Seven conformers of L-threonine in the gas phase: a LA-MB-FTMW study†
José L.
Alonso
*,
Cristóbal
Pérez
,
M.
Eugenia Sanz
,
Juan C.
López
and
Susana
Blanco
Grupo de Espectroscopía Molecular (GEM), Departamento de Química Física y Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, E-47005, Valladolid, Spain. E-mail: jclopez@qf.uva.es; jlalonso@qf.uva.es; Fax: +34 983![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 423204; Tel: +34 983423204
423204; Tel: +34 983423204
First published on 30th October 2008
Abstract
Rotational spectroscopy in combination with molecular beams and laser ablation (laser-ablation molecular-beam Fourier transform microwave (LA-MB-FTMW) spectroscopy) has proved to be successful in characterizing the conformers of natural amino acids. The procedure usually followed to assign and identify the different conformers of an amino acid from the rotational spectrum is described through the study of the natural amino acidL-threonine. The solid sample of L-threonine was vaporized by laser pulses, diluted in Ne and supersonically expanded between the mirrors of a Fabry–Pérot resonator where it was spectroscopically probed by microwave radiation. The rotational and nuclear quadrupole coupling constants extracted from the analysis of the rotational spectrum are directly compared with those predicted by ab initio methods to achieve the conclusive identification of seven different conformers. A complex hydrogen bonding network arises as a consequence of the polar side chain of threonine.
 José L. Alonso José L. Alonso | José Luis Alonso obtained BSc and PhD degrees in Chemistry from Universidad de Valladolid. He was appointed Research and Postdoctoral Fellow at Harvard University before becoming Assistant Professor at Universidad de Valladolid in 1984. He was appointed to a Chair in Physical Chemistry in 1988. He was an Alexander von Humbolt-Stiftung Fellow at Kiel University in 1990. His research focuses on gas-phase studies of molecular complexes and biomolecules using microwave spectroscopy with molecular beams and vaporization techniques, including laser ablation. His contributions to rotational spectroscopy were rewarded with a medal from the Real Sociedad Española de Química in 2003. |
 Cristóbal Pérez Cristóbal Pérez | Cristóbal Pérez obtained his BSc in Chemistry in 2003. He is currently a FPI predoctoral fellow at Universidad de Valladolid under the supervision of Prof. J. L. Alonso and J. C. López. His work has focused on the characterization of the different conformers of several amino acids in gas phase through the analysis of their rotational spectra. |
 M. Eugenia Sanz M. Eugenia Sanz | María Eugenia Sanz received her BSc in Chemistry from Universidad de Valladolid in 1995 and her PhD in 1999 under the supervision of Prof. J. L. Alonso and J. C. López. During 2000–2002, she was visiting scientist at the Harvard-Smithsonian Center for Astrophysics. In 2003 she was the recipient of a Ramón y Cajal Fellowship, and moved back to Universidad de Valladolid where she was appointed Professor Contratado Doctor of Physical Chemistry in 2007. Her research interests focus on the structural characterization of hydrogen-bonded complexes, and molecules of astrophysical and biological interest using rotational spectroscopy. |
 Juan C. López Juan C. López | Juan Carlos López holds a Chair in Physical Chemistry at Universidad de Valladolid since 2002. He received his BSc and PhD degrees in Chemistry from Universidad de Valladolid. In 1982 he obtained a faculty appointment at Universidad de Valladolid and was a postdoctoral fellow at the CNR Bologna, Italy, in 1984–1985. He is currently the Head of the Physical Chemistry and Inorganic Chemistry Department at Universidad de Valladolid. His research interests involve the structural characterization of molecular complexes and biomolecules in gas phase by means of microwave spectroscopy in combination with molecular beams and different vaporization techniques. |
 Susana Blanco Susana Blanco | Susana Blanco obtained her BSc in Chemistry from Universidad de Valladolid and her PhD under the supervision of Prof. J. L. Alonso and J. C. López. She was a postdoctoral fellow at Oxford University during 1997, and visiting scientist at Bologna University in 2006. She was later appointed as a Professor Titular of Physical Chemistry at Universidad de Valladolid. Her main research interests include experimental studies in microwave spectroscopy over a wide range of systems such as intermolecular complexes and biomolecules. |
1. Introduction
Gas-phase conditions are far from the in vivo or condensed-phase environments in which biological reactions occur, but provide the isolation required to observe the intrinsic molecular properties of biochemical systems. In physiological media or crystals, protonation in basic groups or deprotonation in acidic groups gives rise to charged species. As it is well known, amino acids in neutral solutions exist as doubly-charged zwitterions (+NH3–CH(R)–COO−).1 In the gas phase the perturbing agents like solvent or interacting molecules are removed and the observation of the neutral species is possible. Furthermore, most biochemical molecules are characterized by a great torsional flexibility, which results in manifolds of conformational varieties relatively close in energy. Since studies in condensed phases are affected considerably by multiple intermolecular interactions, conformational preferences can be biased by the environment and only gas-phase studies can reveal the inherent structural minima of bare molecules.Despite these advantages, the number of biomolecules studied to date in the gas phase is still very small compared with the overwhelming number of condensed-phase studies. Supersonic jets2 in combination with different spectroscopic techniques have been instrumental to provide conformational information on neutral biomolecules. Modern supersonic-jet laser spectroscopy3–6 combined with double resonance techniques (hole burning UV–UV and ion-dip IR–UV) and mass detection (through resonance-enhanced multiphoton ionization-REMPI) give electronic and vibrational information with mass and conformer selectivity. Since rotational resolution is not usually attained, this kind of experiments must be interpreted with the aid of high level ab initio quantum chemical calculations allowing the assignment of discrete conformational structures to experimental features with a reasonable degree of confidence. However, laser spectroscopy is hampered by the need of an absorbing chromophore that possesses sharp vibronic structure, such as an aromatic ring. Only three of the twenty natural amino acids have aromatic side chains that meet this condition: phenylalanine,7–10tyrosine10,11 and tryptophan.12–14
Unlike the aforementioned techniques, molecular beam Fourier transform microwave (MB–FTMW) spectroscopy,15–17 the most developed rotational spectroscopic technique, can be applied to any gaseous molecular system with the only requirement of having an electric dipole moment.18 By virtue of its extremely high resolution (sub-Doppler) and sensitivity all populated species in the jet, either tautomers, conformers or isotopomers, can be analysed independently. Moreover, small hyperfine effects arising from electric or magnetic interactions, like nuclear electric quadrupole or nuclear spin–spin couplings, can be fully resolved, giving insight on electronic properties of the molecule. Finally, the observation of tunneling doublings in the case of large amplitude motions, like internal rotation or inversion, can give information on the intramolecular dynamics associated to these motions. Summing up, MB-FTMW spectroscopy can be considered a definitive gas-phase structural probe of a molecular system, providing accurate structural information directly comparable to the in vacuo theoretical predictions. This technique has been widely used to study gaseous or easily vaporizable compounds, weakly bounded complexes,19 and even short-lived species generated “in situ” by electric discharges.20 In our laboratory it has been used to study axial/equatorial equilibria21 in hydrogen-bonded complexes, weak C–H··O bonds,22 and has been combined with electric discharges to generate and characterize new unstable compounds.23–26
Nevertheless, investigation of complete series of molecules of biological interest escaped MB–FTMW spectroscopy studies due to their high melting points and associated very low vapor pressures. Several laboratories have incorporated heatable nozzles27,28 to their spectrometers to overcome the problem, but in many cases thermal unstability constituted an important drawback. Laser ablation is an efficient method for vaporizing solid samples which otherwise would decompose upon heating.29 Laser-based vaporization techniques coupled with spectroscopic techniques30 have been used for gas-phase studies of solid compounds. However, there have been only a few attempts to incorporate laser ablation to MB–FTMW spectrometers.28,31–33 Those devoted to organic solids32,33 were discontinued due to poor experimental results.
