Study of the Beckmann rearrangement of acetophenone oxime over porous solids by means of solid state NMR spectroscopy
Ana Belen Fernandez, Ines Lezcano-Gonzalez, Mercedes Boronat, Teresa Blasco* and Avelino Corma
Instituto de Tecnología Química (UPV-CSIC), Avda. de los Naranjos s/n, 46022, Valencia, Spain. E-mail: tblasco@itq.upv.es; Fax: +34 96 387 9444; Tel: +34 96 387 7812
First published on 2nd April 2009
Abstract
The Beckmann rearrangement of acetophenone oxime using zeolite H-beta and silicalite-N as catalysts has been investigated by means of 15N and 13C solid state NMR spectroscopy in combination with theoretical calculations. The results obtained show that the oxime is N-protonated at room temperature on the acid sites of zeolite H-beta. At reaction temperatures of 423 K or above, the two isomeric amides, acetanilide and N-methyl benzamide (NMB) are formed, and interact with the Brønsted acid sites of zeolite H-beta through hydrogen bonds. The presence of residual water hydrolyzes the two amides, while larger amounts inhibit the formation of NMB and cause the total hydrolysis of the acetanilide. Over siliceous zeolite silicalite-N, containing silanol nests as active sites, the oxime is adsorbed through hydrogen bonds and only acetanilide is formed at reaction temperatures of 423 K or above. In the presence of water, the reaction starts at 473 K, still being very selective up to 573 K, and the amide is partially hydrolyzed only above this temperature .
Introduction
The Beckmann rearrangement of ketoximes into amides is a common reaction used in organic chemistry, and has been widely studied for many years.1 One of the most important industrial applications of this reaction is the transformation of cyclohexanone and cyclododecanone oximes into ε-caprolactam and ω-laurolactam, respectively, which are raw materials in the fabrication of fibers (nylon). The industrial production of lactams by the Beckmann rearrangement of cyclic oximes is classically catalyzed by liquid acids and much work has been carried out with the objective of substituting this homogeneous process for an environmentally friendly, heterogenous one.2–4 The research efforts of the last fifteen years have led to the commercialization of a cleaner process by using silica-rich MFI-type zeolite, consisting of a three-dimensional network of 10-membered ring channels (average pore diameter 5.5 Å), as catalysts. Sumitomo Chemical Co. has constructed a production plant which has been working in Japan since 2003.5–7Another potential industrial application of the Beckmann rearrangement reaction is the synthesis of paracetamol (N-acetyl-p-aminophenol). Conventionally, this product is commercially synthesized by acetylating p-aminophenol with acetic anhydride, but this process presents some difficulties because of the occurrence of unwanted reactions. An alternative, new process involves the Beckmann rearrangement of 4-hydroxyacetophenone oxime with liquid acids,8 however, the use of these catalysts requires extensive purification of the final reaction product for human use.9 Therefore, environmentally friendly solid acids such as zeolites have been successfully used as catalysts in the liquid-phase Beckmann rearrangement of 4-hydroxyacetophenone oxime to paracetamol.10–11
Acetanilide is an amide related to paracetamol which has also been synthesized by the Beckmann rearrangement of the corresponding (acetophenone-) oxime using solid acids as catalysts.12–14 Acetanilide possesses industrial interest as an intermediate in the synthesis of pharmaceuticals and as an additive in hydrogen peroxide, varnishes, polymers and rubbers. However, there are only a few publications on the heterogeneous rearrangement of acetophenone oxime and most of them in the liquid phase.12–14
The aim of this work is to investigate the reaction mechanism of the Beckmann rearrangement of acetophenone oxime using zeolites containing active sites of differing nature as catalysts. This knowledge is essential to the design of catalysts with adequate properties for the potential development of green technologies to produce linear amides of pharmacological interest, containing benzyl groups with different ring substituents. For this purpose, we have studied the rearrangement of acetophenone oxime over two different zeolites: Al-containing zeolite beta with Brønsted acid sites, and silicalite with silanol nests as active sites, combining in situ solid state NMR and theoretical calculations. Zeolite beta possesses a three-dimensional network of 12-membered ring channels with an average pore diameter of ca. 6.7 Å. Defective silicalite zeolite was chosen because of its good catalytic performance in the rearrangement of cyclic oximes.7,15,16
Experimental
Materials
