Raman spectroscopy of strained single-walled carbon nanotubes
Zhongfan
Liu
*,
Jin
Zhang
and
Bo
Gao
Center for Nanochemistry, Beijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, MOE Key Laboratory for the Physics and Chemistry of Nanodevices, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, P. R. China. E-mail: zfliu@pku.edu.cn; Fax: 86-10-6275-7157; Tel: 86-10-6275-7157
First published on 7th October 2009
Abstract
Due to remarkable rolling structure and distinct rolling direction, the chirality-dependent Raman spectra of single-walled carbon nanotubes (SWNTs) show two characteristic features: the radial breathing mode (RBM) and the G-band. Rich information about SWNTs presented by these Raman features makes Raman spectroscopy a general and common tool for characterizing structures and properties of SWNTs and their changes. When exerted by external forces, the geometrical structures of SWNTs will change, which further affects the electronic structures and phonon properties of SWNTs. In this article, emphasis is given to how Raman frequency and resonant-Raman intensity evolve under distinct strains, including uniaxial strain, torsional strain, radial deformation and bending deformation. It is found that depending on different structural variations, Raman spectra of SWNTs have different responses to each strain, showing that resonant-Raman spectroscopy is a suitable tool to characterize and study strains in SWNTs.
 Zhongfan Liu | Dr Zhongfan Liu received his PhD from the University of Tokyo in 1990. After a postdoctoral fellowship at Institute for Molecular Science (IMS), Japan, he became an associate professor (1993), full professor (1993) and Cheung Kong Chair professor (1999) of Peking University. He is now the director of Institute of Physical Chemistry and the director of Center for Nanoscale Science and Technology of Peking University. He also serves as the President of the Chinese Electrochemical Society and is the chief scientist of the national basic research program for nanotechnology “Quasi-1D Semiconducting Nanomaterials”. His research is devoted to developing nano/molecular electronic devices using carbon nanotubes, graphenes and other unique nanomaterials, including controlled CVD growth, chemical tailoring and device fundamentals. Dr Liu has published over 280 peer reviewed journal articles. |
 Jin Zhang | Dr Jin Zhang received his PhD from Peking University and Lanzhou University in 1997. After taking a two-year postdoctoral fellowship at University of Leeds, UK, he became associate professor (2000) and full professor (2006) of Peking University. He now serves as a member of the Editorial Advisory Board of Carbon. His research is focused on the controlled chemical vapor deposition growth of carbon nanomaterials, including carbon nanotubes and graphene, and their Raman spectroscopy. Dr. Zhang has published over 100 peer reviewed journal articles. |
 Bo Gao | Dr Bo Gao received his PhD in physical chemistry from Peking University under the supervision of Professors Jin Zhang and Zhongfan Liu. Then he joined in Professor Gang-yu Liu’s group as a postdoctoral fellow. His current research interests include Raman spectroscopy, AFM high-resolution imaging, and AFM nanolithography. |
1. Introduction
Single-walled carbon nanotubes (SWNTs) are considered as the ideal one-dimensional (1-D) system for basic research and have attracted wide attention since Iijima’s report in 1991.1 The most prominent feature of SWNTs is their metallic or semiconducting character:2 roughly 2/3 are semiconducting and 1/3 are metallic, depending on their rolling vector. They were reported3 to carry electrical currents up to 109 A cm−2. Meanwhile, they possess extraordinary mechanical strength with an elastic modulus4–6 on the order of 1 TPa and a shear modulus6 of approximately 1 GPa. Therefore, they are extremely stiff along their axis but easy to bend perpendicular to the axis. Such kinds of excellent physical properties of SWNTs will probably bring a lot of practical applications in areas of composites, nanoelectronic devices,7 nanoelectromechanical systems (NEMS), chemical and biological sensors,8etc.Due to the chirality-dependent physical properties of SWNTs, it is crucial to find an easy and precise way to determine the chiral indices of SWNTs. A number of methods, such as scanning tunneling microscopy (STM), photoluminescence (PL) and resonant-Raman spectroscopy have been developed for this purpose. Among them, resonant-Raman spectroscopy has become a general and common tool for characterization of SWNTs,9–12 since the Raman experiment is simple, quick, non-destructive and non-invasive, and can be done at room temperature and under ambient pressure. Especially the observation of Raman spectrum from just one individual SWNT13,14 allows us to study phonon properties at single SWNT level and to understand the dependence of phonon properties on their distinct structures (chiralities). Raman spectroscopy can also be used to obtain information about conductivity,15 doping,16,17 strains,18 and other physical properties of SWNTs.19–21
In practice, strains of SWNTs can not be avoided. Naturally, if placed on a substrate, there is radial deformation, especially for large-diameter tubes;22 If suspended in air, there is uniaxial strain; even during growth, gas flow can stretch SWNTs. When fabricated into devices or incorporated into composites, SWNTs may also experience various mechanical strains. Studies on strain-induced changes in structures and properties of SWNTs are of great help for understanding various experimental observations and for improving the performances of SWNTs-based materials and devices. Raman spectroscopy, closely related to the structures of SWNTs, is undoubtedly the most powerful tool for strains-related characterizations in bundled and isolated carbon nanotubes.
The contents of this article are as follows. Section 2 briefly presents Raman features of individual SWNTs, telling what information is provided by these features. Section 3 presents the effects of four kinds of strains on Raman spectra of individual SWNTs, including uniaxial strain, torsional strain, radial deformation and bending deformation. Section 4 presents concluding remarks, summarizing past achievements and pointing out promising directions for future developments.
2. Raman spectra of SWNTs
Due to their remarkable 1-D rolling structure, SWNTs have unusual and characteristic electronic structures2 and phonon properties.9 Because of the strong coupling between electrons and phonons in resonant-Raman scattering, Raman spectra of SWNTs can be observed and can be used to probe both phonon properties and electronic structures. This section will introduce Raman features of SWNTs and why we can observe these features.2.1 Structure
A SWNT can be obtained by rolling up a monolayer graphene sheet into a seamless cylinder, and its structure can be described by a pair of indices (n, m) or by its diameter and chiral angle. Due to 1-D confinement of electronic and phonon states, along with distinct geometric structures, unique and distinct electronic structures and phonon properties are observed, both of which can be obtained by using the zone folding approach.9,23The electronic dispersion relations of SWNTs can be determined from those of a two-dimensional (2-D) graphene sheet theoretically using the zone folding approach on zero-order approximation (eqn (1): −π/T < k < π/T; μ = 1, 2, …):23
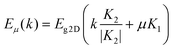 | (1) |
The phonon dispersion relations in SWNTs have been obtained from those of the 2-D graphene sheet by using the same zone folding approach (eqn (2); m = 1, 2, …, 6; μ = 0, 1, …, N− 1; −π/T < k < π/T):9,23
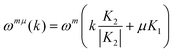 | (2) |
| armchair: 2A1g + 2E1g + 4E2g |
| zigzag: 2A1g + 3E1g + 3E2g |
| chiral: 3A1g + 5E1g + 6E2 |
2.2 Raman features of SWNTs
Despite the large number of Raman active modes, only a few modes have a large Raman scattering cross section, which greatly simplifies the Raman spectra of SWNTs. Therein, the most studied Raman active modes in SWNTs are the radial breathing mode (RBM) and the tangential mode (G-band).As shown in Fig. 1, the RBM is originated from the out-of-plane tangential acoustic modes of monolayer graphene sheet and all carbon atoms vibrate in phase along the radial direction. RBM is the characteristic mode in SWNTs, and ranges from 120 to 300 cm−1 for SWNTs with diameter between 0.9 and 2.0 nm. From RBM, one can obtain important information about geometric structure and electronic structure. The RBM frequency (ωRBM) is inversely proportional to diameter (d), satisfying the relation ωRBM = A/d + B, where A and B are sample and environment dependent, and are given by experiment.31,32 RBM intensity profile with a tunable laser can directly give the electron transition energy Eii with accuracy of ±3 meV.33,34 A much simpler experiment, measuring both Stokes and anti-Stokes Raman signals with a single laser, can be used to calculate Eii within ±10 meV precision.35,36 By considering diameter and Eii obtained from the RBM, the RBM can be used to assign chiral index (n, m). The assignment is performed by comparing characteristic patterns between theory and experiment,34 which does not need a quantitative agreement between theory and experiment.
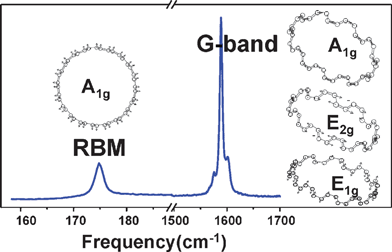 | ||
| Fig. 1 Typical Raman spectrum of an individual (10, 10) SWNT on SiO2 substrate acquired with a laser of 1.96 eV. The left one and the three right atomic displacements are, respectively, for the RBM and the G-band of the (10, 10) SWNT.9 | ||
The G-band, ranging from 1500 to 1600 cm−1 in SWNTs (shown in Fig. 1), is originated from the Raman active optical phonon mode E2g of 2-D graphene by zone folding the 2-D graphene Brillouin zone into the 1-D SWNT Brillouin zone. Due to the phonon wave vector confinement along the SWNT circumferential direction and due to symmetry-breaking effects associated with SWNT curvature, the G-band is composed of several peaks, which can be assigned to different Raman modes by polarized Raman experiment. Based on a simple analysis, there are two intense peaks: G+ and G−. For semiconducting SWNTs, G+ and G− stand for atomic displacements along the tube axis and the circumferential direction, respectively, which is opposite for metallic SWNTs.37–39 The two intense peaks can be used to characterize diameter, although it is less accurate than the RBM. The G-band can also be used to distinguish metallic SWNTs from semiconducting ones through strong differences in lineshape, to characterize doping level and strains in SWNTs.