Over the last years we have configured several MB–FTMW spectrometers which implement laser ablation sources to vaporize molecules in the throat of the nozzle. This approach, laser-ablation molecular-beam Fourier transform microwave (LA–MB–FTMW) spectroscopy,34 was initially tested with several organic solids.35,36 With the initial design34,37 and progressive improvements38 we undertook the study of relevant small biomolecules, such as natural amino acids and nucleic acid bases.39 Aliphatic amino acids have received special attention38,40–49 due to the lack of previous experimental information. We intended to identify and characterize their most stable conformations in the gas phase, where the amino acids exhibit an unsolvated neutral form which represents the best approximation to the electronic environment of an amino acid residue in a polypeptide chain.
In this paper we explain with certain detail the procedure we usually follow in our studies of amino acids. We report the results on the natural α-amino acid L-threonine [CH3–CH(OH)–CH(NH2)–COOH], as an example of the wealth of information that can be extracted from LA-MB-FTMW spectroscopy.
2. Experimental
Threonine has two chiral centres, so it can exist in four possible stereoisomers. The most common diastereomer in nature is the (2S,3R)-2-amino-3-hydroxybutanoic acid, called L-threonine. We used a commercial sample of the parent species of L-threonine (Aldrich, 99%) so that only the conformers arising from this stereoisomer are present in the rotational spectrum.A description of our experiment has been given elsewhere.34,36 Three main blocks can be distinguished: a laser ablation system, an evacuated Fabry–Pérot cavity, and a FTMW spectrometer (see Fig. 1). To efficiently accomplish vaporization, we found necessary to modify the backside of one of the mirrors of the Fabry–Pérot resonator so that it can hold a pulsed-jet nozzle especially designed for laser ablation. The amino acid samples are prepared as solid rods by pressing a fine powder of pure L-threonine (m.p. 256 °C) mixed with minimum quantities of a commercial binder. The rod is then placed vertically in the laser ablation nozzle where a focused laser beam impinges the sample laterally and produces its vaporization. The second (532 nm) or third (355 nm) harmonic from a nanosecond (ca. 50 mJ per pulse) or picosecond (ca. 200 ps, 25 mJ per pulse) Q-switched Nd:YAG laser have been used in the experiments reported here. The use of shorter (ps) laser pulses has been found to be more efficient for the laser ablation. Reproducibility of the ablation process is improved by a continuous translation and rotation of the sample rod, exposing new sample surface to each laser pulse. The laser-ablation nozzle has been modified from its original design34 so that there is a smoother transition between the aperture at the solenoid valve and that at the Fabry–Pérot mirror (see insert in Fig. 1); the shape is now similar to that of a Laval nozzle.50
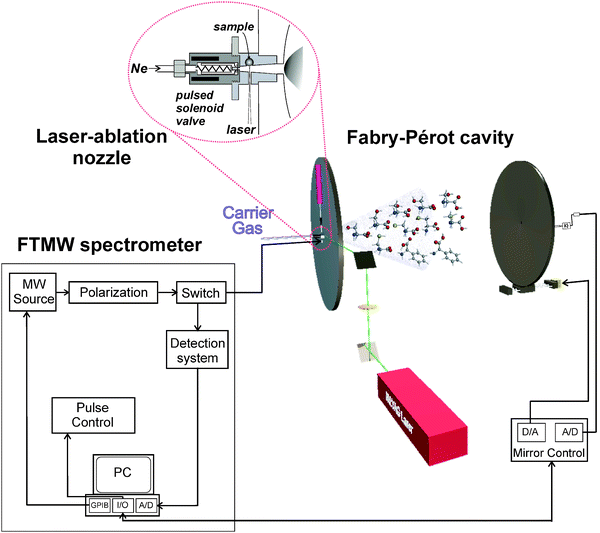 | ||
| Fig. 1 Scheme of the LA-MB-FTMW spectrometer. | ||
An experimental cycle starts (see Fig. 2) with a gas pulse of a noble carrier gas (stagnation pressures 6–20 bar, gas pulse typically 500 ms) (see Fig. 2a). After an adequate delay, a laser pulse hits the sample rod producing the vaporization of the solid. The delay between the laser and the molecular pulse is crucial for an optimum signal. The ablated amino acids are then seeded in the carrier gas and supersonically expanded between the two mirrors of the Fabry–Pérot resonator. In the supersonic expansion the amino acids suffer a strong cooling of the rotational and vibrational degrees of freedom, and individual conformers are frozen into the ground vibrational state of the corresponding well. Thus, the conformer distribution before the expansion may be preserved provided that the interconversion barriers between conformers are high enough. Molecular collisions gradually disappear as the expansion proceeds, so that the different species can be probed in a virtually isolated environment by Fourier transform microwave (FTMW) spectroscopy. Hence, a microwave pulse (typically 0.3–0.4 μs) is applied which produces a macroscopic polarization of the species in the jet (Fig. 2b). Once the excitation ceases, molecular relaxation gives rise to a transient emission signal (free induction decay) at microwave frequencies (Fig. 2c), which is captured in the time domain. Its Fourier transformation to the frequency domain yields the rotational transitions, that appear as Doppler doublets because the supersonic jet travels parallel to the resonator axis. The molecular rest frequencies are calculated as the arithmetic mean of the Doppler doublets, and are obtained with an accuracy better than 3 kHz. A new experimental cycle can start once the vacuum cavity has been evacuated. A repetition rate of 2 Hz is normally employed. For very weak signals hundreds of cycles must be added coherently. In order to probe the jet at different microwave frequencies or conduct a frequency scan, the Fabry–Pérot resonator is tuned mechanically under computer control.