Zeolite H-beta (Si/Al = 12.5, crystal sizes of 0.1–0.2 μm), produced by Zeolyst, is commercially available (CP811) and presents only minor amounts of extra-framework (octahedral) aluminium as detected by solid state 27Al NMR. Silicalite was synthesized following the method described in previous publications,16,17 and subsequently submitted to basic treatment to generate silanol nests (silicalite-N) as follows: 5 g of zeolite calcined at 823 K for 12 h were impregnated with 20 g of an aqueous solution of ammonia (25 wt%) and ammonium nitrate (7.5 wt%) and heated in a stainless steel autoclave at 373 K overnight.16 The final silicalite-N (crystal sizes around 0.5 μm) catalyst was recovered after washing with deionized water, filtering and drying at 383 K for 4 h. (α-13C, 15N)-Acetophenone oxime was prepared mixing 14.4 mmol of 15N-hydroxylamine hydrochloride (98% 15N, Cambridge Isotope Laboratories) with 4.3 mmol of α-13C-acetophenone, 24.4 mmol of sodium acetate and 222.2 mmol of water, and heating at 368 K for 1 h. Before use, the 15N-acetophenone oxime was re-crystallized in ether and identified by NMR.Solid state NMR spectroscopy
Solid state NMR spectra were recorded at room temperature on a Bruker AV 400 WB spectrometer. Samples were dehydrated at 673 K overnight reaching a final pressure of 10−5 mbar. To record 1H NMR spectra, a portion of the sample was transferred into a rotor within a glove box under an atmosphere of N2. The spectra were recorded with a BL4 probe spinning the sample at 10 kHz, by using 90° pulses of 5 μs and recycle delays of 15 s. To study the reaction, 300 mg of catalyst outgassed at 673 K was mixed with 15 mg of (α-13C, 15N)-acetophenone oxime and homogenized under an inert atmosphere in a glove box. A portion of the mixture was introduced into a glass insert, which was sealed after outgassing at room temperature. The 1H to 15N (1H/15N) cross-polarization (CP) MAS spectra were recorded with a 1H 90° pulse of 5 μs, a contact time of 5 ms and a recycle delay of 5 s. The 1H to 13C (1H/13C) CP-MAS spectra were recorded with a 90° pulse for 1H of 5 μs, a contact time of 5 ms and a recycle delay of 5 s.Computational details
Silanol defects and Brønsted acid sites were simulated by means of the Si(OSiH3)3OH and Al(OSiH3)3(OH)SiH3 cluster models, respectively, as described in previous work.18 The geometries of the two zeolitic clusters and the complexes resulting from the adsorption of acetophenone oxime, and of the possible reaction products (acetanilide, N-methyl benzamide, aniline,…) were optimized using the density functional B3PW91 method and the standard 6-31G(d,p) basis set.19–21 In these calculations, the coordinates of all atoms except the terminal H of the SiH3 groups were fully optimized. Isotropic absolute chemical shielding constants were calculated with the B3PW91/6-31G(d,p) method on geometries optimized at the same level, using the gauge including atomic orbitals (GIAO) approach.22,2315N and 13C chemical shifts were calculated as δ = σref−σ, and corrected with the equations obtained from a preliminary study of the performance of B3PW91 functional.18 All calculations in this work were performed using the GAUSSIAN98 computer program.24Results and discussion
Theoretical calculations
The calculations on the adsorption of acetophenone oxime over silanol and Brønsted acid sites in zeolite beta has been previously reported by our group.18 Our results indicated that the N-protonated oxime is readily formed upon adsorption at Brønsted acid sites, whereas the oxime is adsorbed over silanol groups forming H-bonds.18 These conclusions agree with theoretical and experimental results reported by other groups.25–27 Here, we focus our study on the interaction of the reaction products of the Beckmann rearrangement of acetophenone oxime with these same types of zeolite sites i.e., silanols and bridging hydroxyl groups.The reaction product of the Beckmann rearrangement of acetophenone oxime is acetanilide, which results from the migration of the phenyl group in the anti position with respect to the hydroxyl group. Although this reaction is highly stereospecific, we have also considered the N-methyl benzamide (NMB) isomer, as it has been reported to appear as a by-product of the rearrangement of acetophenone oxime.12 Accordingly, we have optimized the adsorption complexes of the two amides, acetanilide and NMB, on silanol (Acetanilide/Silanol and NMB/Silanol) and bridging Si–OH–Al (Acetanilide/Brønsted and NMB/Brønsted) zeolite groups, and obtained the models depicted in Scheme 1. The 15N and 13C (the 13C-carbonyl group) NMR chemical shifts and adsorption energies, calculated using density functional methods, are summarized in Table 1. The distances between the amides and zeolite hydroxyl groups, depicted in the models of Scheme 1, are consistent with the formation of hydrogen bonds, which are shorter with Brønsted acid sites, resulting in more stable complexes (models Acetanilide/Brønsted and NMB/Brønsted in Scheme 1). Regarding chemical shifts, the results summarized in Table 1 show that δ15Ncalc of acetanilide and NMB experience larger shifts to low field when they interact with bridging hydroxyl groups, where they are more strongly adsorbed. We must note that the 15N chemical shifts calculated for NMB/Brønsted and the two acetanilide complexes, i.e., Acetanilide/Brønsted and Acetanilide/Silanol, are in the range −235 to −256 ppm, while displacement to high field is calculated for NMB/Silanol (δ15Ncalc = −284.6 ppm, see Table 1). The structural models calculated for acetanilide and NMB suggest that neither of the two amides becomes protonated on Brønsted acid sites (Scheme 1), in contrast with the formation of O-protonated lactams (cyclic amides) reported previously.28,29 This difference suggests that acetanilide and NMB are less basic than cyclic caprolactam and ω-laurolactam.