2.3 Resonant-Raman spectra of individual SWNTs
The major breakthrough is the observation of Raman spectra from individual SWNTs because of the strong coupling between electrons and phonons in resonant-Raman scattering.13 Usually Raman spectra only depend on phonons, being independent of the electronic structure of the material and the laser excitation energy, and the Raman scattering cross section is very weak. However, the scattering cross section is greatly enhanced when the laser excitation energy matches the optically allowed electronic transition energy of the material. This intensity enhancement process is called resonant-Raman scattering. According to resonant-Raman scattering theory, the resonant-Raman intensity can be described by eqn (3):35,40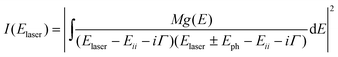 | (3) |
Raman spectra of SWNTs not only depend on their chirality (diameter and chiral angle), but also are affected by external factors, such as temperature,42–49 doping,17 strains, and so on. For example, the frequencies of both RBM and G-band downshift with increase in temperature. Downshift upon n-type doping and the upshift upon p-type doping are also observed.16,50–54 Strains have been used to tune geometric and band structure,55 which is chirality dependent. Research on Raman spectra of SWNTs under strains is not only helpful to understand their structure variation, but also useful to characterize strains in SWNTs, which will be given in the next section.
3. Raman spectra of SWNTs under strain
Strain may exist naturally, be brought about accidentally, or be induced intentionally in SWNTs, which will influence their lattice structure and electronic structure, hence their vibrational frequency and resonant intensity. Furthermore, Raman modes, responding differently to these strains, can be used to characterize these strains in SWNTs. In the following four sub-sections, Raman spectra and electronic transition energy (Eii) of SWNTs under uniaxial strain, torsional strain, radial deformation and bending deformation (shown in Fig. 2) will be reviewed.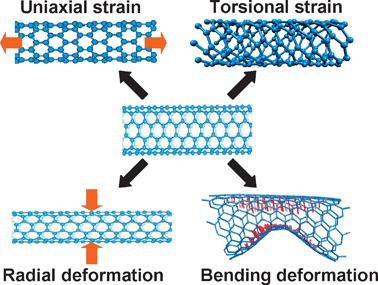 | ||
| Fig. 2 Four types of strains that SWNTs may be subject to. | ||
3.1 Uniaxial strain
When exerted by uniaxial strain, SWNTs will undergo a sudden elongation. Upon high temperature, (5–7–7–5) defects will be produced to release the strain energy56,57 and the theoretical maximum uniaxial strain is almost 20%. Under normal conditions, no energy is provided to form (5–7–7–5) and only a 7% level is observed.58 The changes in lattice and bonds will result in changes in phonon property and electronic structure. It is necessary to study Raman spectral variation of SWNTs under uniaxial strain, and to characterize uniaxial strain in SWNTs by Raman spectra. The relations between uniaxial strain and frequency and intensity will be presented in this subsection.However, force-constant simulation gives different results.59 First, above 3%, the RBM frequency continues to downshift (almost linearly), but the downshift rate of (10, 10) SWNT is different from that of (17, 0) SWNT, which is attributed to the distinct geometric structures of the different SWNTs. Upon compression, the RBM frequency upshifts with increasing compressive uniaxial strain and reaches a plateau. At about −5 and −6%, the RBM frequency shows a sharp reduction for (10, 10) and (17, 0) SWNTs, indicating that the C–C bonds buckle and the volume changes in an unstable manner. Second, Raman modes with different symmetries have different shift rates. The shift rate of E1g is larger than that of A1g and E2g for (10, 10) SWNT, while it is reversed for (17, 0) SWNT; this may cause different shift rates of G+ and G−.
Experimentally, various methods have been utilized to introduce uniaxial strain into SWNTs, e.g. bending SWNTs/epoxy composites,61–68AFM tip manipulation69,70 and stretching suspended SWNTs.71–73 Young et al. bent SWNTs/epoxy composites in a four-point bending rig.61,63–65 Depending on the mode of bending, SWNTs near the top surface and the strain gauge are in tension (bent downward) or in compression (bent upward) (see Fig. 3A).64 A strain gauge is glued on the composite top surface to measure the surface strain, which is used as a measure of the uniaxial strain of SWNTs. Fig. 3B shows that while no shift in the position of the RBMs was detected,65 the G′-band downshifts61 to a lower frequency with increasing uniaxial strain for SWNT-A (arc-discharge prepared with Ni/Y catalyst), at a downshift rate of −1.3 cm−1 %−1, cf. −15 cm−1 %−1 for SWNT-P (prepared by pulsed laser vaporization process) (as shown in Fig. 3C and D). The variations of both the low- and high-frequency modes are consistent with Wu’s calculation. As for the different shift rates of SWNT-A and SWNT-P, it is postulated that SWNT-A has poorer dispersion in the composites, which results in a less efficient reinforcement and poorer adhesion to the epoxy matrix. Therefore, the uniaxial strain of SWNTs in composites may be smaller than that of composites measured by the strain gauge, resulting in that the shift rate of SWNTs measured in composites would be smaller than their inherent shift rates.
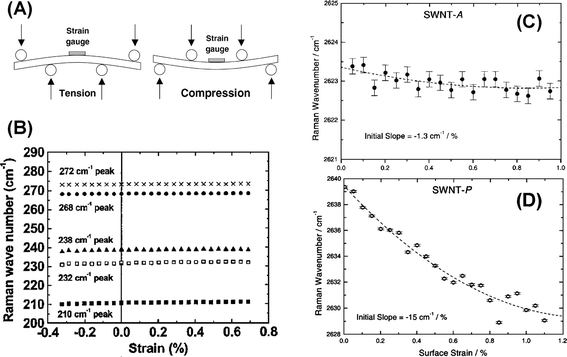 | ||
| Fig. 3 (A) A schematic snapshot of bending SWNTs/epoxy composites in a four-point bending rig.60 (B) Frequency of the RBM for (10, 10) and (17, 0) SWNTs under uniaxial strain. Frequencies of the G-band for (C) (10, 10) and (D) (17, 0) SWNTs under uniaxial strain. | ||
In order to measure the uniaxial strain directly and understand the chirality dependence of the uniaxial strain effect, experiments on individual SWNTs are performed to introduce uniaxial strain into SWNTs.69,70 The amount of uniaxial strain is determined by dividing the elongation by the total unstained length between the fixed electrodes. The elongation is determined by subtracting the deformed length from the local unstrained length in the AFM image. The D-band is downshifted by 16.1 cm−1, the G-band is downshifted by 4.8 and 12.3 cm−1 for G+ and G− respectively, and the G′-band is downshifted by 27.7 cm−1 due to uniaxial strain, when the uniaxial strain is 0.53%.69 Meanwhile, no shift in the RBM frequency is observed within the limit of instrumental accuracy. It is apparent that there is a consistent trend of increasing downshifts in these Raman modes with increasing uniaxial strain amidst the large spread frequencies. There are four points of note: first, the fitted lines give downshift rates of 20.5, 11.7, 16.7 and 37.3 cm−1 %−1 for the D-band, G−, G+ and G′-band respectively, indicating each Raman mode depends on different force constants, which depends on chirality, which is consistent with Yang et al.61 Secondly, the relative shifts of the D-, G- and G′-bands are not in a fixed relation. For some SWNTs the D-band shifts more than the G-band, while for other SWNTs the G-band shifts more than the D-band. This is probably because electrons and phonons are coupled via a double resonance mechanism for the D- and G′-band. The frequencies of the D- and G′-band are strongly dependent on excited electronic wave vectors and the uniaxial strain changes both the spring constant and electronic band structure. However, the relative shifts of the D- and G′-band in all SWNTs scale roughly by a factor of 2, maybe due to ωG′≈ 2ωD for unstrained SWNTs. Third, each set of data is taken from different SWNTs, so variation in diameter and chiral angle are expected to be the main factor for the large spread. The crude estimation of uniaxial strain, or slippage of SWNTs under the metal electrodes, makes the diameter or chiral angle dependence of the shift rates unclear. Finally, the G′-band downshifts more than those in Young’s data when stretching epoxy/SWNTs composites. This is due to the slippage among SWNTs bundles and/or between SWNTs and epoxy when stretching the composite, which makes the strain of SWNTs smaller than the strain of the composite.
To obtain chirality dependence of relations between uniaxial strain and Raman frequency, we devised a novel system.58 This system has three advantages. First, suspended SWNTs can avoid non-uniform uniaxial strain in SWNTs and curved SWNTs, which makes the calculation of uniaxial strain less accurate. Second, the length of uniaxial strained SWNTs can be directly measured by SEM. Third, the SWNTs can be stretched and relaxed many times, which can exclude possible slippage under metal films.
Fig. 4A shows the SEM image of a suspended (18, 5) SWNT, for which the uniaxial strain first increases and then decreases by stretching and relaxing the poly(dimethylsiloxane) (PDMS). The length between two gold particles first increases and then decreases linearly. Raman spectral characterization shows that RBM does not shift (shown in Fig. 4C), but G-bands (both G+ and G−) first downshift and then upshift to the original frequency (shown in Fig. 4D and E). It can be seen that G-band frequencies are linearly proportional to uniaxial strain, satisfying the relations ωG+/cm−1 = 1596.79 − 14.59ε and ωG−/cm−1 = 1577.00 − 11.00ε, for G+ and G− frequencies, respectively. The shift rate of G+ is a little larger than that of G−. It is noted that G-band frequencies overlap at the same strain during stretching and relaxing, which indicates that there is no relative slippage between SWNTs and PDMS during experiment.