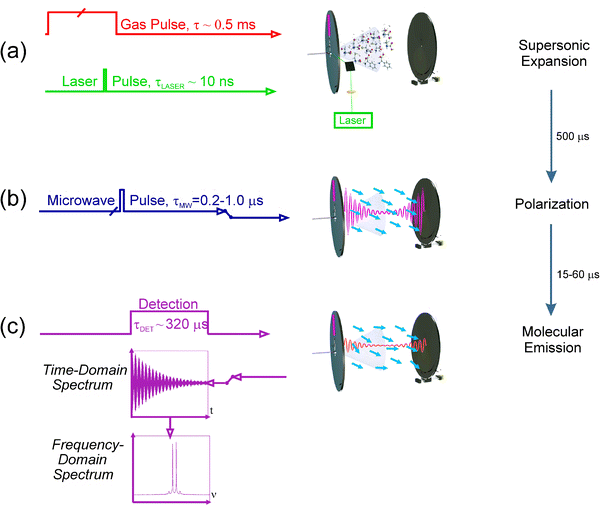 | ||
| Fig. 2 Pulse sequence for a single experimental cycle. | ||
3. Results
The large flexibility of amino acids, which makes folding and unfolding of proteins possible,51 produces the appearance of a significant number of low-energy conformers. Whereas covalent forces determine the molecular skeleton, conformational isomerism is also controlled by weaker nonbonded interactions within the molecule, especially hydrogen bonding. In all α-amino acids three kinds of intramolecular hydrogen bonds can be established between the amino and carboxylic moieties,41 which are denoted as I (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, cis-COOH), II (N⋯H–O, trans-COOH) and III (N–H⋯O–H, cis-COOH) as can be seen in Chart 1. In threonine, the presence of the polar side chain –CH(CH3)–OH is expected to bring about a new set of intramolecular interactions that will greatly increase the number of low-energy forms. This behaviour has been recently shown for the related amino acids serine38 and cysteine,49 for which seven and six structures have been detected, respectively. The conformers observed showed interactions O(S)–H⋯OC and O(S)–H⋯O–H between the side-chain hydroxyl (thiol) group and the oxygen atoms of the carboxylic group, and O(S)–H⋯N and N–H⋯O(S)–H between the side-chain hydroxyl (thiol) and the amino groups. On this basis, a large number of conformers are also expected in L-threonine.
C, cis-COOH), II (N⋯H–O, trans-COOH) and III (N–H⋯O–H, cis-COOH) as can be seen in Chart 1. In threonine, the presence of the polar side chain –CH(CH3)–OH is expected to bring about a new set of intramolecular interactions that will greatly increase the number of low-energy forms. This behaviour has been recently shown for the related amino acids serine38 and cysteine,49 for which seven and six structures have been detected, respectively. The conformers observed showed interactions O(S)–H⋯OC and O(S)–H⋯O–H between the side-chain hydroxyl (thiol) group and the oxygen atoms of the carboxylic group, and O(S)–H⋯N and N–H⋯O(S)–H between the side-chain hydroxyl (thiol) and the amino groups. On this basis, a large number of conformers are also expected in L-threonine.
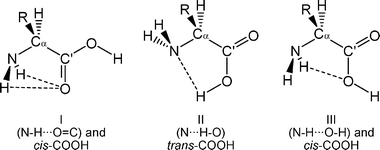 | ||
| Chart 1 Intramolecular hydrogen bonding between the carboxylic and amino groups of α-amino acids. | ||
To treat this conformational problem and identify the different structures in the supersonic jet we follow a three-step procedure (represented in Fig. 3), that is common to all our studies on amino acids. We describe it in the next sections through its application to L-threonine.
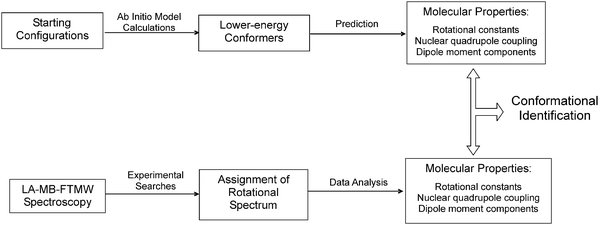 | ||
| Fig. 3 Steps followed in the identification of conformers. | ||
3.1. Model calculations
To have an overall picture of the conformational landscape of L-threonine theoretical predictions are used to find the most stable conformers on the potential energy surface. Starting geometries for ab initio calculations are initially selected by considering all possible rotations around single bonds (see Fig. 4) and identifying plausible intramolecular hydrogen bonds. First, the three configurations of Chart 1 are considered. Next, the orientations of the R = –CH(OH)–CH3 side chain are examined. Rotation around the Cα–Cβ bond allows for the three unstrained staggered configurations of Chart 2, denoted as a, b and c following the notation used for other α-amino acids with polar side chains.38,49 For each of these cases, additional plausible starting configurations were obtained by rotating Cα–NH2 and Cβ–OH bonds considering again the possible intramolecular hydrogen bonds.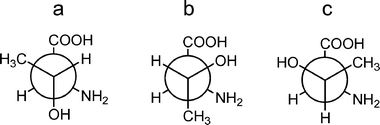 | ||
| Chart 2 Side-chain conformations in L-threonine, generated by rotations around the Cα–Cβ bond. | ||
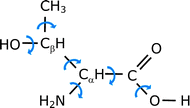 | ||
| Fig. 4 Single bond rotations in L-threonine. | ||
In order to predict the most stable structures, a series of structural optimizations52 is first conducted on the starting configurations using a computationally effective B3LYP density functional model with Pople’s standard 6–31G(d,p) basis set. Because the highest-energy conformational minima are not sufficiently populated in the supersonic expansion of our LA-MB-FTMW experiment, we focus our attention on the predicted lower-energy conformers. These are subjected to full geometry optimizations using second-order Møller–Plesset perturbation theory (MP) in the frozen core approximation and Pople’s 6–311++G(d,p) basis set. This level of theory has been found to behave satisfactorily in previous studies of amino acids. All the predicted conformers are confirmed to be local minima in the potential energy surface. For L-threonine, the ten lower-energy conformers of Fig. 5 were predicted to be within 1000 cm−1.
