, 8.9 and 10.4 ns for MPO- and MPA-capped WS-NCs, respectively, but remains unchanged at 10.7 ns for AET-capped NCs. The lifetime elongation in both ORG-NCs and MPA-capped WS-NCs may be due to the reduction in the nonradiative relaxation rate. As the environment changes from solution to aggregates, stearate forms self-assembled monolayers, while MPA forms hydrogen bond networks between adjacent carboxyl groups upon conversion of the terminal carboxylate moiety in water into the carboxyl group in aggregates. Consequently, the surface layers formed by both stearate and MPA become rigid, thereby reducing the rate of nonradiative relaxation. Meanwhile, relatively small variations in lifetime for both MPO- and AET-capped WS-NCs indicate that the structures of surface layers formed by hydroxyl and amine groups remain unchanged under different conditions.Ligand-dependent blinking
We first obtained the PL image of single NCs dispersed in polymer film, and then acquired the PL spectra from more than 480 bright spots to construct the ensemble average spectra. Finally, we recorded the time trace of blinking at a few isolated bright spots. Images of single ORG-NCs in PMMA film as well as the ensemble average spectra with statistical distributions of the PL maxima are shown in Fig. 3. The statistical average yields the PL maximum at 614 nm for ORG-NCs in PMMA; 618, 620, and 617 nm for AET-, MPO-, and MPA-capped WS-NCs in PVA, respectively. Interestingly, ORG-NCs in PMMA film show a 7 nm blueshift in emission relative to those in solution, whereas MPO-capped NCs in PVA film show a 5 nm redshift. On the other hand, both AET- and MPA-capped NCs in PVA film show no shift. To find out the origin of spectral shifts, we prepared both ORG-NCs (0.5 μM nanocrystals in 0.63 mL of chloroform containing 50 mg of PMMA) and MPO-capped WS-NCs (0.25 μM nanocrystals in 0.95 mL of water containing 50 mg of PVA) in polymer solutions and spin-cast them on a cover glass. The PL maxima of nanocrystals in polymer films remain unchanged from those in solution (0.9 μM ORG-NCs in chloroform and 0.5 μM MPO-capped WS-NCs in water). As the concentration of nanocrystals decreases to 0.1 μM, ORG-NCs are blue-shifted by 5 nm, whereas MPO-capped WS-NCs are red-shifted by 2 nm, following the same trend as the spectral shifts observed in the statistical average. These results suggest that the spectral shift arises from the interaction of single nanocrystals with polymer matrices. Striccoli and co-workers have previously suggested that the strong dielectric confinement by polymeric surroundings causes a blueshift in band-edge absorption of CdS nanocrystals embedded in PMMA.29 Similarly, the blueshift in emission of ORG-NCs in PMMA is ascribed to the coordination of the carbonyls of PMMA to the zinc atoms on the surface of CdSe/ZnS nanocrystals. On the other hand, the absence of such a spectral shift in both AET- and MPA-capped WS-NCs indicates that the PVA hydroxyl group is not so effective for the CdSe/ZnS surface coordination. In the meantime, the redshift in MPO-capped WS-NCs may be due to the solvation of the hydroxyl group of MPO by PVA matrices, which could peel off the ligands and leave the bare nanocrystal surface behind.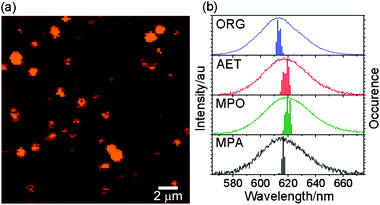 | ||
| Fig. 3 (a) Image of single organic-soluble CdSe/ZnS nanocrystals in PMMA film; (b) the ensemble average spectra of organic- and water-soluble CdSe/ZnS nanocrystals and the statistical distribution of the PL maximum. | ||
The blinking time trace of ORG-NCs in PMMA is shown in Fig. 4a along with an occurrence histogram on the right. A threshold separating “on” from “off” state in a bimodal distribution is set at three times the standard deviation of the average dark count. Both on- and off-time distributions are presented in Fig. 4b and 4c, respectively, and they are fit to a power law of the form:
| P(t) = At−α |
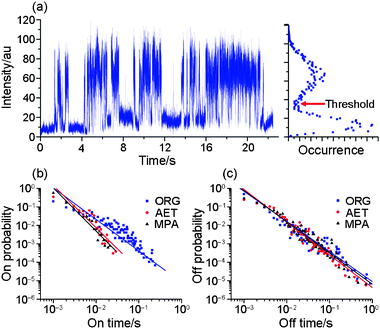 | ||
| Fig. 4 (a) A blinking time trace of a single organic-soluble CdSe/ZnS nanocrystal and a histogram of blinking; (b) the on-time probability distributions and (c) the off-time probability distributions of organic- and water-soluble CdSe/ZnS nanocrystals. The lines denote the least-square fit to a power law. | ||