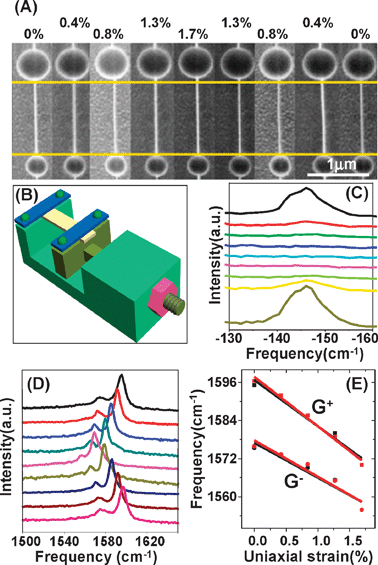 | ||
| Fig. 4 (A) SEM image, (C) RBM spectra and (D) G-band of (18, 5) SWNT when uniaxial strain first increases from 0 to 1.7% and then decreases to 0%, which is shown from top to bottom. (E) G+ and G− frequency variation as a function of uniaxial strain. Black (red) dots and lines correspond to the increase (decrease) of uniaxial strain. Lines are linearly fitted from dots which are obtained by Lorentzian fitting. (B) Schematic diagram of the stretching setup.58 | ||
Table 1 lists the G-band data of seven different SWNTs under uniaxial strain.58 The downshift rates of the G-band range from −1.89 to −31.02 cm−1 %−1. For example, for (7, 5) SWNT, the shift rate of G+ is only −3.95 cm−1 %−1, while for another SWNT is as large as −19.25 cm−1 %−1. Among the six SWNTs with RBM peaks, the shift rates of G+ increase with increasing diameter and decreasing chiral angle. It is thought that the diameter and chiral angle may play joint roles. From Table 1, it can also be seen that, for (7, 5) SWNT, the shift rate of G+ is far larger than that of G−; for (13, 7) and (18, 5) SWNTs, the shift rate of G+ is a little larger than that of G−; for another SWNT, the shift rate of G+ is far smaller than that of G−. The relative shift rate between G+ and G− is related to chiral angle. As we know, G+ and G− correspond to axial and circumferential vibrations for semiconducting SWNTs, respectively. For semiconducting SWNTs close to armchair (e.g. (7, 5) SWNT), G−-dependent C–C bonds are perpendicular to the axis of SWNTs. Therefore G− is less affected than G+. It is the chirality-dependent C-C bonds elongation that causes differences in the relative shift rate between G+ and G−.
| RBM/cm−1 | (n, m) | d/nm | θ/° | Δω(G+)/cm−1 %−1 | Δω(G−)/cm−1 %−1 |
|---|---|---|---|---|---|
| a d = Diameter, θ = chiral angle, (n, m) are assigned from RBM frequency; d and θ are calculated from (n, m). “—” indicates that the peaks are not observed. | |||||
| 310 | (6, 5) | 0.76 | 27.00 | −3.97 | — |
| 281 | (7, 5) | 0.83 | 24.50 | −3.95 | −1.89 |
| 213 | (11, 5) | 1.13 | 17.78 | −6.50 | — |
| 177 | (14, 5) | 1.35 | 14.70 | −6.43 | — |
| 171 | (13, 5) | 1.40 | 20.17 | −13.06 | −11.80 |
| 146 | (18, 5) | 1.66 | 11.93 | −14.59 | −11.00 |
| — | — | — | — | −19.25 | −31.02 |
Due to SERS effect of gold particles on SWNTs, some weak Raman modes are detected.74–77 For instance, for the M-band,78 when uniaxial strain is increased from 0 to 7.1% (shown in Fig. 5A), the M-band frequency (ωM) downshifts linearly with uniaxial strain increasing,58 satisfying the relation ωM(cm−1) = 1695.79 − 2.58ε (shown in Fig. 5C). Furthermore, for the intermediate frequency mode (IFM)79,80 under uniaxial strain from 0 to 7.1%, the IFM frequency (ωIFM) upshifts linearly with uniaxial strain increasing, satisfying the relation ωIFM(cm−1) = 1049.84 + 1.06ε (see Fig. 5E). The shift trend is opposite to other Raman modes (D-, G-, M- and G′-bands). It is known that IFMs relate to the double resonance process and are originated from the combination of two phonon modes, one optical and one acoustic. It is speculated that uniaxial strain induced lattice transformation needs to be considered when some complicated Raman scattering processes are involved, which may also be a minor factor in the shift of G-band frequency.58 Although there is a SERS effect, no D-band81 appears any of the seven uniaxial strained SWNTs even under 7% uniaxial strain. It is known that D-band is centered at ∼1300 cm−1 and is originated from disorders in SWNTs, such as dangling bonds, 5–7 rings, sp3 hybridization or edge effects. The absence of a D-band means that SWNTs possess excellent elastic properties, and uniaxial strain only induces lattice transformation, which can be recovered when uniaxial strain is relaxed.
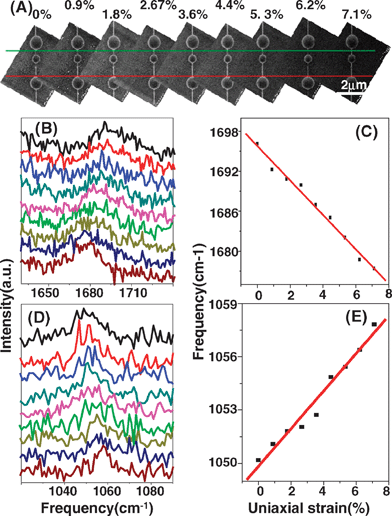 | ||
| Fig. 5 (A) SEM image, (B) M-band spectra and (D) IFM spectra of (7, 5) SWNT when the uniaxial strain is increased from 0 to 7.1% (shown from top to bottom). (C) M-band and (E) IFM frequencies as a function of uniaxial strain; lines are linearly fitted from experimental data which are obtained by Lorentzian fitting.58 | ||
For the same purpose, Lee et al. did a similar study using a combined Raman/AFM setup.71 In their experiment, the RBM frequency does not shift with increasing uniaxial strain within the experimental uncertainty, which is in agreement with above results.58,61,69 No downshift of the G-band is observed but rather an increase in the frequency is observed when the uniaxial strain is increased beyond a certain critical value (∼2%) for two SWNTs, which is not in agreement with all calculations59,60 and other experiments.58,61,69 Further the upshift is irreversible indicating that structural changes occur. Below the critical strain, the G-band frequency remains constant, but there are strong variations in the intensity of the G-band. The authors71 postulate that the SWNTs are very close to the armchair in geometries (11, 10) and (10, 9), where the relevant bond length is not significantly altered by uniaxial strain. However, the upshift of the G-band is still unexpected, which needs more theoretical studies. Furthermore, a much broader D-band becomes clearly visible for large strain. When uniaxial strain is increasing beyond ∼0.5%, the broad D-band increases significantly in intensity, but does not change position in frequency. The introduction of defects in the SWNTs under uniaxial strain is suggested. So many surprising and different results indicate the complicated processes when SWNTs are subjected to uniaxial strain.
ΔEiiS = (−1)i+1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sgn(2p + 1)3t0[(1 + ν)σcos3θ] sgn(2p + 1)3t0[(1 + ν)σcos3θ] | (4) |
| ΔEiiM = ±3t0[(1 + ν)σcos3θ] (+ and − for low and high branches of EiiM) | (5) |
Young et al. studied the intensity variation of RBM in epoxy/SWNT composites induced by uniaxial strain from −0.3% (compression) to 0.7% (tension).65 The intensities of the 210, 232 and 238 cm−1 peaks increase in tension, whereas the intensities of the 268 and 272 cm−1 peaks decrease. During the compression, the RBM intensities vary the opposite way; the intensities of the 210, 232 and 238 cm−1 peaks decrease in tension, whereas the intensities of the 268 and 272 cm−1 peaks increase. For the above semiconducting SWNTs where E22S is in resonance with Elaser, these intensity variations are reversible; the peak intensities returned to their initial values when the uniaxial strain is released. The variations range from 10% for the 210 cm−1 peak to 150% for the 238 cm−1 peak. The direction and amount of intensity change strongly depend on the chirality of the SWNTs.
However, Croninet al. observed no change in RBM intensity70 and anti-Stokes/Stokes intensity ratio when uniaxial strain is induced by pushing semiconducting SWNTs with an AFM tip, where E33S is in resonance with Elaser. One possible reason is that the semiconducting SWNTs are not totally in resonance; therefore a large change in Eii can not affect the intensity strongly. Another possible reason is that the resonance window of E33S is broadened by some uncertain factors. However, for metallic SWNTs, Fig. 6A shows significant changes in their RBM intensities before and after AFM manipulation, where E11M is in resonance with Elaser. Especially one metallic SWNT with RBM at 184 cm−1 (tentatively assigned to (15, 3) SWNT) is brought on resonance from previously off-resonance condition89 (shown in Fig. 6B); while no differences in the intensities of D-, G- and G′-bands are observed for SWNTs where uniaxial strain improves the resonance condition relative to SWNTs and where uniaxial strain spoils the resonance condition. Calculation on (15, 3) SWNT with uniaxial strain of 0.8% shows that this dramatic change in the RBM intensity is due to the constructive interference effect,89 thus enhancing the Raman signal (shown in Fig. 6C). The interference effects arise mainly from the matrix element dependence on strain.
 | ||
| Fig. 6 (A) The changes in intensity of RBM for five metallic SWNTs measured before and after uniaxial strain.70 (B) The Stokes and anti-Stokes RBM spectra of the (15, 3) SWNT measured under different uniaxial strains.89 (C) 2D plot for the calculated resonance-Raman profile for the (15, 3) SWNT under different uniaxial strains.89 The red (blue) areas indicate high (low) intensity. The vertical line stands for Elaser = 1.956 eV used for measuring the spectra shown in (B). | ||
For the (7, 5) semiconducting SWNT shown in Fig. 5A, we observed obvious changes in RBM intensity. As uniaxial strain is increased from 0 to 3.5%, IAS/IS (Anti-Stokes/Stokes) quickly decreases from 0.16 to 0.06; as uniaxial strain is increased from 3.5 to 7.0%, IAS/IS slowly increases from 0.06 to 0.09. Fig. 7B shows the relationship between IAS/IS and E22S for the (7, 5) SWNT calculated by eqn (6), where the thick line corresponds to the change of IAS/IS in the experiment and the arrow indicates the change direction.