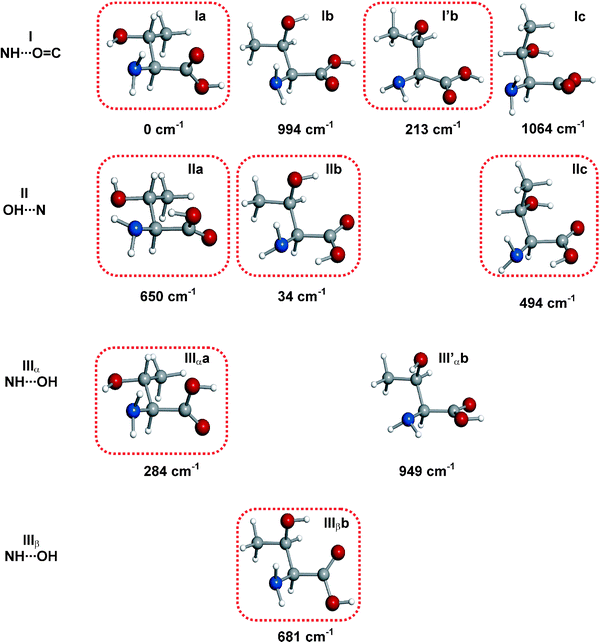 | ||
| Fig. 5 Predicted lower energy conformers of L-threonine and relative energies (MP2/6-311++G(d,p) basis set) with respect to the global minimum in cm−1. Conformers are classified as I, II or III depending on the hydrogen bond established between the amino and carboxylic groups (see text), and as a, b, c depending on the configuration adopted by the –CH(CH3)OH side chain. Additionally, a prime label indicates a down orientation of the –NH2 along with a O–H⋯N intramolecular interaction with the side chain. For configuration III additional α or β notation is used to indicate which H atom of the –NH2 binds to the –OH of the –COOH group. The detected conformers are encircled. | ||
3.2 Prediction of molecular properties
A distinguishing quality of rotational spectroscopy is its capability to generate very accurate spectroscopic parameters directly comparable with in vacuo ab initio predictions to provide unequivocal evidence of the conformers observed. Three molecular properties are relevant for the interpretation of the rotational spectra: rotational constants, nuclear quadrupole coupling constants, and electric dipole moment components. They are predicted in this second step (see Fig. 3).The rotational constants A, B, C (inversely proportional to the moments of inertia) provide information on the mass distribution of the molecules, and they are the key properties to derive molecular structure. Analysis of the rotational constants is normally conclusive in the identification of conformers. From the ab initio optimized structures of the lower-energy conformers of Fig. 5 the rotational constants are calculated and listed in Table 1. All rotamers are prolate asymmetric rotors (A > B≈C)18,53,54 with the Ray’s asymmetry parameter κ = (2B−A−C)/(A−C)18,53,54 ranging between −0.2 and −0.8. Three blocks corresponding to the “a”, “b” and “c” families can be distinguished on the basis of the values of the rotational constants, reflected in the κ values. These values indicate that the “b” family forms are the most extended configurations, while “c” conformers are the most folded ones. The predicted constants will be compared with the experimental ones to identify the conformers present in the supersonic expansion.
| MP2/6-311++G(d,p) | Ia | IIa | IIIαa | Ib | I′b | IIb | III′αb | IIIβb | Ic | IIc |
|---|---|---|---|---|---|---|---|---|---|---|
| a A, B and C are the rotational constants; χaa, χbb, and χcc are the diagonal elements of the 14N nuclear quadrupole coupling tensor; μa, μb and μc are the electric dipole moment components. | ||||||||||
| A a/MHz | 2870 | 2906 | 2885 | 3163 | 3164 | 3233 | 3194 | 3383 | 2488 | 2662 |
| B /MHz | 1608 | 1668 | 1578 | 1542 | 1521 | 1543 | 1498 | 1488 | 1838 | 1793 |
| C /MHz | 1225 | 1201 | 1249 | 1288 | 1324 | 1271 | 1332 | 1239 | 1431 | 1395 |
| κ | −0.53 | −0.45 | −0.60 | −0.73 | −0.79 | −0.72 | −0.82 | −0.77 | −0.23 | −0.37 |
| χ aa/MHz | −4.47 | −0.44 | −4.53 | −2.82 | −0.35 | −3.72 | 0.57 | −2.32 | −2.32 | −3.94 |
| χ bb/MHz | 2.76 | 2.67 | 2.85 | 1.89 | 2.94 | 1.88 | 2.96 | −0.03 | 0.88 | 2.47 |
| χ cc/MHz | 1.71 | −2.23 | 1.69 | 0.93 | −2.59 | 1.84 | −3.52 | 2.44 | 1.43 | 1.46 |
| |μa|/D | 2.3 | 4.3 | 2.1 | 0.3 | 0.1 | 3.4 | 0.2 | 2.3 | 1.0 | 0.1 |
| |μb|/D | 0.1 | 2.2 | 1.5 | 0.5 | 0.6 | 3.3 | 2.8 | 1.1 | 1.0 | 5.6 |
| |μc|/D | 0.8 | 0.3 | 1.3 | 0.9 | 2.9 | 1.2 | 1.9 | 1.0 | 0.3 | 0.3 |
| |μtotal|/D | 2.5 | 4.8 | 2.9 | 1.1 | 3.0 | 4.9 | 3.4 | 2.8 | 1.5 | 5.6 |
Sometimes the difference in the rotational constants between conformers is not large enough to discriminate between them. This happens with some forms that belong to the same family in Table 1 (see, for example, Ia and IIa). In these cases a different and independent way of identifying conformers based on the ubiquitous presence of 14N in amino acids can be utilized. 14N nuclei posses a non-zero quadrupole moment (I = 1) owing to a non-spherical distribution of the nuclear charge, which interacts with the electric field gradient created by the rest of the molecule at the site of those nuclei. This results in a nuclear hyperfine structure in the rotational spectrum18,53,54FTMW spectroscopy provides the high resolution needed to fully distinguish the various quadrupole hyperfine components (see Fig. 6). The associated experimentally-determinable molecular properties are the quadrupole coupling constants χgg (g = a,b,c), that are directly related to the electronic environment of the quadrupolar nucleus referred to the principal inertial axes and strongly depend on the orientation of the amino group. For example, conformers Ia and IIa with similar predicted rotational constants have different orientations of the amino group (Fig. 6) which is reflected in the nuclear quadrupole coupling constants χaa and χcc, as it can be seen by the predicted values of Table 1.
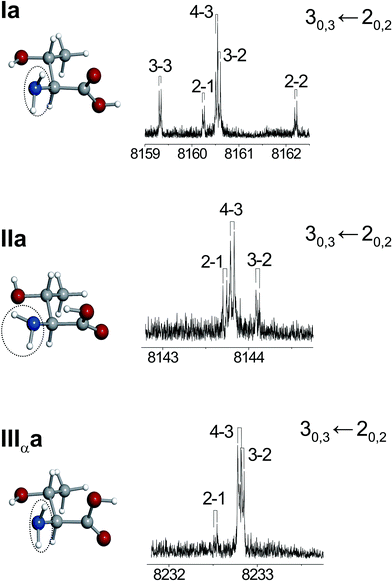 | ||
| Fig. 6 Nuclear quadrupole hyperfine structure of the 30,3← 20,2 rotational transitions of the rotamers M, N, and O, identified as IIa, Ia, and IIIαa, respectively (see text). | ||
A third molecular property to be considered is the electric dipole moment. The selection rules and intensity of the rotational transitions of asymmetric tops depend on the dipole moment components along the principal inertial axes, that is, on μa, μb and μc, which give rise to a-, b- and c-type spectra, respectively. All conformers of an amino acid possess the same connectivity between the atoms but they differ in the orientation of the functional groups, and this necessarily produces diverse charge distributions that are reflected in different values of the dipole moment components as it can be seen in Table 1.
The microwave power necessary for optimal polarization depends on the dipole moment component involved in a rotational transition. Hence, the difference in the values of the dipole moment components of conformers can be exploited to discriminate in some favourable cases between specific conformers just by varying the polarization power. By itself, it cannot be used as a conclusive tool, but it can always corroborate the conformer identification achieved with the previously described molecular properties. Once the rotational spectrum of a conformer is detected, from the microwave power required for optimal polarization it is possible to check whether this is consistent with predictions of the value of the electric dipole moment component of that particular form. The electric dipole moment components are necessarily used in estimating relative conformational abundances, as it will be explained below.