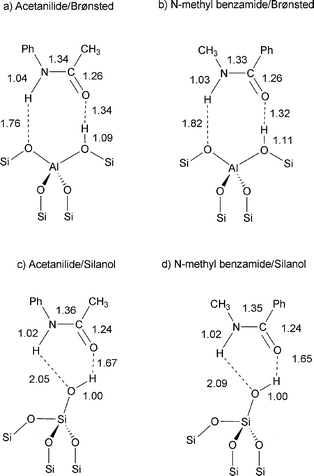 | ||
| Scheme 1 Optimized structures of acetanilide (a, c) and N-methyl benzamide (b, d) adsorbed on bridging Si–OH–Al (a, b) and silanol zeolite groups (c, d). Bond distances are expressed in angstroms. | ||
| Eads/kcal mol−1 | δ15Ncalc/ppm | δ13Ccalc/ppm | δ15Nexp/ppm | δ13Cexp/ppm | |
|---|---|---|---|---|---|
| a NMB is not formed in pure silica solids.b From a 13C NMR spectrum of NMB on H-beta zeolite, not shown. | |||||
| Acetanilide | — | −247.8 | 165.5 | — | 176.0 |
| Acetanilide/Silanol | −9.1 | −248.0 | 174.2 | −246.0 | 173.0 |
| Acetanilide/Brønsted | −22.2 | −235.9 | 175.6 | −230.0 | 177.0 |
| Acetanilide/H2O | −248.2 | 168.2 | −250.0 | 176.0 | |
| NMB | — | −293.2 | 165.8 | — | 166.0 |
| NMB/Silanol | −8.0 | −284.6 | 175.1 | a | a |
| NMB/Brønsted | −21.8 | −255.7 | 177.3 | −245.0 | 168.0b |
The presence of residual water, difficult to completely remove from the reaction medium, can produce some hydrolysis of the oxime and/or the amide, depending on the catalyst properties. Accordingly, we have also theoretically calculated the complexes resulting from the interaction of the hydrolysis products of acetanilide (aniline and acetic acid) and of NMB (benzoic acid and methylamine) with a zeolite Brønsted acid site as depicted in Scheme 2. The simulation of the interaction with silanol groups has been omitted, since no noticeable changes on chemical shifts are expected according to the results for the amides reported in Table 1. The 15N and 13C (of the carboxylate group) NMR chemical shifts calculated for the complexes of Scheme 2 by using ab initio methods, together with the experimental values reported in the bibliography are summarized in Table 2. We must note that, according to Scheme 2, both amines, i.e., aniline and methylamine, become protonated on the Brønsted acid sites of the zeolite; though their δ15N values are only slightly shifted when compared with the neat amine (see Table 2).
