 Open Access Article
Open Access ArticleOligobenzamide proteomimetic inhibitors of the p53–hDM2 protein–protein interaction†
Jeffrey P.
Plante
ab,
Thomas
Burnley
b,
Barbora
Malkova
b,
Michael E.
Webb
ab,
Stuart L.
Warriner
ab,
Thomas A.
Edwards
*b and
Andrew J.
Wilson
*ab
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK LS29JT. E-mail: A.J.Wilson@leeds.ac.uk; Fax: +44 (0)113 3431409; Tel: +44 (0)113 3436565
bAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds, UK LS29JT. E-mail: T.A.Edwards@leeds.ac.uk
First published on 24th June 2009
Although many cellular processes depend on enzymatic reactions, protein–protein interactions (PPIs) mediate many regulatory pathways—the explosion of interest in their study mirrors a key role in diseased states.1 Small molecules that selectively target PPIs are therefore urgently required.2,3 What is not clear is how to achieve inhibition using a small molecule, given that it must cover ∼800–1100 Å of a protein surface and complement the discontinuous projection of hydrophobic and charged domains on a relatively shapeless surface.4 In the proteomimetic5 approach a scaffold is used to project binding functionality in an identical spatial orientation to mimic that presented by a given secondary structure (commonly an α-helix) involved in the interaction.6 In addition to Hamilton’s terphenyl,5,7 a number of proteomimetic scaffolds have been described.8–15 Similarly, foldamers,16 if suitably designed, function as inhibitors of PPIs.17–19Aromatic oligoamides are particularly attractive as foldamers because they exhibit predictable folding patterns controlled largely by the preferred conformation of the Aryl–NHCO–Aryl bond and adjacent ortho interactions;20,21 however, such foldamers have rarely been shown to act as α-helix mimicking antagonists of PPIs.10,22,23
During our studies on aromatic oligoamides,24,25 we reported a modular synthesis of rod shaped aromatic oligobenzamides that incorporate different side chains via an O-alkyl substituent.25 Elsewhere, trimers of this type have been proposed to act as α-helix mimetics23,26,27 and shown to act as inhibitors of PPIs.23 We now report that such compounds act as low μM affinity inhibitors of the p53–hDM2 protein–protein interaction. p53 is the major tumour suppressor in humans and is regulated by binding to hDM2.28 Over-expression of hDM2 prevents p53 from exerting its pro-apoptotic activity and so this PPI is a major target for cancer chemotherapy, with several examples of small molecule inhibitors reported.29–33 The interaction between p53 and hDM2 involves three key hydrophobic residues Phe19, Trp23 and Leu26 from p53 binding in a helical conformation to a hydrophobic cleft on hDM2 (Fig. 1).34
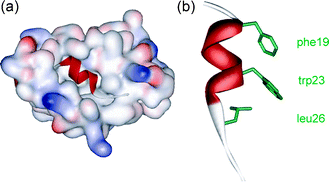 | ||
| Fig. 1 (a) Crystal structure of hDM2 in complex with a p53 peptide (PDB ID: 1YCR) and (b) the p53 peptide showing side chains key for binding. | ||
Our design was driven by two criteria: (i) the scaffold should maintain sufficient flexibility so as to optimise its conformation for maximum binding affinity and (ii) mimetic synthesis should be amenable to library generation. The scaffold should also position side chains to mimic the key residues at i, i + 4 and i + 7 of the p53 helix (Fig. 2a). Shown in Fig. 2b is the minimum energy conformation of one such trimeric benzamide as identified by a Monte Carlo search in Macromodel using the MMFFs force field (see ESI† for details). Pleasingly, the O-alkyl substituents mimic the spatial orientation of the i, i + 4 and i + 7 residues of the p53 α-helix reasonably well. Shown in Fig. 2c is mimetic 1aec superimposed upon the p53 α-helix—the RMSD = (0.1245 Å) indicates a good geometrical match between the oxygen of the scaffold and the α-carbons of the helix. For this amino-terminated tribenzamide, the O-alkyl substituents all reside on the same face—in contrast to the results obtained when a nitro group is at the N-terminus of the tribenzamide as described earlier.25 However, conformational analysis in solution reveals these compounds also adopt an extended conformation with intramolecular hydrogen-bonding fixing rotation about the Ar–NH bond and free rotation about the CO–Ar bond (see ESI† ).