 | (6) |
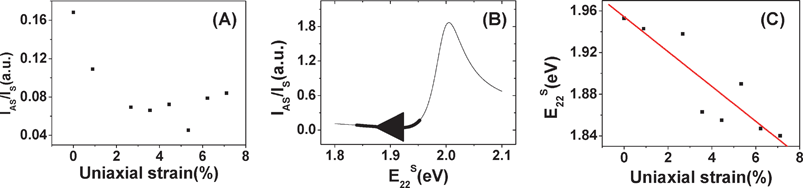 | ||
| Fig. 7 (A) The IAS/IS variation of the (7, 5) SWNT when uniaxial strain is increased from 0 to 7.1%. (B) The relationship between IAS/IS and E22S for the (7, 5) SWNT calculated by resonance theory, where the thick line corresponds to the change of IAS/IS in the experiment and the arrow indicates the change direction. (C) E22S as a function of uniaxial strain for the (7, 5) SWNT. | ||
3.2 Torsional strain
Due to their tubular cross section, SWNTs will rotate when a torque is exerted on their walls. If two ends are fixed, torsional strain will be introduced into SWNTs. Torsional strain was first intentionally introduced into SWNTs by Meyer et al., who built a torsional pendulum based on SWNTs by nanolithography.90 By electrically rotating the metal block, quantitative elastic torsional strain was induced. We manipulated ultra-long SWNTs on a substrate using an AFM tip. Due to the friction between the SWNTs and substrate, SWNTs would roll on the substrate. As a result, a local torsion was also introduced into the SWNT.18 This subsection will focus on how torsional strain affects Raman spectra of SWNTs.Chang et al. observed significant nonlinear response of ωRBM to torsional strain, even at small torsional strain.91ωRBM of achiral SWNTs, always decreases, no matter along which direction the SWNT is twisted. The variation of ωRBM of chiral SWNTs is complicated and dependent significantly on the torsional direction because of geometrical symmetry. With torsional strain increasing along the right hand direction, ωRBM of chiral SWNTs decreases monotonously, while along the left hand direction, ωRBM slightly increases first and then decreases after torsional strain beyond a critical value. For a given diameter, the downshift rate of RBM frequency of a zigzag SWNT is more significant than that of an armchair SWNT.
Experimentally, we introduced torsional strain into SWNTs by AFM manipulation and studied the effect of torsional strain on Raman spectra of SWNTs.18,92Fig. 8A shows a typical result, where the (13, 11) SWNT was dragged away from the manipulation point, about 0.767 μm, revealing that frictional forces between tube and substrate are strong enough to maintain the deformations of SWNTs after the removal of AFM tip. As shown in Fig. 8B, on the left of manipulation point, ωRBM upshifts from 148.5 to 150.2 cm−1, then downshifts to 148.5, forming a “Λ” shaped distribution. The same change occurs on the right of the manipulation point. Therefore, the distribution of ωRBM along the axis of SWNT forms an “M” shape. The “M” shape is induced by the elastic retraction of the nanotube in combination with the friction after the tip has been removed. After excluding the effects of temperature, charge transfer, and radial deformation, bending and uniaxial strain of SWNTs, the formation of torsional strain is the only possibility for the “M” shape distribution of the RBM frequency (dashed lines in Fig. 8B). More importantly, it indicates that the RBM of (13, 11) SWNT upshifts to higher frequency under torsional strain.
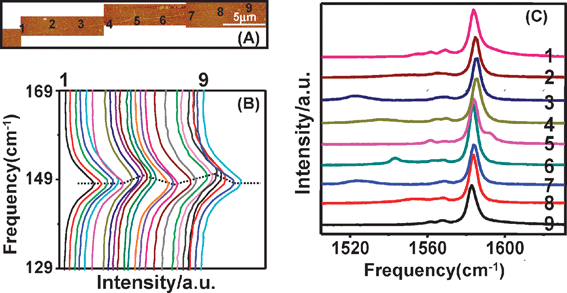 | ||
| Fig. 8 (A) Typical tapping mode AFM image of a SWNT after AFM manipulation. (B) RBM and (C) G-band92spectra of (14, 2) SWNT after AFM manipulation. “1–9” are the post-manipulation spectra of nine points along SWNT indicated in (A). The dashed lines indicate the M-shaped distribution of ωRBM along the axis of the SWNT. | ||
Fig. 8C shows the G-band spectra at nine points (labeled by “1–9” in Fig. 8A) along the tube axis. The G-band spectra vary significantly along the SWNT axis, which reveals a mixing effect of both uniaxial and torsional strain: uniaxial strain makes all the peaks downshift except the peak at the highest frequency; while torsional strain makes the peak at the highest frequency downshift remarkably, but the other peaks upshift a little. It is noteworthy that in the torsional region, G-band spectra at nine points vary gradually and non-monotonously along the tube axis. This indicates that torsional strain is not uniformly distributed, but shows a gradual variation, with an “M” shape along the tube axis, which is due to the balance of torsional strain in the SWNT and frictional forces by the substrate. The G-band modes, 2A1g, 2E1g and 2E2g were assigned by polarized resonant-Raman spectroscopy. The effects of strains on these modes of individual (13, 11) SWNTs are shown in Fig. 9, where the frequencies of E2g+, A1g+, A1g− and E1g− modes vs. the distance away from the manipulation point were plotted. It can be seen92 from Fig. 9A that uniaxial strain has no effect on the E2g+ mode, but torsional strain makes the E2g+ mode downshift by a maximum of about 79.0 cm−1 (shown in Table 2). However, as shown in Fig. 9B, C and D, uniaxial strain makes A1g+, A1g− and E1g− modes downshift by a maximum of about 8.0, 8.0 and 4.5 cm−1, but torsional strain makes them upshift by a maximum of about 1.4, 1.5 and 4.5 cm−1 (shown in Table 2). Table 2 lists G-bands of eight individual ultra-long SWNTs, including four semiconducting tubes and four metallic tubes. Of eight individual SWNTs, G-bands of seven individual SWNTs have a similar response to torsional strain. The E2g+ mode downshifts significantly, by up to 91 cm−1 whereas A1g+, A1g−, E1g+, E1g− and E2g− modes upshift a little, typically by several wavenumbers. For any one SWNT, the upshifts of A1g+, A1g− and E1g+ modes are almost similar, and the E1g− mode upshifts 3–4 times as much as that of A1g+, A1g− and E1g+ modes. This indicates the effect of torsional strain on G-band modes is related to mode symmetries. If SWNTs are assumed to roll on the substrate without slippage, the downshift of the E2g+ mode increases with increasing the diameter and chiral angle, indicating that chiral index plays an important role in torsional strain effect on G-band modes.
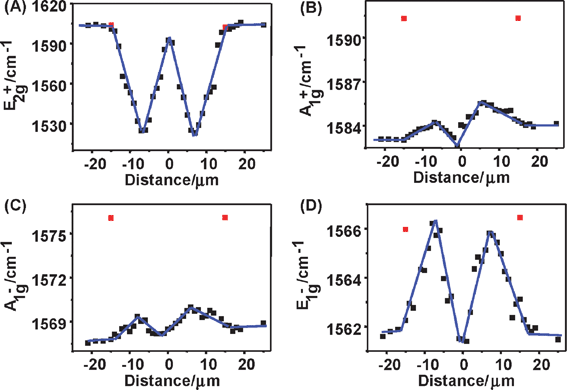 | ||
| Fig. 9 Frequency profiles of (A) E2g+, (B) A1g+, (C) A1g− and (D) E1g− modes along the (13, 11) SWNT axis after manipulation.92 Red and black squares are frequencies before and after manipulation, respectively. Black lines are linear curve-fitted results. The manipulation point is at 0 μm. | ||
| Δω/cm−1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RBM/cm−1 | (n, m) | d t (theor.)/nm | θ/° | d t (exptl.)/nm | Δd/μm | E2g+ | A1g+ | E1g+ | E1g− | A1g− | E2g− | BWF |
| a All the RBM were collected before manipulation and calibrated by Rayleigh scattering. Chiral indices (n, m) are assigned based on frequencies and intensities. Diameter dt and chiral angle θ are calculated based on chiral indices (n, m); Δd refers to the distance from the manipulation point; “—” means the modes were not observed. | ||||||||||||
| 141.4 | (15, 10) | 1.73 | 23.4 | 1.4 | 1.160 | −52.0 | +0.5 | — | +2.0 | +1.0 | — | — |
| 148.5 | (13, 11) | 1.65 | 27.2 | 1.4 | 0.767 | −79.0 | +1.4 | — | +4.5 | +1.5 | + | — |
| 155.9 | (16, 6) | 1.54 | 15.3 | 1.2 | 0.484 | −33.0 | +1.0 | +1.0 | + | +0.8 | +2.0 | — |
| 160.6 | (12, 10) | 1.51 | 27.0 | 1.0 | 0.775 | −66.0 | +1.0 | +1.0 | +3.6 | +1.0 | — | — |
| 198.9 | (14, 2) | 1.20 | 6.59 | 1.7 | 1.142 | −26.0 | −27.0 | — | — | −30.0 | — | −51.0 |
| 211.9 | (11, 5) | 1.13 | 17.8 | 2.1 | 2.428 | −91.0 | +1.2 | — | — | — | — | — |
| 218.6 | (12, 3) | 1.09 | 10.9 | 2.1 | 0.658 | −75.0 | +1.4 | — | — | — | — | — |
| 285.8 | (7, 5) | 0.83 | 24.5 | 1.1 | 0.573 | −29 | +0.5 | — | — | +0.5 | — | — |
Note that in Table 2, G-band modes of (14, 2) SWNT and torsional-strain-induced variation are different from the other seven SWNTs. The G-band has a broad and asymmetric Breit–Wigner–Fano (BWF) line shape at lower frequency, indicating that the quasi-acoustic plasmon mode in SWNT forms hybrid excitations with the phonon mode.93 Under torsional strain, all modes downshift to lower frequencies significantly (tens of wavenumbers). It is probably the interaction between the quasi-acoustic plasmon mode and G-band modes that changes the torsional strain effect on G-band modes.