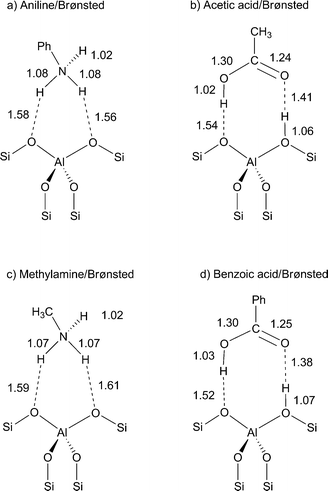 | ||
| Scheme 2 Optimized structures of (a) aniline, (b) acetic acid, (c) methylamine, and (d) benzoic acid interacting with bridging Si–OH–Al zeolite groups. Bond distances are expressed in angstroms. | ||
| δ15Ncalc/ppm | δ13Ccalc/ppm | δ15Nexp/ppm | δ13Cexp/ppm | |
|---|---|---|---|---|
| a Appears in the amide region and, thus, is difficult to detect. | ||||
| Acetic acid/Brønsted | — | 183.3 | — | 182 |
| Benzoic acid/Brønsted | — | 175.8 | — | —a |
| Aniline/Brønsted | −329.7 | — | −330 | |
| Methylamine/Brønsted | −360.7 | — | −360 |
Solid state NMR spectroscopy
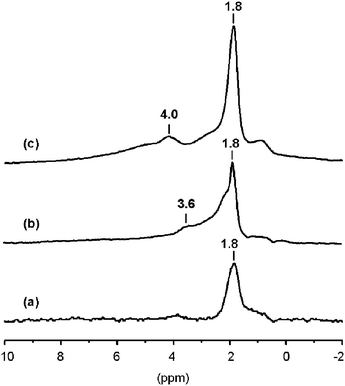 | ||
| Fig. 1 1H MAS NMR spectra of zeolites dehydrated at 673 K overnight: (a) silicalite, (b) silicalite-N and (c) H-beta. | ||
The 1H NMR spectrum of the parent silicalite, which has been included in Fig. 1 for comparison purposes, consists mainly of a peak of isolated SiOH groups at 1.8 ppm (Fig. 1(a)). The spectrum of silicalite-N, shown in Fig. 1(b), evidences the creation of structural defects in the MFI zeolite framework by submitting the sample to a basic treatment. Besides isolated silanols (signal at 1.8 ppm), the spectrum of silicalite-N (Fig. 1(b)) shows a shoulder at 2.2 ppm usually assigned to geminal or vicinal SiOH groups (infrared band at around 3690 cm−1),16,31,32 and an additional band at 3.6 ppm that can be associated with the broad infrared hydroxyl band centred at 3500 cm−1, assigned to silanol nests (see Fig. 1(b)).16,31
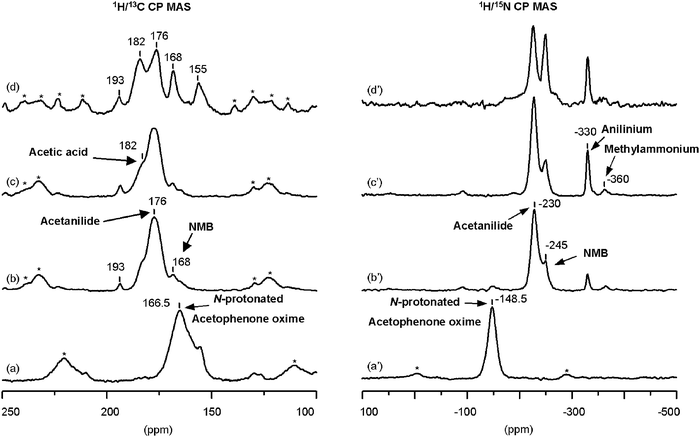 | ||
| Fig. 2 1H/13C (a–d) and 1H/15N (a′–d′) CP-MAS NMR spectra of (α-13C, 15N)-acetophenone oxime: (a) + (a′) adsorbed on zeolite H-beta at room temperature, and subsequently treated during 1 h at (b) + (b′) 423 K, (c) + (c′) 473 K, and (d) + (d′) 623 K. | ||
The oxime is adsorbed at room temperature by capturing the zeolite acidic proton; it becomes 15N-protonated, giving broad 13C and 15N peaks at 166.5 ppm and −148.5 ppm, respectively (Fig. 2(a) and (a′)). A sharper and weaker 13C component at 155 ppm in the spectrum of Fig. 2(a) can indicate the presence of some amounts of non-interacting oxime. Heating the reaction system at 423 K produces the practical disappearance of the 13C and 15N signals of the oxime and the appearance of new resonances as can be observed in the spectra of Fig. 2(b) and (b′). The 15N NMR spectrum of Fig. 2(b′) shows two peaks in the chemical shift range typical of amides, one at −230 ppm and another, weaker, at −245 ppm, which are attributed to 15N-acetanilide and 15N-NMB, respectively. This assignment is made by comparison with the 15N chemical shifts calculated theoretically for the two amides interacting with a bridging hydroxyl group, listed in Table 1. It should be noted that the signal at −245 ppm could also be attributed to 15N-acetanilide interacting with silanol groups or with trace amounts of water present in the system (see Table 1). However, the presence of NMB in the products was confirmed by mass spectroscopy, reinforcing the assignment of the −245 ppm signal to 15N-NMB adsorbed on a Brønsted acid site. Besides these two peaks, the 15N spectrum (Fig. 2(b′)) displays weak signals at −330 ppm and −360 ppm attributed to 15N-protonated aniline and 15N-protonated methylamine on Brønsted acid sites (see Scheme 2), according to the chemical shifts listed in Table 2. These amines must come from the hydrolysis of acetanilide (aniline) and NMB (methylamine) with residual amounts of water present in the reaction system. The 13C NMR spectrum recorded after heating at this same temperature (423 K, Fig. 2(b)) shows a band at 176 ppm due to 13CO-acetanilide and a weaker signal at 168 ppm from 13CO-NMB (see Table 1) with a shoulder at 182 ppm due to 13COOH-acetic acid, again indicating hydrolysis of acetanilide. The signal of benzoic acid (δ13C = 175.8 ppm) coming from the hydrolysis of NMB must be overlapped in the main broad resonance of the spectrum. Besides these signals, a very weak 13CO-acetophenone signal appears at 193 ppm pointing to some hydrolysis of the oxime.
