Zirconocene-mediated ligand-switched selective cleavage of active and inert carbon–carbon bonds in allylcyclopropanes†
Chao
Wang
a,
Liang
Deng
a,
Jun
Yan
a,
Hailin
Wang
a,
Qian
Luo
a,
Wen-Xiong
Zhang
a and
Zhenfeng
Xi
*ab
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, 202, Chengfu Rd, Beijing 100871, China. E-mail: zfxi@pku.edu.cn; Fax: +86 6275 9728; Tel: +86 6275 9643
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 11th June 2009
Abstract
Selective cleavage of both active and inert C–C bonds in substituted allylcyclopropanes was realized via a novel ligand-controlled approach using zirconocene(II)-1-butene or zirconocene(II)-benzyne complexes.
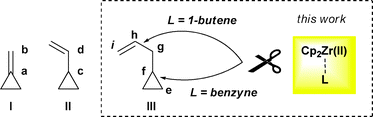
Carbon–carbon bond activation promoted by transition metal complexes has attracted much attention due to its ultimate importance either theoretically or practically.1 Compared with the tremendous difficulties in selective breaking of inert C–C single bonds, much progress in the activation of highly-strained bonds in cyclopropane derivatives,1–3 especially the “cumulative” methylene2 or “conjugated” vinyl substituted cyclopropanes3I and II (Scheme 1), has been reported. Normally, in these cases, the C–C bonds of the cyclopropane ring were successfully cleaved with the preservation of the side chain such as Ca–Cb in I or Cc–Cd in II. On the other hand, in contrast to many reports on I and II, “non-conjugated” analogues possessing a linker between the small ring and the double bond like allylcyclopropane III (Scheme 1) have been rarely studied except in some polycyclic systems,4 although interesting interactions or cooperation between the olefin moiety and the cyclopropane ring can be expected.4,5 As part of our continuous interest in the development of synthetically useful methodologies based on cleavage of C–C bonds,6,7 we would like to report herein a novel case of ligand-controlled selective cleavage of different types of carbon–carbon bonds of cyclopropane derivatives III promoted by zirconocene(II) complexes (Scheme 1, dashed rectangle). Ligands (L) on the zirconium acted as the crucial switch for the selectivity of the reaction, during which a stable Cg–Ch side chain could be cut off under mild conditions ignoring the active contorted Ce–Cf bond in the small ring. To the best of our knowledge this is the first example of breaking this type of carbon–carbon bond with such converted selectivity.
 | ||
| Scheme 1 | ||
We took 1-allyl-1-bromocyclopropane 1 as our initial substrate. When early transition metals were considered to react with 1, two reaction pathways could be expected, including (1) reaction of the metal with the C–Br bond to form a new spiro moiety after coordination or (2) direct opening the cyclopropane ring. (Path A and B, Scheme 2). Based on the above supposition, several experiments were carried out under various conditions using 7-allyl-7-bromobicyclo[4.1.0.]heptane 1a8 and a Negishi reagent, i.e. Cp2ZrnBu2, which was easily prepared in situ at −78 °C and would release Cp2Zr(II)-1-butene complex if the temperature rose.9 Results from these attempts were disappointing because only unknown mixtures were obtained after the reactions were quenched with water (Scheme 3). Other electrophiles such as acid chloride, iodine or allyl bromide were used to terminate the above reaction but no useful information or products were obtained. Since transmetalation is a useful method for applying to carbon–metal bonds, copper(I) chloride was then added to the reaction mixture followed by acid chloride to trap the newly formed carbon–metal bond.7c,10 Surprisingly, we found that butadiene derivative 2 was formed as the major isolable product (Scheme 3). Different acid chlorides, including both aliphatic and aromatic ones, give similar results. Hereby, it is clear that in this reaction, cleavage of C–C bonds of 1a occurred. Modifications such as lowering the temperature or adding a ligand did not increase the yield probably owing to the high activity of the corresponding zirconium-containing intermediate.
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
A plausible mechanism including two steps of C–C bond cleavage is outlined in Scheme 4. The reaction commenced with ligand exchange of zirconocene(II) from 1-butene to 1a. The new zirconium complex 3 or 3′ then rearranged as shown in Path A in Scheme 2 to give spiro intermediate 4.11 The highly strained structure of 4 makes it so unstable that different transformations may be triggered simultaneously, thus producing a complicated mixture if quenched at this point. Among all these conceivable transformations of 4, a possible and relatively major one was the C–C bond cleavage by way of β-carbon elimination to form intermediate 5. If copper(I) chloride was added instead of quenching, organocopper intermediate 6 would form as the result of transmetalation. A second ring-opening isomerization then occurred, presumably via either a nucleophilic pathway or a classic β-carbon elimination. The above obtained intermediate 7 would afford final product 2 by treatment with acid chloride.