3.3 Rotational spectra analysis and conformer identification
According to the values of the electric dipole moment components predicted in Table 1, the rotational spectra of several conformers will show the characteristic pattern of a near-prolate asymmetric top with sets of μa-type, R-branch transitions separated approximately B + C that will take place between the lower rotational levels, preferentially populated due to the cooling produced by the supersonic expansion. Wide frequency scans with low power polarization conditions were conducted to search for such rotational transitions of conformers with relatively large μa. Several sets of R-branch lines ranging from J = 2–4 corresponding to five different rotamers were detected. Apart from the instrumental Doppler doubling, all transitions are observed split into several close hyperfine components (see Fig. 6) arising from the 14N nuclear quadrupole interaction described above, indicative that they arise from a molecule with a nitrogen nucleus. These rotamers are labelled as L, M, N, O and P in the order of their increasing transition frequencies. Successive predictions and new experimental measurements discarded or confirmed the initial assignment until a group of consistent rotational transitions (also including μb-transitions for M, O and P, and μc-type transitions for N and O) was collected for each conformer.All measured transitions (see the ESI, Tables S1–S5†) were analyzed55 using a Hamiltonian H = H(A)R + HQ, where H(A)R represents the Watson’s A-reduced semirigid rotor Hamiltonian56 in the Ir representation and HQ is the nuclear quadrupole coupling interaction term.18 The determinable spectroscopic parameters are the rotational constants and the elements of the nuclear quadrupole coupling tensor χ, linearly related to the electric field gradient by χ = −eQq. Usually only the diagonal elements of the tensor are determined. The experimental rotational constants and the χaa, χbb, and χcc nuclear quadrupole coupling constants for the rotamers L, M, N, O and P have been determined and are collected in Table 2. Centrifugal distortion constants and details of the fit are given in Table S6 of the ESI.†
| Experimental | L | M | N | O | P | Q | R |
|---|---|---|---|---|---|---|---|
| a A, B and C are the rotational constants; χaa, χbb, and χcc are the diagonal elements of the 14N nuclear quadrupole coupling tensor. b Standard error in parentheses in units of the last digit. | |||||||
| A a/MHz | 3379.841(14)b | 2912.6227(20) | 2872.77049(48) | 2889.93352(45) | 3232.4827(12) | 3148.59247(32) | 2670.72096(53) |
| B /MHz | 1482.04984(21) | 1660.21807(34) | 1608.95699(26) | 1572.32152(50) | 1533.71801(32) | 1506.27679(37) | 1784.66894(60) |
| C /MHz | 1237.59121(22) | 1189.31443(34) | 1211.39762(38) | 1241.83423(47) | 1267.88615(34) | 1316.33575(44) | 1383.75384(51) |
| κ | −0.77 | −0.45 | −0.52 | −0.60 | −0.73 | −0.79 | −0.38 |
| χ aa/MHz | −2.201(14) | −0.544(11) | −4.1859(25) | −4.1529(32) | −3.4971(21) | −0.7403(21) | −3.7652(73) |
| χ bb/MHz | −0.157(50) | 2.582(16) | 2.6611(42) | 2.5682(46) | 1.7519(27) | 2.8781(28) | 2.4258(75) |
| χ cc/MHz | 2.358(64) | −2.038(50) | 1.5248(17) | 1.5846(46) | 1.7452(60) | −2.1378(70) | 1.3394(20) |
A first look at the rotational constants of Table 2 allows us to easily clasify rotamers as belonging to different families. Hence, rotamers M, N and O belong to the “a” family while rotamer L and P belong to the “b” family. Conformers belonging to the “a” family have similar mass distributions: their rotational constants are very similar and they do not allow further discrimination. In these cases, as mentioned above, conclusive evidence comes from the values of 14N quadrupole coupling constants because they are very sensitive to the orientation of the –NH2group with respect to the principal inertial axis system (see Table 1). The values of these constants, reflected in the hyperfine structure (see Fig. 6) clearly identify rotamer M as IIa.
Rotamers N and O have very similar quadrupole coupling constants; comparison of their values with those predicted ab initio indicate that they are necessarily conformers Ia and IIIαa (see Tables 1 and 2). Due to the alike orientation of the amino group in these forms (see Fig. 5) they cannot be discriminated on the basis of the quadrupole constants. We can distinguish them from their selection rules and intensities of the observed transitions. The rotational spectrum of rotamer N shows strong μa-type transitions and fairly weak μc-type transitions, while form O presents strong μa-type transitions and medium-strength μb- and μc-type transitions. No μb-type transitions have been detected for conformer N. Considering the predicted dipole moment components of Table 1, these data are consistent with the identification of rotamer N as conformer Ia and O as conformer IIIαa. The microwave power used to optimally polarize the rotational transitions is also in agreement with the predicted values for the dipole moment components of each conformer.
The remaining rotamers, which belong to the “b” family, can be attributed to I′b, Ib, IIb or IIIβb considering the values of the rotational constants. However, detailed comparison of the values of the 14N quadrupole coupling constants conduct to the identification of rotamer L as conformer IIIβb, and rotamer P as IIb. In this case the quadrupole coupling constants are essential for the discrimination of conformers.
All conformers identified so far (Ia, IIa, IIIαa, IIb and IIIβb) have large predicted μa dipole moments, consistent with the low microwave polarization power used experimentally. Hence, a new set of spectral searches were conducted to detect other sets of μa-type, R-branch transitions. Despite using high microwave power for polarization, no other transition sets were observed. This is compatible with the presence of conformers Ib, I′b, IIIαb and IIc with very low predicted μa dipole moments and, consequently, extremely weak μa transitions. Therefore, wide frequency scans were carried out to identify μb- and μc-type transitions. Finally, two more rotamers labelled Q and R were identified. For rotamer Q μc- and μb-type transitions were measured while for rotamer R only μb-type transitions were observed (see Tables S7 and S8 of the ESI†). Following the same analysis described above, their rotational and quadrupole coupling constants were determined and are collected in Table 2. Again, from the rotational constants alone it is only possible to match rotamer Q to a conformer belonging to the “b” family, and form R to a conformer of the “c” family. Further consideration of the nuclear quadrupole coupling constants uniquely identifies form Q as I′b and form R as IIc.
A small number of very weak signals remain unassigned in the wide frequency regions scanned. They probably belong to other conformers of L-threonine predicted to lie at higher energies. Due to the weakness of these lines and their insufficient number it has not been possible to try new assignments.