The increase of the reaction temperature of the acetophenone oxime on zeolite H-beta to 473 K does not appreciably change the 13C and 15N spectra, producing only a better resolution of the peak of 15N-NMB at −245 ppm, and an enhancement of the relative intensity of the anilinium cation, as shown in Fig. 2(c) and (c′). Changes in the product distribution are evident after the reaction temperature of 623 K; the 15N NMR spectrum (Fig. 2(d′)) shows only some increase in the relative intensity of NMB, but new signals are evident in the better resolved 13C spectrum (Fig. 2(d)). The spectrum of Fig. 2(d) clearly shows the 13C peaks of acetanilide (and probably benzoic acid) at 176 ppm, and of NMB at 168 ppm, as well as that of acetic acid at 182 ppm and the very weak peak of acetophenone at 193 ppm. Besides these signals, which were already present at lower reaction temperatures, a new one appears at 155 ppm (Fig. 2(d)). This latter peak is attributed to a secondary product formed from NMB and/or acetanilide at higher reaction temperatures (above 573 K) on zeolites containing Brønsted acid sites; however, we have failed in its identification by different methods.
The main 15N and 13C NMR spectroscopic results of this reaction over siliceous zeolite beta were reported in our previous publication.18 The 15N NMR spectra are quite similar to those depicted in Fig. 3 for the transformation of (α-13C, 15N)-acetophenone oxime over silicalite-N. The spectrum recorded after the adsorption of the oxime at room temperature, displayed in Fig. 3(a), shows a very sharp peak at −21 ppm due to unadsorbed oxime; however, inspection of the spectrum shows a very weak, broad resonance centred at −48 ppm from the 15N-oxime adsorbed on silanol groups. Comparison of this spectrum with that recorded when the same oxime is mixed with zeolite beta containing defective silanol groups suggests a weaker interaction of the oxime with silicalite-N,18 which can be due to a lower concentration of connectivity defects. However, although the oxime enters the pores of Al-containing zeolite ZSM-5 (spectrum not shown) to become protonated, we cannot completely discard the limitations of diffusion of the oxime into the pores of silicalite.
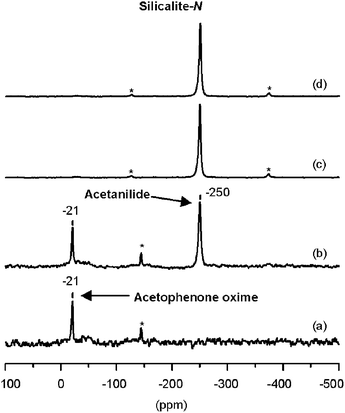 | ||
| Fig. 3 1H/15N CP-MAS NMR spectra of (α-13C, 15N)-acetophenone oxime: (a) adsorbed on zeolite silicalite-N at room temperature and subsequently treated during 1 h at (b) 423 K, (c) 473 K and (d) 523 K. | ||
As shown in Fig. 3(b), when the (α-13C, 15N)-oxime–silicalite-N system is heated at 423 K, a new signal grows at −250 ppm. Although the position of this peak is close to that of 15N-NMB on zeolite H-beta, the δ15Ncalc of the two amides interacting with silanol groups allows its assignment to 15N-acetanilide (δ15Ncalc = −248 ppm for the model Acetanilide/Silanol, Scheme 1, Table 1). When the reaction temperature is increased to 473 K, the spectrum of Fig. 3(c) is obtained, which shows a unique signal of acetanilide at δ15N = −250 ppm, indicating that the reaction is complete at this temperature.