 | ||
| Scheme 4 | ||
Considering that if the C–Zr bond of 5 is converted into a comparatively inert one, the resulting compound should be more stable and not undergo further skeleton breakage, we then used carbon monoxide to capture intermediate 5. After hydrolysis, the expected aldehyde9a was obtained if 1a was used (Scheme 5), suggesting the existence of 5, in which insertion of CO into the C–Zr bond formed the carbonyl zirconium intermediate 8.12 Additionally, when 1b was treated via the same procedure, similar aldehyde9b was obtained and characterized by X-ray analysis.‡ Hence, the reaction of 1a mediated sequentially by Cp2ZrnBu2 and copper(I) chloride in fact achieved an efficient tandem activation of different types of C–C bonds via ring-opening reactions of cyclopropanes.
 | ||
| Scheme 5 | ||
A sharp but intriguing difference was observed in the reaction between 1a and Cp2Zr(II) when the ligand at the zirconium center changed from 1-butene to benzyne. Cp2Zr(II)–benzyne complex can be easily prepared in situ from Cp2ZrPh2.13,14 Unlike the Cp2ZrnBu2 case, reaction between 1a and Cp2ZrPh2 gave cyclopropane derivative 10 as the only product in good isolated yields (Scheme 6). It is interesting that, in addition to the survival of the C–Br bond in the whole course, a new strained ring was generated despite the thermodynamic disadvantage. The appearance of a phenyl group in 10 indicates that benzyne has taken part in the reaction rather than being substituted like in the manner of 1-butene. Deuterated product 10D implies that zirconacycle 11 might be the final product before quenching.
 | ||
| Scheme 6 | ||
In summary, we have developed a novel zirconium(II)-mediated ligand-controlled selective cleavage of different C–C bonds in bromoallylcyclopropane, which contains multiple reactive sites. This reaction represents a new way leading to selective cleavage of both reactive and inert C–C bonds. Further investigation on the modification, substrate diversity, metal reagent variety, mechanism and patterns of C–C bond cleavage are in progress.
This work was supported by the National Natural Science Foundation of China (20632010, 20521202, 20702003) and the Major State Basic Research Development Program (2006CB806105). The Cheung Kong Scholars Programme, Qiu Shi Science & Technologies Foundation, Dow Corning Corporation and BASF are gratefully acknowledged.
Notes and references
- For representative reviews, see: (a) K. C. Bishop III, Chem. Rev., 1976, 76, 461 CrossRef; (b) M. Murakami and Y. Ito, Top. Organomet. Chem., 1999, 3, 97; (c) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (d) C.-H. Jun, C. W. Moon and D.-Y. Lee, Chem.–Eur. J., 2002, 8, 2422 CrossRef CAS; (e) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (f) T. Takahashi and K. Kanno, Top. Organomet. Chem., 2005, 8, 217 CAS; (g) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS; (h) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS.
- For representative reviews and recent results on transition-metal mediated reactions of methylenecyclopropanes (MCPs), see: (a) S. Bräse and A. de Meijere, Angew. Chem., Int. Ed. Engl., 1995, 34, 2545 CrossRef; (b) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS; (d) S. Ma, L. Lu and J. Zhang, J. Am. Chem. Soc., 2004, 126, 9645 CrossRef CAS; (e) A. I. Siriwardana, M. Kamada, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2005, 70, 5932 CrossRef CAS; (f) M. Shi, L. Liu and J. Tang, J. Am. Chem. Soc., 2006, 128, 7430 CrossRef CAS; (g) A. Fürstner and C. Aïssa, J. Am. Chem. Soc., 2006, 128, 6306 CrossRef; (h) C. Aïssa and A. Fürstner, J. Am. Chem. Soc., 2007, 129, 14836 CrossRef CAS.
- For representative reviews and recent results on transition-metal mediated reactions of vinylcyclopropanes, see: (a) H. U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS; (b) J. E. Baldwin, Chem. Rev., 2003, 103, 1197 CrossRef CAS; (c) P. A. Wender, L. O. Haustedt, J. Lim, J. A. Love, T. J. Williams and J. Yoon, J. Am. Chem. Soc., 2006, 128, 6302 CrossRef CAS , and reference therein; (d) X. Deng, P. Cai, S. Ye, X. Sun, W. Liao, K. Li, Y. Tang, Y. Wu and L. Dai, J. Am. Chem. Soc., 2006, 128, 9730 CrossRef CAS; (e) V. Bagutski, N. Moszner, F. Zeuner, U. K. Fischer and A. de Meijere, Adv. Synth. Catal., 2006, 348, 2133 CrossRef CAS; (f) Z.-X. Yu, P. H.-Y. Cheong, P. Liu, C. Y. Legault, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 2378 CrossRef CAS; (g) P. Mauleón, J. L. Krinsky and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 4513 CrossRef CAS.
- (a) W. J. Leigh and R. Srinivasan, Acc. Chem. Res., 1987, 20, 107 CrossRef CAS; (b) T. Matsuda, T. Tsuboi and M. Murakami, J. Am. Chem. Soc., 2007, 129, 12596 CrossRef CAS; (c) M. A. A. Walczak and P. Wipf, J. Am. Chem. Soc., 2008, 130, 6924 CrossRef CAS.