In the present experiment, both ORG- and WS-NCs are derived from the same batch of TOPO-free colloidal synthesis, so that the only difference lies in the surface ligands. Moreover, the surface ligands and optical properties of both ORG- and WS-NCs are well characterized. In ensemble measurements, the spectral features of WS-NCs are almost identical to that of ORG-NC, and the PL lifetimes of WS-NCs are of the same order of magnitude as that of ORG-NC. However, the blinking of single nanocrystals presents remarkable ligand-dependent phenomena. The on-state durations strongly depend on surface ligand, as noted by others.10,20,30 Apparently, the zinc-thiolate linkage in WS-NCs reduces the on-state durations more than the stearate and/or PMMA carbonyl coordination in ORG-NCs does. Thus we can draw a meaningful correlation between the mode of surface conjugation and blinking. The zinc-thiolate linkage forms a coordinate covalent bond, whereas stearate and/or PMMA carbonyls noncovalently coordinate onto the surface zinc atom. The zinc-thiolate linkage effectively uses up the surface dangling bonds of the zinc atom, which removes the electron-trap states, but at the same time the two nonbonding electron pairs of the sulfur atom become the hole-trap states. To the contrary, the stearate and/or PMMA carbonyl coordination perturbs the energy levels of the band structure, but they do not remove or add any new trap states. As a result, the zinc-thiolate linkage on WS-NCs, which increases the density of hole-trap states, ends up suppressing the on-state durations. This interpretation is in accord with a report supposing that thiolate deactivates electron-trap states at low concentrtions and introduces new hole-trap states at high concentrations.31 It is worth mentioning the previous finding of near-complete suppression of blinking in streptavidin-coated CdSe/ZnS nanocrystals by β-mercaptoethanol in pH 7.4 buffer.32 Because β-mercaptoethanol hardly dissociates to thiolate at neutral pH,31 we think their suppression of blinking is not due to thiolates. In our system, hydrophilic thiols are shifted to thiolates by equilibrium under basic conditions. Thus, the thiolates conjugated onto the surface zinc atom are the cause of on-time variation.
Conclusion
Colloidal syntheses free of trioctylphosphine oxide allow the preparation of zinc-blende CdSe/ZnS core/shell nanocrystals with well-defined surface ligands in the absence of unknown impurities. Both organic-soluble CdSe/ZnS nanocrystals covered with stearates and water-soluble CdSe/ZnS nanocrystals covered with bifunctional hydrophilic thiolates offer a unique opportunity of investigating the effects of density of hole-trap states on the blinking of single nanocrystals. The off-time distributions show almost no dependence on surface ligand, whereas the on-time distributions display strong dependence. As the number of surface hole-trap states increases upon zinc-thiolate conjugation, the number of paths to the charge-separated dark states increases, and so does the rate of photoinduced charge separation process. The blinking model should consider the multiple competing pathways from the bright to the charge-separated state that randomly proceed back and forth. Further studies are warranted to assess the effects of shell thickness on ligand-dependent blinking of single nanocrystals.Acknowledgements
We acknowledge supports from POSTECH Biotech Center, Advanced Scientific Analysis Instruments Development Project (Grant No. RH0-2005-000-01004-0), Korea Research Foundation (Grant No. KRF-2006-005-J01202), Korea Foundation for International Cooperation of Science and Technology (K20501000002-07-E0100-00210), Pioneer Research Program for Converging Technology (Grant No. M10711250003-08M1125-00310) and Korea Science and Engineering Foundation (Grant No. R11-2007-007-03003-0).References
- S. K. Shin, H.-J. Yoon, Y. J. Jung and J. W. Park, Curr. Opin. Chem. Biol., 2006, 10, 423–429 CrossRef CAS.