Since torsional strain can cause symmetry breaking, it is predicted that torsion would induce mode splitting.59 How are original peaks, plus new peaks, affected by torsional strain? Fig. 10A shows G′-band spectra94 of one SWNT from three positions at the torsional region, which originates from a strong coupling between phonons and electrons, and their frequency is strongly sensitive to electronic band structure. From bottom to top, they are spectra without torsional strain, with small torsional strain and with large torsional strain, respectively. Clearly new peaks appear upon application of torsional strain. Fig. 10B shows the peak frequencies at different torsional regions. The data in red is the frequency of the original peak and is upshifted a little under torsional strain. As torsional strain is increased, a new peak appears, and it is downshifted significantly under torsional strain (data in green). As the SWNT is twisted further, another new broad peak appears, which is also downshifted significantly under torsional strain (data in blue). This interesting observation is caused by the combinational effects of bond change and symmetry breaking. Torsional strain induced symmetry breaking and led to new peaks; while bond changes made peaks upshift or downshift. Such symmetry breaking-induced new peaks have also been observed in G-band spectra.18
 | ||
| Fig. 10 (A) G′-band spectra of one SWNT from three positions at the torsional region. From bottom to top, the spectra correspond to no torsional strain, small torsional strain and large torsional strain, respectively. (B) G′-band frequencies vs. position along the axis of the SWNT. Red data corresponds to the original peak while green and blue data are from new peaks as a result of torsional strain. | ||
| ΔEiiS = (−1)isgn(2p + 1)3γt0sin3θ | (7) |
| ΔEiiM = ±3γt0sin3θ (+ and − for high and low branches of EiiM) | (8) |
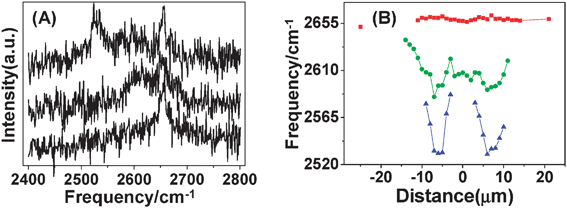
In addition to frequency upshift, changes in intensity of RBM are observed.95 For the SWNT shown in Fig. 8A, in torsional strained region, IS decreased and formed a W shape along the SWNT axis; accordingly IAS/IS increased and formed an M shape (shown in Fig. 11A). The optimal Γ value of 30 meV was estimated by theoretically fitting experimental IS and IAS/IS data (inset of Fig. 11B). Using eqn (6), E33S along the SWNT axis was obtained (shown in Fig. 11B). As torsional strain γ continuously increased from −15 to −8 μm, E33S decreased continuously from 1.865 to 1.823 eV. That is, for semiconducting (13, 11) SWNT, dE33S/dγ is negative. As for the asymmetrical Eii profile at the two sides of the manipulation points, it may be the opposite twisting direction that causes some difference in the Eii variation and results in the asymmetrical Eii profile.
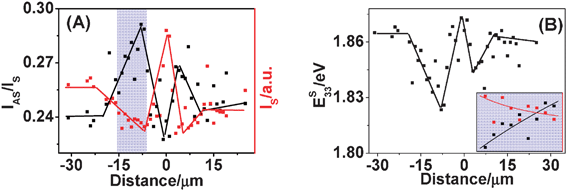 | ||
| Fig. 11 (A) IS (red dots and lines) and IAS/IS (black dots and lines) profiles of RBM spectra along (13, 11) SWNT axis after manipulation. Dots are experimental data and lines are linear curve-fitted results. The blue-shaded region is used to estimate Γ as shown in the inset of (B). (B) E33S along the SWNT axis after manipulation. Dots are values calculated using eqn (6) in the text and lines are linear curve-fitted results. Inset: the Γ value of 30 meV was obtained by fitting between experimental IS and IAS/IS data and theoretical calculation. The experimental data are from the blue-shaded region of (B), from −15 to −8 μm.95 | ||
Table 3 lists the data of 10 individual ultra-long SWNTs, including six semiconducting tubes and four metallic tubes. It can be seen that the variation of Eii induced by torsional strain is related to q. Under torsional strain, for semiconducting SWNTs, E33S increases for q = +1, E33S decreases and E22S increases for q = −1, and for metallic SWNTs, E11M always increases. Theoretically, the torsional strain-induced variation of Eii is related to the sign of q and the van Hove singularity index i (1, 2, 3 …). The relative positions of semiconducting SWNTs with q = +1 and q = −1 in k space are opposite relative to the Brillouin zone vertices. Under torsional strain, perturbations to the Fermi wave vector kF act in opposite directions and consequently cause shifts of Eii in different directions. E22S and E33S are generated where two cutting lines on the two sides of Brillouin zone vertices cross with π and π* electronic states, so that they have opposite responses under torsional strain. For metallic SWNTs, dE11M/dγ is always positive. This is because the resonance with the higher energy component of E11M has a much lower intensity than that of the lower energy component and only the lower energy component of E11M can be detected by resonant-Raman scattering.
| ω RBM /cm−1 | (n, m) | ΔEiimax/meV | dEii/dγ | q |
|---|---|---|---|---|
| a All the ωRBM were collected before manipulation and calibrated by Rayleigh scattering. Chiral indices (n, m) are assigned based on ωRBM and IAS/IS. The inverse scattering lifetime Γ is obtained by fitting experimental IS and IAS/IS data to theoretical calculation. ΔωRBMmax and ΔEiimax are, respectively, the maximum variation of ωRBM and Eii after SWNTs were broken. γ is the torsional strain. “—” means that anti-Stokes spectra were too weak and were not completely collected. | ||||
| 141.4 | (15, 10) | −10.1 | dE33S/dγ < 0 | −1 |
| 148.5 | (13, 11) | −39.1 | ||
| 160.6 | (12, 10) | −9.0 | ||
| 155.9 | (16, 6) | +1.0 | dE33S/dγ > 0 | +1 |
| 159.4 | (13, 9) | +4.5 | ||
| 285.8 | (7, 5) | — | dE22S/dγ < 0 | −1 |
| 177.1 | (14, 5) | +4.9 | dE11M/dγ < 0 | 0 |
| 198.9 | (14, 2) | +15.8 | ||
| 211.9 | (11, 5) | — | ||
| 218.6 | (13, 1) | +6.5 | ||
After stretching to breakage, the maximal upshifts of ωRBM ranged from 0.55 to 2.13 cm−1 and the maximal variation of Eii were between 1.0 and 39.1 meV, which has some relation to the chiral indices (n, m). Moreover, the ωRBM of SWNTs close to armchair upshifted more than those close to zigzag. This also applied to the variation of Eii. It is possible that SWNTs close to armchair are more sensitive to torsional strain than those close to zigzag.82 Comparing semiconducting SWNTs with q = −1 to +1, the upshift of ωRBM and the variation of E33S are more obvious for q = −1. Therefore, for SWNTs with q = −1, torsional strain can result in larger phonon variation and E33S variation for the same shift of kF. This could be understood as follows: near the K point at the corner of hexagonal Brillouin zone, due to a trigonal warping effect, equi-energy contours along the Γ–K line in the inner region of the Brillouin zone are denser than those along the K–M line at the outer regions. That is, Eii varies more rapidly at the inner than that at the outer regions.96 The third nearest cutting line of SWNTs with q = −1 is at the inner region of the Brillouin zone, and q = +1 at the outer region. Thus under equal torsional strain E33S of SWNTs with q = −1 varies more. It is the chirality-dependent Eii variation that makes the variation of resonant-Raman intensity chirality-dependent.
3.3 Radial deformation
Since radial deformation was first discovered in two closely parallel carbon nanotubes due to van der Waals interaction,97 more theoretical98–103 and experimental98–100,103–106 studies have touched this field. There are mainly two ways to introduce radial deformation into SWNTs: hydrostatic pressure98 and AFM indentation.107 With the former, SWNTs are loaded into a gasketed diamond anvil cell (DAC) using gas or liquid as the pressure medium, where SWNTs under hydrostatic pressure are easily characterized by Raman spectroscopy. With the latter technique, SWNTs are compressed by an AFM tip under lithography mode, where radial deformed SWNTs can not be easily characterized by Raman spectroscopy. Hence, this subsection mainly focuses on Raman spectra of SWNTs under hydrostatic pressure.Similar studies have been performed at a higher pressure, in different pressure media and with different laser excitation. Regardless of individual SWNTs or SWNT bundles, there are no obvious differences in all these upshift rates, generally ranging from 4.5 to 10.1 cm−1GPa−1, which is chirality dependent. For pressures higher than 15 GPa, a D-band appeared and the original spectra could not be recovered completely, indicating damage to SWNTs under high pressure. When excited by different laser energies, different upshift rates were observed. For instance, when excited by a 633 nm laser, the upshift rate of G+ is about 6.5 cm−1GPa−1, while by the 514 or 785 nm laser, it is about 8.0 cm−1GPa−1.111RBM signals disappear around 5 GPa with the 514.5 nm laser and at 10 GPa with the 632.8 nm laser.112
Different solvents have been used as pressure medium, namely, methanol, 4 : 1 and 2 : 1 methanol–water mixtures. It is found that the upshift rate is 7.2 cm−1GPa−1 for pure methanol and 80 mol% methanol while it is 11.4 cm−1GPa−1 for 60 mol% methanol. Similar observation has been found for G-band spectra. The reason is given as follows.113 Upon increasing pressure, methanol molecules are preferentially adsorbed forming a mobile phase on the surface of SWNTs, and this causes an upshift in the Raman spectra. As the pressure increases, the number of adsorbed molecules increases leading to the observed linear upshift. Changing the chemical composition of the pressure medium thus affects the phonon deformation potentials of Raman modes.