- (a) A. de Meijere, C. Weitemeyer and O. Schallner, Chem. Ber., 1977, 110, 1504 CrossRef CAS; (b) D. Zhou, J. Chou and H. D. Roth, J. Phys. Org. Chem., 1999, 12, 867 CrossRef CAS; (c) J. Storsberg, M.-L. Yao, N. Ocal, A. de Meijere, A. E. W. Adam and D. E. Kaufmann, Chem. Commun., 2005, 5665 RSC.
- (a) Z. Xi, K. Sato, Y. Gao, J. Lu and T. Takahashi, J. Am. Chem. Soc., 2003, 125, 9568 CrossRef CAS; (b) X. Sun, C. Y. Wang, Z. Li, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2004, 126, 7172 CrossRef CAS; (c) T. Yu, X. Sun, C. Wang, L. Deng and Z. Xi, Chem.–Eur. J., 2005, 11, 1895 CrossRef CAS; (d) C. Wang, J. Yuan, G. Li, Z. Wang, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2006, 128, 4564 CrossRef CAS.
- (a) T. Takahashi, Z. Xi, Y. Obora and N. Suzuki, J. Am. Chem. Soc., 1995, 117, 2665 CrossRef CAS; (b) T. Takahashi, Z. Xi, R. Fischer, S. Huo, C. Xi and K. Nakajima, J. Am. Chem. Soc., 1997, 119, 4561 CrossRef CAS; (c) Z. Xi, R. Fischer, R. Hara, W. Sun, Y. Obora, N. Suzuki, K. Nakajima and T. Takahashi, J. Am. Chem. Soc., 1997, 119, 12842 CrossRef CAS.
- K. Kitatani, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1975, 97, 949 CrossRef CAS.
- E. Negishi, F. E. Cederbaum and T. Takahashi, Tetrahedron Lett., 1986, 27, 2829 CrossRef CAS.
- Bruce H. Lipshutz, Acc. Chem. Res., 1997, 30, 277 CrossRef CAS , and reference therein.
- For transformations similar to this step, see: (a) T. Takahashi, M. Kotora, R. Fischer, Y. Nishihara and K. Nakajima, J. Am. Chem. Soc., 1995, 117, 11039 CrossRef CAS; (b) T. Takahashi, D. Y. Kondakov and N. Suzuki, Tetrahedron Lett., 1993, 34, 6571 CrossRef CAS; (c) N. Suzuki, D. Y. Kondakov, M. Kageyama, M. Kotora, R. Hara and T. Takahashi, Tetrahedron, 1995, 51, 4519 CrossRef CAS.
- For examples of insertion of CO into C–Zr bonds, see: (a) G. Erker, Acc. Chem. Res., 1984, 17, 103 CrossRef CAS; (b) T. Takahashi, Z. Xi, Y. Nishihara, S. Huo, K. Kasai, K. Aoyagi, V. Denisov and E. Negishi, Tetrahedron, 1997, 53, 9123 CrossRef CAS; (c) F. De Angelis, A. Sgamellotti and N. Re, Organometallics, 2000, 19, 4904 CrossRef CAS; (d) T. Takahashi, M. Ishikawa and S. Huo, J. Am. Chem. Soc., 2002, 124, 388 CrossRef CAS; (e) M. Lutz, M. Haukka, T. A. Pakkanen and L. H. Gade, Organometallics, 2002, 21, 3477 CrossRef CAS; (f) H. Shen and R. F. Jordan, Organometallics, 2003, 22, 2080 CrossRef CAS.
- (a) S. L. Buchwald, B. T. Watson and J. C. Hoffman, J. Am. Chem. Soc., 1986, 108, 7411 CrossRef CAS; (b) G. Erker and K. Kropp, J. Am. Chem. Soc., 1979, 101, 3659 CrossRef CAS.
- For representative cases of recent applications on other types of Cp2Zr(II) reagents, see: (a) A. D. Miller, J. F. Tannaci, S. A. Johnson, H. Lee, J. L. McBee and T. Don Tilley, J. Am. Chem. Soc., 2009, 131, 4917 CrossRef CAS; (b) T. Beweries, C. Fischer, S. Peitz, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg, D. Heller and U. Rosenthal, J. Am. Chem. Soc., 2009, 131, 4463 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and spectra data for all new compounds. CCDC 730462. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b908803b |
| ‡ Crystal data for 9b: C19H18O, Mw = 262.33 g mol−1, T = 293(2) K, monoclinic, space group Cc, a = 14.631(3), b = 12.773(3), c = 9.3737(19) Å, α = 90°, β = 121.93(3)°, γ = 90° V = 1486.6(5) Å3, Z = 4, ρcalcd = 1.172 Mg m3, μ = 0.071 mm−1, reflections collected: 5430, independent reflections: 1699 (Rint = 0.0644), final R indices [I > 2σI]: R1 = 0.0412, wR2 = 0.0743, R indices (all data): R1 = 0.0777, wR2 = 0.0835. CCDC 730462 (9b).† |
| This journal is © The Royal Society of Chemistry 2009 |