A remarkable result here is the formation of the amide isomer NMB over Brønsted acid sites, which is not expected according to the high stereospecificity of the migration of the alkyl group in the anti position to the hydroxyl group, though we must note that 5% of NMB has previously been reported in the Beckmann rearrangement of acetophenone oxime on zeolite HY.12 The formation of the NMB isomer probably requires the prior isomerization of the oxime to place the methyl group in the anti position with respect to the hydroxyl group. This process involves rotation around the C–N bond and must be hindered for the neutral oxime having a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond (see Scheme 3). However, this rotation can occur to some extent in the N-protonated acetophenone oxime possessing a single C–N bond. Accordingly, no NMB is formed over siliceous zeolites, whereas some is formed over acidic H-beta.
N double bond (see Scheme 3). However, this rotation can occur to some extent in the N-protonated acetophenone oxime possessing a single C–N bond. Accordingly, no NMB is formed over siliceous zeolites, whereas some is formed over acidic H-beta.
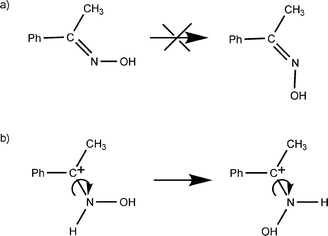 | ||
| Scheme 3 Rotation of the C–N bond for (a) the neutral acetophenone oxime, and (b) the N-protonated oxime. | ||
Fig. 4 depicts the results obtained by 15N and 13C NMR spectroscopy for the reaction of (α-13C, 15N)-acetophenone oxime over zeolite H-beta. Despite the presence of water, the acetophenone oxime adsorbed on the acidic zeolite becomes N-protonated at room temperature, as evidenced in the 15N NMR spectrum of Fig. 4(a′). As shown in the spectrum of Fig. 4(b′), when the reaction temperature is raised to 473 K, the 15N peak of the 15N-protonated oxime disappears, while two new signals appear, one at −250 ppm, in the amide region, and another one at −332 ppm from 15N aniline protonated on Brønsted acid sites (see Scheme 2 and Table 2). The corresponding 13C NMR spectrum (Fig. 4(b)) shows a peak for acetanilide at 176 ppm and another at 183 ppm due to acetic acid. The presence of anilinium and acetic acid clearly indicates the hydrolysis of acetanilide. The amide resonance is slightly high-field shifted and appears at a similar position to that of 15N-NMB on Brønsted acid sites (−250 ppm, see Scheme 1 and Table 1). However, neither the 15N nor the 13C spectra (Fig. 4(b) and (b′)) give any evidence for the formation of NMB or any of its hydrolysis products. This suggests that the 15N peak at −250 ppm in the spectrum of Fig. 4(b′) must be assigned to acetanilide, and that its chemical shift is changed by the presence of water. This assignment is supported by the 15N chemical shift calculated for the complex formed by the interaction of acetanilide with one molecule of water (see Table 1). The absence of NMB in the reaction products, further confirmed by mass spectrometry, suggests that water inhibits the isomerization of the oxime (or the migration of the syn-methyl group) during rearrangement. Further increase of the reaction temperature favours the hydrolysis of acetanilide, which is almost complete at 473 K, as can be seen in the spectra shown in Fig. 4(c) and 4(c′).