In practice, radial deformation exists in sitting-on-substrate (SOS) SWNTs due to van der Waals interactions between SWNTs and the substrate, which may deform SWNTs sitting on the substrate in the radial deformation.22,114–117 By comparing Raman spectra of the same individual SWNTs between SOS segments and suspended (SUS) segments (shown in Fig. 12), we studied the effect of radial deformation on Raman spectra of individual SWNTs.22,117 The RBM frequencies from the SUS and the SOS segments of the same SWNTs exhibit an observable shift, up to 12 cm−1. It is also found that the shift is more obvious for SWNTs with larger diameter and appears to be chirality dependent. For SWNTs with similar diameter, the shift tends to increase with decrease in chiral angle, which is consistent with calculated results.83,118 The linewidth of the RBM is reduced significantly in the SUS segments compared with the SOS segments for about half of the studied SWNTs, while a few SWNTs exhibit wider linewidth in the SUS segments. It is expected that the change in Eii affects the resonance condition and increases or decreases the linewidth in the SUS segments. In the same way, the observed IAS/IS changes are explained. The variation of Eii is consistent with the predicted family behavior of Eii variation for SWNTs under radial deformation.82,119 The G-band of semiconducting SWNTs shows little change except for a couple of SWNTs which exhibit a small shift of about 5 cm−1 in the G− frequency. This is consistent with the curvature dependent of the G− frequency.
 | ||
| Fig. 12 Raman spectra of the same SWNT with the laser spot moving along the tube from the middle of the trench to the left side. The left inset is the schematic figure and the right inset is the RBM spectra when the laser beam moves from the middle of the trench to the right side.22 | ||
To explain their experiment, Yao et al. performed first principles calculation using the local-density approximation (LDA) in density functional theory.110 The optimized structure under hydrostatic pressure changes from circular to elliptical and to a peanut-shaped structure, in agreement with other simulations (shown in Fig. 13B).102,121 The frequency of the G-band first increases and then becomes almost constant, followed by even a slight decrease. The intensity of the RBM noticeably drops almost simultaneously and then becomes weak, while the frequency of the RBM increases monotonously; the RBM mode can still exist until the peanut structure forms. The general tendencies of the frequency of both the RBM and G-band under hydrostatic pressure are in good qualitative agreement with their experiment, indicating that the plateaus in G-band frequency shift originate from a structural change in SWNTs. In addition, the calculated decrease in the RBM intensity with the increasing strain is at the onset of the G-band plateau (shown in Fig. 13C), which is also observed in their experiment and Yang’s study.101 Yan et al. found the RBM transition pressures scale with the diameter,101,122 as P∼ 1/d3. As for the G-band, the variation depends on Raman modes symmetry: above 5.3 GPa, the E1g mode frequency upshifts, the A1g mode frequency keeps constant and the E2g mode frequency downshifts, which are assumed to be originated from the symmetry degradation.
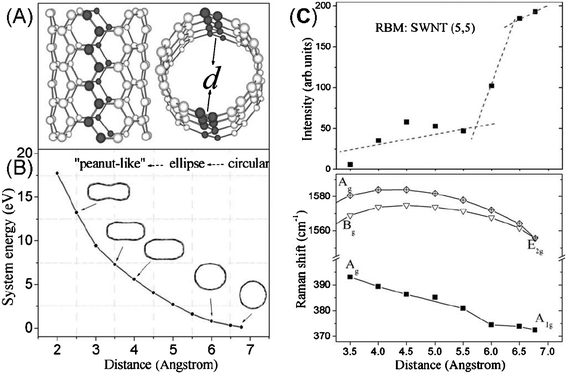 | ||
| Fig. 13 (A) Sketch of a (5, 5) SWNT model, and (B) the system energy of deformed SWNTs with different optimized structural shapes. The distance (x axis) is the shortest distance between two carbon atoms located opposite each other on different sides of the axis of the SWNT (marked with black atoms). (C) Calculated relative intensities and frequencies for the same SWNT.110 | ||
From experiments and models, pressure induced structural transitions in SWNTs are well supported and are identified in Raman measurements. The formation of a peanut structure may be the onset of collapse, which causes irreversible variation of Raman spectra.
3.4 Bending deformation
Because SWNTs have large aspect ratio, from 103 up to 106, they are easily subject to bending deformation. Under bending deformation, the inner side is compressed and the bonds are shortened; while the outer side is stretched and the bonds are elongated, which makes the system more complex. Bending is ubiquitous in experiments. It is both theoretically123–126 and experimentally5,125,127–129 found that there two stages when bending SWNTs: first delocalized effects, then buckling. However, when bending SWNTs, uniaxial strain (tension or compression) can not be avoided. It is difficult to detect electronic structures and physical properties under pure bending deformation. Most bending related experimental studies are focused on geometric structure evolution under bending deformation, which ordinarily is accompanied by small uniaxial strain. Meanwhile, bending with small curvature radius, less than tens of nanometers, can affect electronic structures and physical properties of SWNTs. Experimentally, such small and uniform curvature radius over a large region can hardly be induced, and therefore its effect can hardly be detected.Theoretically, there is only one paper concerning the effect of bending on Raman spectra of SWNTs by a density-function-based tight-binding method.130Raman spectra are calculated by a non-resonant bond polarization method. A dimensionless variable (Θ) is defined by Θ = D/2R, where D is the diameter, and R is the curvature radius. For simplicity, only the first stage is considered. RBM does not vary, because bending mainly affects bonds parallel to the axis of SWNTs. However, as shown in Fig. 14 there are obvious changes in the G-band spectra. First, new peaks appear, e.g. the E1+ mode of (6, 6) SWNT. It is thought that bending breaks the symmetry in the x direction and causes E1+ to resemble A0+ and E2+ modes which are Raman active in straight SWNTs. Due to this resemblance, E1+ and A0+ have nearly equal Raman intensities. Second, significant frequency shifts are observed. For example, the A0+ and E2+ modes upshift; this is because the antinodes concentrate in the inner side of the bending, where the spring constants are larger. At the same time, the E1+ and E3+ modes downshift because the antinodes concentrate in the outer side of the bending, where the spring constants are smaller. If antinodes are more equally distributed, Raman modes are less affected. Finally, some peaks are split into smaller ones. Bending modifies two-dimensional Raman modes pairwise the same way (antinodes migrate in same direction) and does not lift the degeneracy of Raman modes because vibrational energy is increased or decreased for both Raman modes. Hence the appearance of peak splitting is due to originally different Raman modes, and not due to lifted degeneracy. Fig. 14 also shows that effects of bending deformation on Raman modes of SWNTs are related to chirality.130
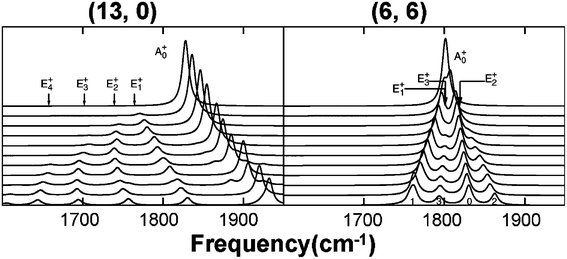 | ||
| Fig. 14 Raman spectra of bending (13, 0) and (6, 6) SWNTs. Bending deformation increases linearly from zero (upmost lines) to Θ = 4.2% for (13, 0) and Θ = 2.6% for (6, 6) SWNTs (lowest lines).130 | ||
4. Summary and outlook
Resonant Raman spectra of SWNTs and their application to strains have been reviewed, showing that strains can affect/modulate the geometric structure and hence resonant-Raman spectra of SWNTs depending on the strain type, and that Raman spectroscopy is a powerful and useful tool for the study and characterization of strains in SWNTs.Strains, either naturally existing or deliberately induced, can affect/modulate the structures of SWNTs. From this review we can see that a lot of work has been done to investigate the Raman spectra of strained SWNTs. On the one hand, the strain-induced structure transformation tends to modulate the phonon properties of SWNTs, causing frequency shift and even new peaks. On the other hand, the strain-induced structure transformation tends to modulate the electronic structure (Eii), changing the resonant intensity. These studies will be useful for using Raman spectroscopy as a tool to determine the strain type and to measure the strain amount in SWNTs. For SWNTs-based composites, it may provide information on the strain level and the adhesion between SWNTs and the matrix. Also quantitative studies on frequency shifts of SWNTs embedded in composites may be used as a sensing device. Since Raman spectra of SWNTs are closely related to structure and symmetry, it is a good tool to correlate the structure and symmetry with strains. For example, assigning chiral indices by strains has been attempted.131 Also the study of the structure of SWNTs in response to strains may help understand structure evolution at the nanoscale under strain.
Some of the conclusions are expected to extend to graphene,132–134 which is another new promising carbon nanomaterial. For example, the chirality dependence of uniaxial strain in SWNTs will probably exist in graphene when stretching graphene along different directions. Raman spectroscopy has been used to show strain uniformity in large-area epitaxial graphene, which is crucial for clearly understanding the graphene process/property relationship and ultimately controlling the graphene/substrate interface properties.135
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 50821061, 20833001, 20828004, 20725307) and the Ministry of Science and Technology of China (Grants 2007CB936203, 2006CB932701, 2006CB932403).References
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS
.