In all cases, the calculated rotational constants are in good agreement with the experimentally determined values with a maximum deviation of approximately 1.1%. We can infer that the actual geometries of the conformers are very close to those predicted ab initio given in Tables S9–S15 of the ESI.†
4. Conformational preferences and intramolecular forces
Assuming that the cooling in the supersonic expansion brings all conformers to their vibrational ground state, the post-expansion abundances of the different conformers can be estimated from relative intensity measurements performed on rotational transitions. For this purpose, it is assumed that transition intensities for a conformer i are proportional to μi·Ni, where μi is the corresponding electric dipole moment component and Ni is the number density of species i in the jet. In this manner, from selected μa- and μb-type transitions the conformational population ratios have been estimated to be Ia > IIb ∼ I′b > IIa ∼ IIIαa ∼ IIc ∼ IIIβb. These relative abundances can be considered as indicative of the conformational stability trend, and are in a reasonable qualitative agreement with the ab initio relative energies (see Fig. 5). Conformer Ia is clearly the global minimum, followed in stability by conformer IIb, a preference also displayed by the related amino acid serine.38 This behaviour has also been found in all α-amino acids without polar side chains40–48 The only exception so far is the α-amino acid with a polar side chain cysteine,49 where a type II conformer was found to be the global minimum.Type III conformers have not been observed in the simplest α-amino acids,40–48 a fact that has been explained in terms of their relaxation to the lower-energy type I forms through relatively low interconversion barriers. This process occurs in the early stages of the supersonic expansion as a result of collisions with the carrier gas.57,58 In serine,38 a low-energy barrier for the interconversion IIIαa → Ia was predicted (see Fig. 7a), which explained the non observation of conformer IIIαa in the supersonic expansion. In contrast, interconversion IIIαa → Ia for L-threonine is hindered by a barrier high enough (approximately 1000 cm−1, see Fig. 7b) to prevent relaxation,58–60 thus allowing the detection of a conformer IIIαa for the first time. Threonine differs from serine in the presence of an additional methyl group in the side chain [–CH(CH3)OH]. This seems to increase the steric interactions and thus affects IIIαa → Ia interconversion barrier, making possible the observation of the IIIαa conformer that has not been detected for the other α-amino acids with polar side chains studied so far.
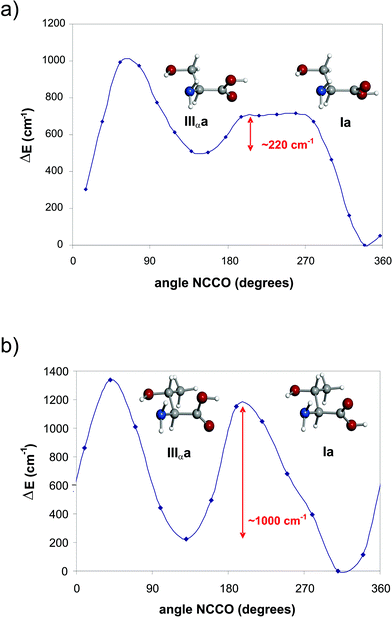 | ||
| Fig. 7 Interconversion barrier between conformers Ia ↔ IIIαa calculated at the MP2/6-311++G(d,p) level of theory for (a) serine and (b) L-threonine. | ||
Another type III conformer, IIIβb, has also been detected for L-threonine. In this case the behaviour of L-threonine is the same as that found in serine for the “b” family: the interaction of the side chain with the other polar groups in the amino acid backbone causes the type III form to be more stable that the type I conformer. Hence conformer IIIβb cannot relax to Ib and thus it exists in the molecular beam.
All these experimental results corroborate the understanding that the conformers observed in the supersonic expansion are the consequence of their relative energies and the interconversion barriers between them.60
The characterization of the preferred conformers of L-threonine provides information on the intramolecular interactions that occur in this natural amino acid. Conformers Ia and I′b both exhibit a hydrogen bond involving a hydrogen atom of the –NH2group and the carbonyl oxygen at the –COOH group, and they have a cis-COOH arrangement. Both Ia and I′b also show an O–H⋯N interaction between the hydroxyl group side chain and the lone pair of the N atom, but the different arrangement of the side chain in conformer I′b obliges the amino group to rotate clockwise with respect to Ia for this interaction to occur. Conformers IIa, IIb and IIc are stabilized by an O–H⋯N hydrogen bond between the hydroxyl group of the –COOH group and the nonbonding electron pair at the nitrogen atom. In conformers IIa and IIb there is also a hydrogen bond between a H atom of the amino group and the oxygen of the hydroxyl side chain. Besides, in conformer IIb the hydrogen atom of the hydroxyl group in the side chain interacts with the carbonyl oxygen of the –COOH group. This latter O–H⋯O interaction is also present in conformer IIc, where, in contrast with IIa and IIb, the –NH2 and side-chain –OH groups cannot interact since they are in anti disposition. The type III conformers observed are stabilized by a N–H⋯O–H hydrogen bond. In conformer IIIαa there is also an O–H⋯N bond involving the hydroxyl side chain and the nonbonding electron pair of the N atom. In conformer IIIβb the H atom of the side chain –OH group interacts with the carbonyl oxygen of the –COOH group.
Like in all the other α-amino acids,37,38,40–43,46–49 the intramolecular hydrogen bonds are the forces which drive the conformational preferences in L-threonine. However, other intramolecular interactions start to become important in β- and γ-amino acids. Specifically, conformers stabilized by a new n–π* interaction, established between the lone electron pair at the nitrogen of the –NH2group and the electronic density at the π* orbital of the carbonyl in the –COOH group, have been characterized for β-alanine45 and γ-aminobutyric acid.61
5. Conclusions
The results obtained for L-threonine confirm LA-MB-FTMW spectroscopy as a matchless tool to investigate the conformational landscape of solid biomolecules in the gas phase. The molecular properties extracted from the analysis of the rotational spectrum, such as the rotational and quadrupole coupling constants, can be directly compared with those predicted by ab initio methods to achieve a conclusive identification of the most abundant conformers in the supersonic expansion. Besides, rotational data provide a benchmark against which quantum theory calculations can be checked.The presence of a polar side chain has been found to have an influential role in all the α-amino acids studied, which is reflected in the increased number of low-energy conformers. The interaction of the side chain with the other the other polar groups in the amino acid backbone reverses the relative stabilities of type I and type III forms or increases the III ↔ I interconversion barrier.38,49 In L-threonine, this has allowed the characterization of a IIIαa form that had not been detected before.
Thanks to the LA-MB-FTMW technique, knowledge of the structural properties of neutral amino acids in the gas phase expanded considerably in recent years. Future work involves further improvement in our experiment in two directions. In order to study larger systems it is necessary to work at lower frequencies. We have recently reached 3 GHz just by reducing the radius of curvature of the Fabry–Pérot resonator. Changes in the laser ablation conditions (pulse width, laser frequency and energy) are crucial to minimize photofragmentation problems. Using a ps laser we have just observed for the first time the rotational spectrum of tryptophan61 and one conformer has been identified. Other devices combining laser ablation and microwave spectroscopy in molecular beams are under development and hopefully will be applied to the study of larger biologically relevant molecules, as well as clusters with water and acceptor–receptor adducts.