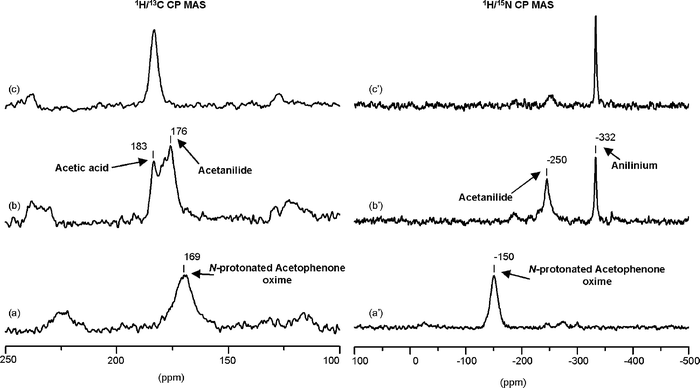 | ||
| Fig. 4 1H/13C (a–d) and 1H/15N (a′–d′) CP-MAS NMR spectra of (α-13C, 15N)-acetophenone oxime: (a) + (a′) adsorbed on non-dehydrated zeolite H-beta at room temperature and subsequently treated during 1 h at (b) + (b′) 423 K, and (c) + (c′) 473 K. | ||
Fig. 5 shows the 15N NMR spectra obtained after heating a mixture of zeolite silicalite-N without previous dehydration and 15N-acetophenone oxime. The results reported in Fig. 5 indicate that the presence of water has no dramatic influence on the Beckmann rearrangement reaction. The main effects of water are: (i) to increase the temperature at which the rearrangement starts to 473 K (from 423 K), and (ii) to hydrolyse some acetanilide at reaction temperatures of 573 K or higher.
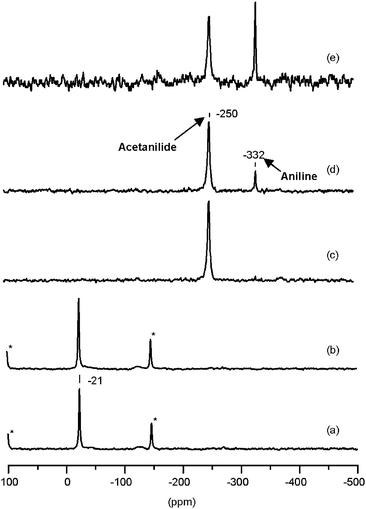 | ||
| Fig. 5 1H/15N CP-MAS NMR spectra of (α-13C, 15N)-acetophenone oxime: (a) adsorbed on zeolite silicalite-N at room temperature, and subsequently treated during 1 h at (b) 373 K, (c) 473 K, (d) 573 K and (e) 623 K. | ||
Conclusions
The results obtained in the Beckmann rearrangement reaction of acetophenone oxime over acidic zeolite H-beta and silicalite-N, dehydrated and in the presence of water, have led us to propose the reaction pathways shown in Schemes 4(a) and (b).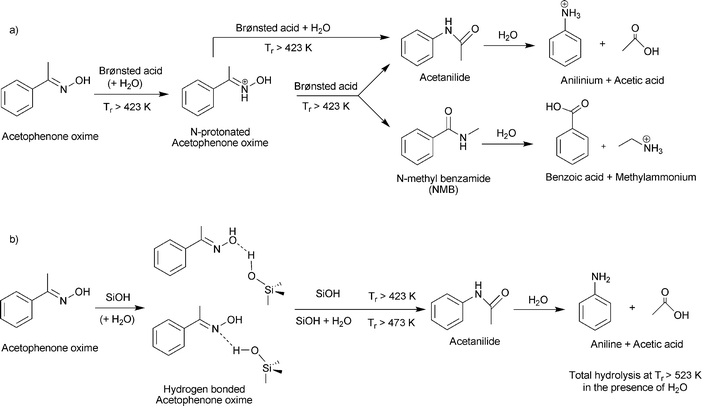 | ||
| Scheme 4 Reaction pathway for the Beckmann rearrangement of acetophenone oxime into acetanilide on zeolite hydroxyl groups: (a) bridging Si–OH–Al and (b) silanol. | ||
N-protonated acetophenone oxime is readily formed on the Brønsted acid sites at room temperature even in the presence of water. After heating at 423 K or above, both acetanilide and N-methyl benzamide (NMB) are formed, and a new unidentified secondary product formed from the amides appears at reaction temperatures above 573 K. Residual water produces partial hydrolysis of acetanilide to give aniline and acetic acid, and of NMB to give benzoic acid and methylamine. Both amines (methylamine and aniline) are protonated on the zeolite Brønsted acid sites. The presence of water in the reaction medium inhibits the formation of NMB; only acetanilide is formed, which is practically completely hydrolyzed above 473 K.
Pure siliceous silicalite-N is more selective to acetanilide than the acidic zeolite both in presence and absence of water in the reaction medium. Only part of acetophenone oxime forms hydrogen bonds with silanol groups at room temperature and reacts above 423 K to selectively give acetanilide. When water is present in the reaction medium (even if there is an excess with respect to the oxime) there is only partial hydrolysis of acetanilide above 523 K.