-
R. Saito, G. Dresselhaus and M. S. Dresselhaus, Physical Properties of Carbon Nanotubes, Imperial College Press, London, 1998 Search PubMed
- Z. Yao, C. L. Kane and C. Dekker, Phys. Rev. Lett., 2000, 84, 2941 CrossRef CAS
- M. M J. Treacy, T. W. Ebbesen and J. M. Gibson, Nature, 1996, 381, 678 CrossRef CAS
- E. W. Wong, P. E. Sheehan and C. M. Lieber, Science, 1997, 277, 1971 CrossRef CAS
- J. P. Salvetat, G. A. D Briggs, J. M. Bonard, R. R. Bacsa, A. J. Kulik, T. Stockli, N. A. Burnham and L. Forro, Phys. Rev. Lett., 1999, 82, 944 CrossRef CAS
- V. Sgobba and D. M. Guldi, Chem. Soc. Rev., 2009, 38, 165 RSC
- A. Popp, O. Yilmazoglu, O. Kaldirim, J. J. Schneider and D. Pavlidis, Chem. Commun., 2009, 3205 RSC
- M. S. Dresselhaus and P. C. Eklund, Adv. Phys., 2000, 49, 705 CrossRef CAS
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47 CrossRef
- M. S. Dresselhaus, G. Dresselhaus and A. Jorio, J. Phys. Chem. C, 2007, 111, 17887 CrossRef CAS
- J. L. Sauvajol, E. Anglaret, S. Rols and L. Alvarez, Carbon, 2002, 40, 1697 CrossRef CAS
- A. Jorio, R. Saito, J. H. Hafner, C. M. Lieber, M. Hunter, T. McClure, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. Lett., 2001, 86, 1118 CrossRef CAS
- A. Mews, F. Koberling, T. Basche, G. Philipp, G. S. Duesberg, S. Roth and M. Burghard, Adv. Mater., 2000, 12, 1210 CrossRef CAS
- H. Kataura, Y. Kumazawa, Y. Maniwa, I. Umezu, S. Suzuki, Y. Ohtsuka and Y. Achiba, Synth. Met., 1999, 103, 2555 CrossRef CAS
- A. M. Rao, P. C. Eklund, S. Bandow, A. Thess and R. E. Smalley, Nature, 1997, 388, 257 CrossRef CAS
- M. Terrones, A. G. Souza and A. M. Rao, Top. Appl. Phys., 2008, 111, 531 CAS
- X. J. Duan, H. B. Son, B. Gao, J. Zhang, T. J. Wu, G. G. Samsonidze, M. S. Dresselhaus, Z. F. Liu and J. Kong, Nano Lett., 2007, 7, 2116 CrossRef CAS
- D. A. Heller, P. W. Barone, J. P. Swanson, R. M. Mayrhofer and M. S. Strano, J. Phys. Chem. B, 2004, 108, 6905 CrossRef CAS
- A. G. Souza, A. Jorio, G. G. Samsonidze, G. Dresselhaus, M. S. Dresselhaus, A. K. Swan, M. S. Unlu, B. B. Goldberg, R. Saito, J. H. Hafner, C. M. Lieber and M. A. Pimenta, Chem. Phys. Lett., 2002, 354, 62 CrossRef
- A. Jorio, C. Fantini, M. A. Pimenta, D. A. Heller, M. S. Strano, M. S. Dresselhaus, Y. Oyama, J. Jiang and R. Saito, Appl. Phys. Lett., 2006, 88, 23109 CrossRef
- Y. Y. Zhang, J. Zhang, H. B. Son, J. Kong and Z. F. Liu, J. Am. Chem. Soc., 2005, 127, 17156 CrossRef CAS
-
M. S. Dresselhaus, G. Dresselhaus and P. Avouris, Carbon Nanotubes: Synthesis, Structure, Properties, and Applications, Springer, Berlin, 2001 Search PubMed
- A. M. Rao, E. Richter, S. Bandow, B. Chase, P. C. Eklund, K. A. Williams, S. Fang, K. R. Subbaswamy, M. Menon, A. Thess, R. E. Smalley, G. Dresselhaus and M. S. Dresselhaus, Science, 1997, 275, 187 CrossRef CAS
- S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361 CrossRef CAS
- V. N. Popov, New J. Phys., 2004, 6, 17 CrossRef
- G. G. Samsonidze, R. Saito, N. Kobayashi, A. Gruneis, J. Jiang, A. Jorio, S. G. Chou, G. Dresselhaus and M. S. Dresselhaus, Appl. Phys. Lett., 2004, 85, 5703 CrossRef CAS
- P. C. Eklund, J. M. Holden and R. A. Jishi, Carbon, 1995, 33, 959 CrossRef CAS
- M. Damnjanovic, I. Milosevic, T. Vukovic and R. Sredanovic, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 2728 CrossRef CAS
- T. Vukovic, I. Milosevic and M. Damnjanovic, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 45418 CrossRef
- J. Kürti, G. Kresse and H. Kuzmany, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R8869 CrossRef CAS
- G. D. Mahan, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235402 CrossRef
- A. Jorio, A. G. Souza, G. Dresselhaus, M. S. Dresselhaus, R. Saito, J. H. Hafner, C. M. Lieber, F. M. Matinaga, M. S. S. Dantas and M. A. Pimenta, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245416 CrossRef
- J. Maultzsch, H. Telg, S. Reich and C. Thomsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 205438 CrossRef
- A. G. Souza, A. Jorio, J. H. Hafner, C. M. Lieber, R. Saito, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 241404 CrossRef
- A. Jorio, M. A. Pimenta, A. G. Souza, R. Saito, G. Dresselhaus and M. S. Dresselhaus, New J. Phys., 2003, 5, 139 CrossRef
- B. Gao, Y. Y. Zhang, J. Zhang, J. Kong and Z. F. Liu, J. Phys. Chem. C, 2008, 112, 8319 CrossRef CAS
- S. Piscanec, M. Lazzeri, J. Robertson, A. C. Ferrari and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 35427 CrossRef
- H. Farhat, H. Son, G. G. Samsonidze, S. Reich, M. S. Dresselhaus and J. Kong, Phys. Rev. Lett., 2007, 99, 145506 CrossRef CAS
- J. Jiang, R. Saito, A. Gruneis, S. G. Chou, G. G. Samsonidze, A. Jorio, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 205420 CrossRef
- R. Saito, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Appl. Phys. Lett., 1992, 60, 2204 CrossRef CAS
- F. M. Huang, K. T. Yue, P. H. Tan, S. L. Zhang, Z. J. Shi, X. H. Zhou and Z. N. Gu, J. Appl. Phys., 1998, 84, 4022 CrossRef CAS
- P. Corio, P. S. Santos, M. A. Pimenta and M. S. Dresselhaus, Chem. Phys. Lett., 2002, 360, 557 CrossRef CAS
- H. D. Li, K. T. Yue, Z. L. Lian, Y. Zhan, L. X. Zhou, S. L. Zhang, Z. J. Shi, Z. N. Gu, B. B. Liu, R. S. Yang, H. B. Yang, G. T. Zou, Y. Zhang and S. Iijima, Appl. Phys. Lett., 2000, 76, 2053 CrossRef CAS
- P. H. Tan, Y. M. Deng, Q. Zhao and W. C. Cheng, Appl. Phys. Lett., 1999, 74, 1818 CrossRef CAS
- Y. Y. Zhang, L. M. Xie, J. Zhang, Z. Y. Wu and Z. F. Liu, J. Phys. Chem. C, 2007, 111, 14031 CrossRef CAS
- A. Jorio, C. Fantini, M. S. S. Dantas, M. A. Pimenta, A. G. Souza, G. G. Samsonidze, V. W. Brar, G. Dresselhaus, M. S. Dresselhaus, A. K. Swan, M. S. Unlu, B. B. Goldberg and R. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 115411 CrossRef
- Z. P. Zhou, X. Y. Dou, L. J. Ci, L. Song, D. F. Liu, Y. Gao, J. X. Wang, L. F. Liu, W. Y. Zhou, S. S. Xie and D. Y. Wan, J. Phys. Chem. B, 2006, 110, 1206 CrossRef CAS
- Y. Y. Zhang, H. Son, J. Zhang, J. Kong and Z. F. Liu, J. Phys. Chem. C, 2007, 111, 1988 CrossRef CAS
- A. S. Claye, N. M. Nemes, A. Janossy and J. E. Fischer, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R4845 CrossRef CAS
- J. L. Sauvajol, N. Bendiab, E. Anglaret and P. Petit, C. R. Phys., 2003, 4, 1035 Search PubMed
- N. Bendiab, L. Spina, A. Zahab, P. Poncharal, C. Marliere, J. L. Bantignies, E. Anglaret and J. L. Sauvajol, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 153407 CrossRef
- S. B. Cronin, R. Barnett, M. Tinkham, S. G. Chou, O. Rabin, M. S. Dresselhaus, A. K. Swan, M. S. Unlu and B. B. Goldberg, Appl. Phys. Lett., 2004, 84, 2052 CrossRef CAS
- A. Claye, S. Rahman, J. E. Fischer, A. Sirenko, G. U. Sumanasekera and P. C. Eklund, Chem. Phys. Lett., 2001, 333, 16 CrossRef CAS
- E. D. Minot, Y. Yaish, V. Sazonova, J. Y. Park, M. Brink and P. L. McEuen, Phys. Rev. Lett., 2003, 90, 156401 CrossRef CAS
- M. B. Nardelli, B. I. Yakobson and J. Bernholc, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, R4277 CrossRef
- Q. Z. Zhao, M. B. Nardelli and J. Bernholc, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 144105 CrossRef
- B. Gao, L. Jiang, X. Ling, J. Zhang and Z. F. Liu, J. Phys. Chem. C, 2008, 112, 20123 CrossRef CAS
- G. Wu, J. Zhou and J. M. Dong, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 115411 CrossRef
- W. Yang, R. Z. Wang and H. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195440 CrossRef
- C. A. Cooper, R. J. Young and M. Halsall, Composites, Part A, 2001, 32, 401 CrossRef
- V. G. Hadjiev, M. N. Iliev, S. Arepalli, P. Nikolaev and B. S. Files, Appl. Phys. Lett., 2001, 78, 3193 CrossRef CAS
- C. C. Kao and R. J. Young, Compos. Sci. Technol., 2004, 64, 2291 CrossRef CAS
- M. Lucas and R. J. Young, Compos. Sci. Technol., 2004, 64, 2297 CrossRef CAS
- M. Lucas and R. J. Young, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 85405 CrossRef
- N. Lachman, C. Bartholome, P. Miaudet, M. Maugey, P. Poulin and H. D. Wagner, J. Phys. Chem. C, 2009, 113, 4751 CrossRef CAS
- T. K. Leeuw, D. A. Tsyboulski, P. N. Nikolaev, S. M. Bachilo, S. Arepalli and R. B. Weisman, Nano Lett., 2008, 8, 826 CrossRef CAS