- D. E. Gómez, M. Califano and P. Mulvaney, Phys. Chem. Chem. Phys., 2006, 8, 4989–5011 RSC.
- F. Cichos, C. von Borczyskowski and M. Orrit, Curr. Opin. Colloid Interface Sci., 2007, 12, 272–284 CrossRef CAS.
- P. Frantsuzov, M. Kuno, B. Jankó and R. A. Marcus, Nat. Phys., 2008, 4, 519–522 CrossRef.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- P. K. Chattopadhyay, D. A. Price, T. F. Harper, M. R. Betts, J. Yu, E. Gostick, S. P. Perfetto, P. Goepfert, R. A. Koup, S. C. De Rosa, M. P. Bruchez and M. Roederer, Nat. Med., 2006, 12, 972–977 CrossRef CAS.
- M. Nirmal, B. O. Dabbousi, M. G. Bawendi, J. J. Macklin, J. K. Trautman, T. D. Harris and L. E. Brus, Nature, 1996, 383, 802–804 CrossRef CAS.
- A. L. Efros and M. Rosen, Phys. Rev. Lett., 1997, 78, 1110–1113 CrossRef CAS.
- J. R. Krogmeier, H. G. Kang, M. L. Clarke, P. Yim and J. Hwang, Opt. Commun., 2008, 281, 1781–1788 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2000, 112, 3117–3120 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028–1040 CrossRef CAS.
- V. Fomenko and D. J. Nesbitt, Nano Lett., 2008, 8, 287–293 CrossRef CAS.
- R. Verberk, A. M. van Oijen and M. Orrit, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 233202 CrossRef.
- A. Issac, C. von Borczyskowski and F. Cichos, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 161302 CrossRef (R).
- K. T. Shimizu, W. K. Woo, B. R. Fisher, H. J. Eisler and M. G. Bawendi, Phys. Rev. Lett., 2002, 89, 117401 CrossRef CAS.
- M. Kuno, D. P. Fromm, S. T. Johnson, A. Gallagher and D. J. Nesbitt, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 125304 CrossRef.
- P. A. Frantsuzov and R. A. Marcus, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 155321 CrossRef.
- J. Tang and R. A. Marcus, Phys. Rev. Lett., 2005, 95, 107401 CrossRef.
- M. Pelton, G. Smith, N. F. Scherer and R. A. Marcus, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14249–14254 CrossRef.
- X. Xia and R. J. Silbey, arXiv:cond-mat/0601092v4 [cond-mat.mes-hall], 2006.
- K. T. Shimizu, R. G. Neuhauser, C. A. Leatherdale, S. A. Empedocles, W. K. Woo and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 205316 CrossRef.
- S. J. Lim, B. Chon, T. Joo and S. K. Shin, J. Phys. Chem. C, 2008, 112, 1744–1747 CrossRef CAS.
- H. Min, Y. Kim, H. Yu, D. W. Moon, S. J. Lim, H.-J. Yoon, T. G. Lee and S. K. Shin, Chem.–Eur. J., 2008, 14, 8461–8464 CrossRef CAS.
- I. Rasnik, S. A. McKinney and T. Ha, Nat. Methods, 2006, 3, 891–893 CrossRef CAS.
- A. Hoshino, K. Fujioka, T. Oku, M. Suga, Y. F. Sasaki, T. Ohta, M. Yasuhara, K. Suzuki and K. Yamamoto, Nano Lett., 2004, 4, 2163–2169 CrossRef CAS.
- C. A. Leatherdale and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 165315 CrossRef.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- M. Tamborra, M. Striccoli, R. Comparelli, M. L. Curri, A. Petrella and A. Agostiano, Nanotechnology, 2004, 15, S240–S244 CrossRef CAS.
- D. E. Gómez, J. van Embden, J. Jasieniak, T. A. Smith and P. Mulvaney, Small, 2006, 2, 204–208 CrossRef CAS.
- S. Jeong, M. Achermann, J. Nanda, S. Ivanov, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2005, 127, 10126–10127 CrossRef CAS.
- S. Hohng and T. Ha, J. Am. Chem. Soc., 2004, 126, 1324–1325 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: High-resolution transmission electron microscope images, hydrodynamic size distributions obtained from dynamic light scattering and variation profiles of photoluminescence decay components. See DOI: 10.1039/b822351c |
| This journal is © the Owner Societies 2009 |