The results presented here suggest that, as for cyclic oximes, acid zeolites are more active and less selective catalysts than siliceous zeolites containing structural defects, i.e. silanol groups, in the Beckmann rearrangement of linear oximes involving benzyl groups. However, they may require higher reaction temperatures, and, therefore, it may be necessary to carry out the reaction in the gas phase.
Acknowledgements
The authors acknowledge the Spanish CICYT (MAT2006-14274-C02-01 y CTQ2006-09358) for financial support. I.L.-G. thanks UPV for a FPI-UPV grant.References
- T. Tatsumi, in Fine chemicals through heterogeneous catalysis, ed. R. A. Sheldon and H. Van Bekkum, Wiley-VCH, New York, 2001, p. 185 Search PubMed.
- H. Sato, Catal. Rev.-Sci. Eng, 1997, 39, 395 CrossRef CAS.
- G. Dahlhoff, J. P. M. Niederer and W. F. Hölderich, Catal. Rev., 2001, 43, 381 CrossRef CAS.
- A. Corma, J. Catal., 2003, 216, 298 CrossRef CAS.
- H. Ichihashi and M. Kitamura, Catal. Today, 2002, 73, 23 CrossRef.
- H. Ichihashi, M. Ishida, A. Shiga, M. Kitamura, T. Suzuki, K. Suenobu and K. Sugita, Catal. Surv. Asia, 2003, 7, 261 CrossRef CAS.
- Y. Izumi, H. Ichihashi, Y. Shimazu, M. Kitamura and H. Sato, Bull. Chem. Soc. Jpn., 2007, 80, 7.
- K. G. Davenport and C. B. Milton, US Patent 4 524 217, 1985, to Celanese Corp.
- J. R. Fritch, D. A. Aguila, T. Horlenko and O. S. Fruchey, EP. 469 742, 1991, to Celanese Corp.
- Y. M. Chung and H. K. Rhee, J. Mol. Catal. A: Chem., 2000, 159, 389 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, J. Catal., 2005, 233, 308 CrossRef CAS.
- P. S. Landis and P. B. Venuto, J. Catal., 1966, 6, 245 CrossRef CAS.
- M. A. Camblor, A. Corma, H. García, V. Semmer-Herlédan and S. Valencia, J. Catal., 1998, 177, 267 CrossRef CAS.
- E. Gutierrez, A. J. Aznar and E. Ruiz-Hitzky, Stud. Surf. Sci. Catal., 1991, 59, 539 CAS.
- G. P. Heitmann, G. Dahlhoff, J. P. M. Niederer and W. F. Hölderich, J. Catal., 2000, 194, 122 CrossRef CAS.
- G. P. Heitmann, G. Dahlhoff and W. F. Hölderich, J. Catal., 1999, 186, 12 CrossRef CAS.
- M. Kitamura, H. Ichihashi and H. Tojima, EP. 494.535, 1991, to Sumitomo Chemical Co.
- A. B. Fernandez, M. Boronat, T. Blasco and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 2370 CrossRef CAS.
- D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789 CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A. 7), Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed.
- V. R. R. Marthala, Y. Jiang, J. Huang, W. Wang, R. Gläser and M. Hunger, J. Am. Chem. Soc., 2006, 128, 14812 CrossRef CAS.
- T. Bucko, J. Hafner and L. Benco, J. Phys. Chem. A, 2004, 108, 11388 CrossRef CAS.
- J. Sirijaraensre, T. N. Truong and J. Limtrakul, J. Phys. Chem. B, 2005, 109, 12099 CrossRef CAS.
- A. B. Fernández, I. Lezcano-Gonzalez, M. Boronat, T. Blasco and A. Corma, J. Catal., 2007, 249, 116 CrossRef CAS.
- V. R. Reddy Marthala, R. Rabl, J. Huang, S. A. S. Rezai, B. Thomas and M. Hunger, J. Catal., 2008, 257, 134 CrossRef.
- G. P. Heitmann, G. Dahlhoff and W. F. Hölderich, Appl. Catal., A, 1999, 185, 99 CrossRef CAS.
- A. B. Fernández, A. Marinas, T. Blasco, V. Fornés and A. Corma, J. Catal., 2006, 243, 270 CrossRef CAS.
- M. Hunger, S. Ernst, S. Steuernagel and J. Weitkamp, Microporous Mesoporous Mater., 1996, 6, 349 CAS.
| This journal is © the Owner Societies 2009 |