Acknowledgements
We would like to thank the Dirección General de Investigación—Ministerio de Ciencia y Tecnología (grant CTQ2006-05981/BQU) and the Junta de Castilla y León-Fondo Social Europeo (grant VA012 C05) for financial support. CP gratefully acknowledges a FPI grant from the Ministerio de Educación y Ciencia.References
- T. J. Kistenmacher, G. A. Rand and R. E. Marsh, Acta Crystallogr., Sect. B, 1974, 30, 2573–2578 CrossRef; C. H. Görbitz and B. Dalhus, Acta Crystallogr., Sect. C, 1996, 52, 1754 CrossRef.
- D. H. Levy, Annu. Rev. Phys. Chem., 1980, 31, 197 CrossRef CAS; G. Scoles, in Atomic and Molecular Beam Methods, ed. G. Scoles, Oxford University Press, New York & Oxford, 1988, vol. 1 Search PubMed.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1 RSC , and references therein.
- T. S. Zwier, J. Phys. Chem. A, 2001, 105, 8827 CrossRef CAS.
- R. P. Weinkauf, J.-P. Schermann, M. S. de Vries and K. Kleinermanns, Eur. Phys. J. D, 2002, 20, 309 CrossRef CAS.
- D. H. Levy, Jet Spectroscopy and Molecular Dynamics, ed. J. M. Hollas, Phillips, Blackie, London, 1995 Search PubMed.
- L. C. Snoek, E. G. Robertson, R. T. Kroemer and J. P. Simons, Chem. Phys. Lett., 2000, 321, 49 CrossRef CAS.
- K. T. Lee, J. Sung, K. J. Lee, S. K. Kim and Y. D. Park, J. Chem. Phys., 2002, 116, 8251 CrossRef CAS.
- Y. H. Lee, J. Jung, B. Kim, P. Butz, L. C. Snoek, R. T. Kroemer and J. P. Simons, J. Phys. Chem. A, 2004, 108, 69 CrossRef CAS.
- S. J. Martinez, J. C. Alfano and D. H. Levy, J. Mol. Spectrosc., 1992, 156, 421 CrossRef.
- L. I. Grace, R. Cohen, T. M. Dunn, D. M. Lubman and M. S. de Vries, J. Mol. Spectrosc., 2002, 215, 204 CrossRef CAS.
- T. R. Rizzo, Y. D. Park, L. A. Peteanu and D. H. Levy, J. Chem. Phys., 1986, 84, 2534 CrossRef CAS.
- L. C. Snoek, R. T. Kroener, M. R. Hockridge and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1819 RSC.
- J. M. Bakker, L. M. Aleese, G. Meijer and G. von Helden, Phys. Rev. Lett., 2003, 91, 203003 CrossRef.
- T. J. Balle and W. H. Flygare, Rev. Sci. Instrum., 1981, 52, 33 CrossRef CAS.
- J.-U. Grabow and W. Stahl, Z. Naturforsch., A, 1990, 45, 1043.
- J. L. Alonso, F. J. Lorenzo, J. C. López, A. Lesarri, S. Mata and H. Dreizler, Chem. Phys., 1997, 218, 267 CrossRef CAS.
- W. Gordy and R. L. Cook, Microwave Molecular Spectra, Wiley, New York, 1984 Search PubMed.
- K. R. Leopold, G. T. Fraser, S. E. Novick and W. Klemperer, Chem. Rev., 1994, 94, 1807 CrossRef CAS; A. Bauder, in Low Temperature Molecular Spectroscopy, ed. R. Fausto, Kluwer Academic, Netherlands, 1996, pp. 271–289 Search PubMed; A. C. Legon and D. J. Millen, Chem. Rev., 1986, 86, 635 Search PubMed.
- Y. Endo, H. Kohguchi and Y. Ohshima, Faraday Discuss., 1994, 97, 341 RSC; M. C. McCarthy and P. Thaddeus, Chem. Soc. Rev., 2001, 30, 177 RSC; D. H. Sutter and H. Dreizler, Z. Naturforsch., A, 2001, 56, 425 CAS; M. C. L. Gerry, Chem. Phys. Lett., 1992, 188, 213 CrossRef CAS; B. Brupbacher and T. Brupbacher, J. Chem. Phys., 1999, 111, 6300 CrossRef CAS.
- S. Antolínez, J. C. López and J. L. Alonso, Angew. Chem., Int. Ed., 1999, 38, 1772 CrossRef CAS; M. E. Sanz, J. C. López and J. L. Alonso, Chem.–Eur. J., 1999, 5, 3293 CrossRef CAS; M. E. Sanz, A. Lesarri, J. C. López and J. L. Alonso, Angew. Chem., Int. Ed., 2001, 40, 935 CrossRef CAS; S. Antolínez, J. C. López and J. L. Alonso, ChemPhysChem, 2001, 2, 114 CrossRef CAS.
- J. L. Alonso, S. Antolínez, S. Blanco, A. Lesarri, J. C. López and W. Caminati, J. Am. Chem. Soc., 2004, 126, 3244 CrossRef CAS; W. Caminati, J. C. López, J. L. Alonso and J.-U. Grabow, Angew. Chem., Int. Ed., 2005, 44, 2; P. Ottaviani, W. Caminati, L. B. Favero, S. Blanco, J. C. López and J. L. Alonso, Chem.–Eur. J., 2006, 12, 915 CrossRef CAS; J. C. López, W. Caminati and J. L. Alonso, Angew. Chem., Int. Ed., 2006, 45, 290 CrossRef CAS.
- S. Blanco, M. E. Sanz, S. Mata, A. Lesarri, J. C. López, H. Dreizler and J. L. Alonso, Chem. Phys. Lett., 2003, 375, 355 CrossRef CAS.
- S. Blanco, M. E. Sanz, A. Lesarri, J. C. López and J. L. Alonso, Chem. Phys. Lett., 2004, 397, 379 CrossRef CAS.
- S. Blanco, J. C. López, M. E. Sanz, A. Lesarri, H. Dreizler and J. L. Alonso, J. Mol. Spectrosc., 2004, 227, 202 CrossRef CAS.
- M. E. Sanz, J. L. Alonso, S. Blanco, A. Lesarri and J. C. López, Astrophys. J. Lett., 2004, 621, L157.
- R. D. Suenram, F. J. Lovas and G.T. Fraser, J. Mol. Spectrosc., 1988, 127, 472 CAS; A. Welzel and W. Stahl, Phys. Chem. Chem. Phys., 1999, 1, 5109 RSC; F. J. Lovas, D. F. Plusquellic, A. Lesarri, Y. Kawashima, J. O. Jensen and A.C. Samuels, J. Mol. Spectrosc., 2001, 211, 110; S. Blanco, J. C. López, J. L. Alonso, P. Ottaviani and W. Caminati, J. Chem. Phys., 2003, 119, 880 CrossRef CAS; S. Brünken, M. C. McCarthy, P. Thaddeus, P. D. Godfrey and R. D. Brown, Astron. Astrophys., 2006, 459, 317 CrossRef CAS.