- R. Kumar and S. B. Cronin, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 155421 CrossRef
- S. B. Cronin, A. K. Swan, M. S. Unlu, B. B. Goldberg, M. S. Dresselhaus and M. Tinkham, Phys. Rev. Lett., 2004, 93, 167401 CrossRef CAS
- S. B. Cronin, A. K. Swan, M. S. Unlu, B. B. Goldberg, M. S. Dresselhaus and M. Tinkham, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 35425 CrossRef
- S. W. Lee, G. H. Jeong and E. E. B. Campbell, Nano Lett., 2007, 7, 2590 CrossRef CAS
- H. Maki, T. Sato and K. Ishibashi, Nano Lett., 2007, 7, 890 CrossRef CAS
- M. Y. Huang, Y. Wu, B. Chandra, H. Yan, Y. Shan, T. F. Heinz and J. Hone, Phys. Rev. Lett., 2008, 100, 136803 CrossRef
- T. Fujimori, K. Urita, Y. Aoki, H. Kanoh, T. Ohba, M. Yudasaka, S. Iijima and K. Kaneko, J. Phys. Chem. C, 2008, 112, 7552 CrossRef CAS
- S. Lefrant, I. Baltog and M. Baibarac, J. Raman Spectrosc., 2005, 36, 676 CrossRef CAS
- L. M. Tong, Z. P. Li, T. Zhu, H. X. Xu and Z. F. Liu, J. Phys. Chem. C, 2008, 112, 7119 CrossRef CAS
- R. Kumar, H. Zhou and S. B. Cronin, Appl. Phys. Lett., 2007, 91, 223105 CrossRef
- V. W. Brar, G. G. Samsonidze, M. S. Dresselhaus, G. Dresselhaus, R. Saito, A. K. Swan, M. S. Unlu, B. B. Goldberg, A. G. Souza and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155418 CrossRef
- Z. T. Luo, F. Papadimitrakopoulos and S. K. Doorn, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 205438 CrossRef
- C. Fantini, A. Jorio, M. Souza, L. O. Ladeira, A. G. Souza, R. Saito, G. G. Samsonidze, G. Dresselhaus, M. S. Dresselhaus and M. A. Pimenta, Phys. Rev. Lett., 2004, 93, 87401 CrossRef CAS
- S. D. M. Brown, A. Jorio, M. S. Dresselhaus and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 73403 CrossRef
- L. Yang and J. Han, Phys. Rev. Lett., 2000, 85, 154 CrossRef CAS
- W. Fa, F. L. Hu and J. M. Dong, Phys. Lett. A, 2004, 331, 99 CrossRef CAS
- L. Yang, M. P. Anantram, J. Han and J. P. Lu, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 13874 CrossRef CAS
- R. Heyd, A. Charlier and E. McRae, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 6820 CrossRef CAS
- T. W. Tombler, C. W. Zhou, L. Alexseyev, J. Kong, H. J. Dai, L. Lei, C. S. Jayanthi, M. J. Tang and S. J. Wu, Nature, 2000, 405, 769 CrossRef CAS
- J. Cao, Q. Wang and H. J. Dai, Phys. Rev. Lett., 2003, 90, 157601 CrossRef
- R. J. Grow, Q. Wang, J. Cao, D. W. Wang and H. J. Dai, Appl. Phys. Lett., 2005, 86, 93104 CrossRef
- A. G. Souza, N. Kobayashi, J. Jiang, A. Gruneis, R. Saito, S. B. Cronin, J. Mendes, G. G. Samsonidze, G. G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. Lett., 2005, 95, 217403 CrossRef CAS
- J. C. Meyer, M. Paillet and S. Roth, Science, 2005, 309, 1539 CrossRef CAS
- T. C. Chang, Appl. Phys. Lett., 2008, 93, 61901 CrossRef
- B. Gao, X. J. Duan, J. Zhang, G. Wu, J. M. Dong and Z. F. Liu, J. Phys. Chem. C, 2008, 112, 10789 CrossRef CAS
- M. Paillet, P. Poncharal, A. Zahab, J. L. Sauvajol, J. C. Meyer and S. Roth, Phys. Rev. Lett., 2005, 94, 237401 CrossRef CAS
- A. G. Souza, A. Jorio, A. K. Swan, M. S. Unlu, B. B. Goldberg, R. Saito, J. H. Hafner, C. M. Lieber, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 85417 CrossRef
- B. Gao, X. J. Duan, J. Zhang, T. J. Wu, H. B. Son, J. Kong and Z. F. Liu, Nano Lett., 2007, 7, 750 CrossRef CAS
- R. Saito, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 2981 CrossRef CAS
- R. S. Ruoff, J. Tersoff, D. C. Lorents, S. Subramoney and B. Chan, Nature, 1993, 364, 514 CrossRef CAS
- U. D. Venkateswaran, A. M. Rao, E. Richter, M. Menon, A. Rinzler, R. E. Smalley and P. C. Eklund, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 10928 CrossRef
- J. Tang, L. C. Qin, T. Sasaki, M. Yudasaka, A. Matsushita and S. Iijima, Phys. Rev. Lett., 2000, 85, 1887 CrossRef CAS
- M. J. Peters, L. E. McNeil, J. P. Lu and D. Kahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 5939 CrossRef CAS
- W. Yang, R. Z. Wang, X. M. Song, B. Wang and H. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 45425 CrossRef
- X. P. Yang, G. Wu, J. Zhou and J. M. Dong, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235403 CrossRef
- J. Sandler, M. S. P. Shaffer, A. H. Windle, M. P. Halsall, M. A. Montes-Moran, C. A. Cooper and R. J. Young, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 35417 CrossRef
- S. A. Chesnokov, V. A. Nalimova, A. G. Rinzler, R. E. Smalley and J. E. Fischer, Phys. Rev. Lett., 1999, 82, 343 CrossRef CAS
- P. V. Teredesai, A. K. Sood, S. M. Sharma, S. Karmakar, S. K. Sikka, A. Govindaraj and C. N. R. Rao, Phys. Status Solidi B, 2001, 223, 479 CrossRef CAS
- C. Thomsen and S. Reich, Phys. Status Solidi B, 2001, 225, R18 CrossRef CAS
- H. Y. Wang, M. Zhao and S. X. Mao, Appl. Phys. Lett., 2006, 89, 211906 CrossRef
- M. H. F. Sluiter and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 224111 CrossRef
- S. Rols, I. N. Goncharenko, R. Almairac, J. L. Sauvajol and I. Mirebeau, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 153401 CrossRef
- M. G. Yao, Z. G. Wang, B. B. Liu, Y. G. Zou, S. D. Yu, W. Lin, Y. Y. Hou, S. F. Pan, M. X. Jin, B. Zou, T. Cui, G. T. Zou and B. Sundqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 205411 CrossRef
- S. Lebedkin, K. Arnold, O. Kiowski, F. Hennrich and M. M. Kappes, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 94109 CrossRef
- A. Merlen, N. Bendiab, P. Toulemonde, A. Aouizerat, A. San Miguel, J. L. Sauvajol, G. Montagnac, H. Cardon and P. Petit, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 35409 CrossRef
- M. S. Amer, M. M. El-Ashry and J. F. Maguire, J. Chem. Phys., 2004, 121, 2752 CrossRef CAS
- T. Hertel, R. E. Walkup and P. Avouris, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13870 CrossRef CAS
- S. Komura, K. Tamura and T. Kato, Eur. Phys. J. E, 2004, 13, 73 CrossRef CAS
- T. Hertel, R. Martel and P. Avouris, J. Phys. Chem. B, 1998, 102, 910 CrossRef CAS
- Y. Y. Zhang, H. Son, J. Zhang, M. S. Dresselhaus, J. Kong and Z. F. Liu, J. Phys. Chem. C, 2007, 111, 1983 CrossRef CAS
- P. E. Lammert, P. H. Zhang and V. H. Crespi, Phys. Rev. Lett., 2000, 84, 2453 CrossRef CAS
- D. Karaiskaj, C. Engtrakul, T. McDonald, M. J. Heben and A. Mascarenhas, Phys. Rev. Lett., 2006, 96, 106805 CrossRef CAS
- J. A. Elliott, J. K. W. Sandler, A. H. Windle, R. J. Young and M. S. P. Shaffer, Phys. Rev. Lett., 2004, 92, 95501 CrossRef
- M. Hasegawa and K. Nishidate, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 115401 CrossRef
- W. Yang, R. Z. Wang, Y. F. Wang, X. M. Song, B. Wang and H. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 33402 CrossRef
- A. Pantano, M. C. Boyce and D. M. Parks, Phys. Rev. Lett., 2003, 91, 145504 CrossRef
- B. I. Yakobson, C. J. Brabec and J. Bernholc, Phys. Rev. Lett., 1996, 76, 2511 CrossRef CAS
- S. Iijima, C. Brabec, A. Maiti and J. Bernholc, J. Chem. Phys., 1996, 104, 2089 CrossRef CAS
- T. C. Chang, W. L. Guo and X. M. Guo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 64101 CrossRef
- P. Poncharal, Z. L. Wang, D. Ugarte and W. A. de Heer, Science, 1999, 283, 1513 CrossRef CAS
- X. J. Duan, C. Tang, J. Zhang, W. L. Guo and Z. F. Liu, Nano Lett., 2007, 7, 143 CrossRef CAS
- M. R. Falvo, G. J. Clary, R. M. Taylor, V. Chi, F. P. Brooks, S. Washburn and R. Superfine, Nature, 1997, 389, 582 CrossRef CAS
- S. Malola, H. Hakkinen and P. Koskinen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 153409 CrossRef
- L. J. Li, R. J. Nicholas, R. S. Deacon and P. A. Shields, Phys. Rev. Lett., 2004, 93, 156104 CrossRef
- T. M. G. Mohiuddin, A. Lombardo, R. R. Nair, A. Bonetti, G. Savini, R. Jalil, N. Bonini, D. M. Basko, C. Galiotis, N. Marzari, K. S. Novoselov, A. K. Geim and A. C. Ferrari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 205433 CrossRef
- Z. H. Ni, T. Yu, Y. H. Lu, Y. Y. Wang, Y. P. Feng and Z. X. Shen, ACS Nano, 2008, 2, 2301 CrossRef CAS
- M. Y. Huang, H. G. Yan, C. Y. Chen, D. H. Song, T. F. Heinz and J. Hone, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7304 CrossRef CAS
- J. A. Robinson, C. P. Puls, N. E. Staley, J. P. Stitt, M. A. Fanton, K. V. Emtsev, T. Seyller and Y. Liu, Nano Lett., 2009, 9, 964 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2009 |