- S. Brünken, M. C. McCarthy, P. Thaddeus, P. D. Godfrey and R. D. Brown, Astron. Astrophys., 2006, 459, 317 CrossRef CAS.
- R. J. Levis, Ann. Rev. Phys. Chem., 1994, 45, 483 CrossRef CAS.
- B. Simard, S. A. Mitchell, M. R. Humphries and P. A. Hackett, J. Mol. Spectrosc., 1988, 129, 186 CrossRef CAS; J. R. Cable, M. J. Tubergen and D. H. Levy, J. Am. Chem. Soc., 1989, 111, 9032 CrossRef CAS; M. Barnes, M. M. Fraser, P. G. Hajigeorgiou, A. J. Merer and S. D. Rosner, J. Mol. Spectrosc., 1995, 170, 449 CrossRef CAS; J. W. Elam and D. H. Levy, J. Phys. Chem. B, 1998, 102, 8113 CrossRef CAS.
- R. D. Suenram, F. J. Lovas, G. T. Fraser and K. Matsumura, J. Chem. Phys., 1990, 92, 4724 CrossRef CAS; K. A. Walker and M. C. L. Gerry, J. Mol. Spectrosc., 1997, 182, 178 CrossRef CAS; Y. Oshima and Y. Endo, Chem. Phys. Lett., 1993, 213, 95 CrossRef CAS; R. J. Low, T. D. Varberg, J. P. Sonnely, A. R. Auty, B. J. Howard and J. M. Brown, J. Mol. Spectrosc., 1993, 161, 499 CrossRef CAS; D. Banser, M. Schnell, J. U. Grabow, E. J. Cocinero, A. Lesarri and J. L. Alonso, Angew. Chem., Int. Ed., 2005, 44, 6311 CrossRef CAS.
- U. Kretschmer, D. Consalvo, A. Knaack, W. Schade, W. Stahl and H. Dreizler, Mol. Phys., 1996, 87, 1159 CrossRef CAS.
- F. J. Lovas, Y. Kawashima, J.-U. Grabow, R. D. Suenram, G. T. Fraser and E. Hirota, Astrophys. J. Lett., 1995, 455, L201 CAS.
- A. Lesarri, S. Mata, J. C. López and J. L. Alonso, Rev. Sci. Instrum., 2003, 74, 4799 CrossRef CAS.
- S. Antolinez, A. Lesarri, S. Mata, S. Blanco, J. C. López and J. L. Alonso, J. Mol. Struct., 2002, 612, 125 CrossRef CAS.
- A. Lesarri, S. Mata, S. Blanco, J. C. López and J. L. Alonso, J. Chem. Phys., 2004, 120, 6191 CrossRef CAS.
- A. Lesarri, S. Mata, E. J. Cocinero, S. Blanco, J. C. López and J. L. Alonso, Angew. Chem., Int. Ed., 2002, 41, 4673 CrossRef CAS.
- S. Blanco, M. E. Sanz, J. C. López and J. L. Alonso, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20183 CrossRef CAS.
- V. Vaquero, M. E. Sanz, J. C. López and J. L. Alonso, J. Phys. Chem. A., 2007, 111, 3443 CrossRef CAS; J. C. López, M. I. Peña, M. E. Sanz and J. L. Alonso, J. Chem. Phys., 2007, 126, 191103 CrossRef.
- S. Blanco, A. Lesarri, J. C. López and J. L. Alonso, J. Am. Chem. Soc., 2004, 126, 11675 CrossRef CAS.
- A. Lesarri, E. J. Cocinero, J. C. López and J. L. Alonso, Angew. Chem., Int. Ed., 2004, 43, 605 CrossRef CAS.
- A. Lesarri, R. Sánchez, E. J. Cocinero, J. C. López and J. L. Alonso, J. Am. Chem. Soc., 2005, 127, 12952 CrossRef CAS.
- E. J. Cocinero, A. Lesarri, J.-U. Grabow, J. C. López and J. L. Alonso, ChemPhysChem., 2007, 8, 599 CrossRef CAS.
- A. Lesarri, E. J. Cocinero, J. C. López and J. L. Alonso, J. Am. Chem. Soc., 2005, 127, 2572 CrossRef CAS.
- M. E. Sanz, A. Lesarri, I. Peña, V. Vaquero, V. Cortijo, J. C. López and J. L. Alonso, J. Am. Chem. Soc., 2006, 128, 3812 CrossRef CAS.
- M. E. Sanz, V. Cortijo, W. Caminati, J. C. López and J. L. Alonso, Chem.–Eur. J., 2006, 12, 2564 CrossRef CAS.
- E. J. Cocinero, A. Lesarri, M. E. Sanz, J. C. López and J. L. Alonso, ChemPhysChem., 2006, 7, 1481 CrossRef CAS.
- E. J. Cocinero, P. Villanueva, A. Lesarri, M. E. Sanz, S. Blanco, S. Mata, J. C. López and J. L. Alonso, Chem. Phys. Lett., 2007, 435, 336 CrossRef CAS.
- M. E. Sanz, S. Blanco, J. C. López and J. L. Alonso, Angew. Chem., Int. Ed., 2008, 47, 6216 CrossRef CAS.
- D. B. Atkinson and M. A. Smith, Rev. Sci. Instrum., 1995, 66, 4434 CrossRef CAS.
- K. A. Dill and H. S. Chan, Nat. Struct. Biol., 1997, 4, 10 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision B.04), Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- C. H. Townes and A. L. Schawlow, Microwave Spectroscopy, Dover, 1975 Search PubMed.
- H. W. Kroto, Molecular Rotation Spectra, Wiley, London, 1975 Search PubMed.
- H. M. Pickett, J. Mol. Spectrosc., 1991, 148, 371 CrossRef CAS.
- J. K. G. Watson, in Vibrational Spectra and Structure, ed. J. R. Durig, Elsevier, New York/Amsterdam, 1977, vol. 6, pp. 1–78 Search PubMed.
- R. S. Ruoff, T. D. Klots, T. Emilson and H. S. Gutowski, J. Chem. Phys., 1990, 93, 3142 CrossRef CAS.
- P. D. Godfrey, R. D. Brown and F. M. Rodgers, J. Mol. Struct., 1996, 376, 65 CrossRef CAS.
- P. D. Godfrey and R. D. Brown, J. Am. Chem. Soc., 1998, 120, 10724 CrossRef CAS.
- G. M. Florio, R. A. Christie, K. D. Jordan and T. S. Zwier, J. Am. Chem. Soc., 2002, 124, 10236 CrossRef CAS.
- M. E. Sanz, S. Blanco, S. Mata, J. C. López and J. L. Alonso, contribution M7, Isolated Biomolecules and Biomolecular Interactions IBBI08, Valladolid (Spain), 13–18 April 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Observed frequencies and residuals; experimental spectroscopic constants; ab initio Cartesian coordinates. See DOI: 10.1039/b810940k |
| This journal is © the Owner Societies 2009 |