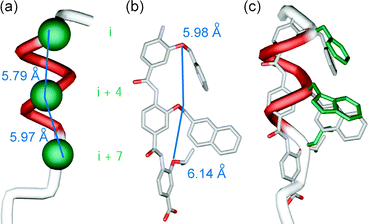 | ||
| Fig. 2 (a) p53 helix depicting key side chains (in green); (b) minimised structure of an aromatic oligoamide with R3 = Bn, R2 = Me-2-Napth and R3 = iPr (carbon in grey, oxygen in red and nitrogen in light purple); (c) p53 α-helix superimposed onto minimised aromatic oligoamide. | ||
We prepared a series of compounds (Fig. 3) and initially tested compound 1aec in a fluorescence anisotropyassay described previously.35 The assay involves displacement of a fluorescein labelled analogue of p53 : p5315-31Flu, from hDM2 with concomitant loss of anisotropy. In our hands, direct titration of p5315-31Flu with hDM2 resulted in the expected increase in fluorescence anisotropy and a dissociation constant of 74 nM was determined based on a 1 : 1 isotherm (Fig. S2, ESI† ). Upon titration with mimetic 1aec, the expected decrease in anisotropy was observed and an IC50 of 1.0 μM was determined (Fig. 4 and Table 1). Similarly, a decrease in anisotropy was observed for the positive control; unlabelled p53 peptide (p5315-31Flu) gave a IC50 of 1.2 μM. We then tested a further series of mimetics 1 and 2 containing different side chains to probe for side chain specificity. Compounds 2aa and 2ba are negative controls, which do not possess a sufficient number of side chains to mimic two turns of an α-helix. Compounds 1aaa, 1bca, 1acd, 1ace and 1adc contain a selection of side chains matched and mismatched to the sequence of p53 (naphthyl acting as a mimetic of indole). The results of our screening are summarised in Table 1 (see also Fig. S4, ESI† ). As predicted, compounds 2aa and 2ba were both inactive under the conditions of our assay . The remaining compounds were found to inhibit the interaction with low μM affinity: compounds with more and larger aromatic groups tended to be more potent although this effect was only subtle. We could not perform a titration with a nitro terminated compound because our synthetic approach results in hydrolysis of the 4-nitro benzamide terminus rather than the methyl ester.25
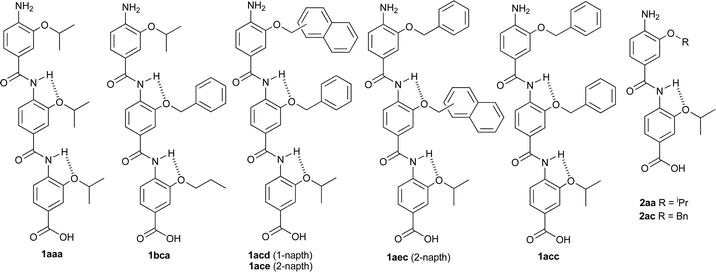 | ||
| Fig. 3 Compounds tested. | ||
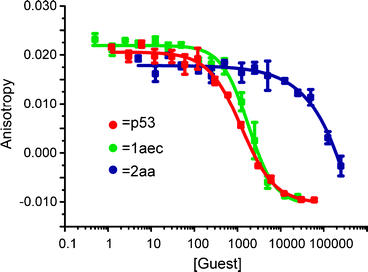 | ||
| Fig. 4 Representative FA competition titration data (40 mM sodium–potassium phosphate buffer pH 7.5, 54 nM p5315-31Flu, 42 nM hDM2). | ||
IC50 values from competition assays have been used to extract a direct measure of binding affinity (Ki) for a given target.36 Upon careful analysis of our data, we observed that in the competition experiment, anisotropy values for ligands 1 and p5315-31 decreased below the original value observed at the start of the direct titration of hDM2 into p5315-31Flu. This points to a more complex equilibrium that precludes determination of Ki. One possibility is that p5315-31Flu is present in an aggregated/dimeric form before titration with hDM2 and then becomes bound to the competitor upon displacement. Indeed, direct titration of p5315-31Flu with both p5315-31 and our compounds resulted in a decrease in anisotropy value comparable to that observed in the competition experiments where hDM2 is also present. Data fitting to a 1 : 1 association model provided Kd values in the μM range (see ESI† ) for the interaction between p5315-31Flu and p5315-31/ligand. We therefore derived a model to extract a binding constant for interaction of the competitors with hDM2 (see ESI† ). Not surprisingly, as the Kd values for p5315-31Flu binding to competitors are in the μM range and the concentration of competitor is far in excess of all other components in the experiment (μM vs. nM), no meaningful data can be extracted and it is not possible to confirm that the compounds bind to hDM2. We therefore derivatised one of the more soluble compounds (1acc) with a fluorescein label to give FITC-Gly-1acc and performed a direct binding experiment with hDM2—this afforded a dissociation constant of 760 (±140) nM and confirmed strong interaction with the target protein.
In summary, we have described the design, synthesis and binding studies of potent μM inhibitors of the p53–hDM2 protein–protein interaction and highlighted complications associated with interpretation of data from competition experiments. The generality of the syntheses reported by us and others25 represents a powerful starting point for generation of libraries for screening against a plethora of α-helix mediated PPIs. Our own investigations will focus on optimising the current compounds for binding to hDM2 and targeting other PPIs.
This work was supported by EPSRC (EP/D077842/1) and the Wellcome Trust (080709/Z/06/Z) through a PhD studentship (BM). We would also like to thank John Robinson (University of Zurich) for providing the hDM2 plasmid.
Notes and references
- W. E. Stites, Chem. Rev., 1997, 97, 1233–1250 CrossRef CAS
.
- M. R. Arkin and J. A. Wells, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS
- H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 4130–4163 CrossRef CAS
- A. J. Wilson, J. Hong and A. D. Hamilton, Org. Biomol. Chem., 2007, 5, 276–285 RSC
- B. P. Orner, J. T. Ernst and A. D. Hamilton, J. Am. Chem. Soc., 2001, 123, 5382–5383 CrossRef CAS
- I. Saraogi and A. D. Hamilton, Biochem. Soc. Trans., 2008, 36, 1414–1417 CrossRef CAS
- H. Yin, G.-I. Lee, K. A. Sedey, O. Kutzki, H. S. Park, B. P. Orner, J. T. Ernst, H.-G. Wang, S. M. Sebti and A. D. Hamilton, J. Am. Chem. Soc., 2005, 127, 10191–10196 CrossRef CAS
- H. Yin, G.-i. Lee, K. A. Sedey, J. M. Rodriguez, H.-G. Wang, S. M. Sebti and A. D. Hamilton, J. Am. Chem. Soc., 2005, 127, 5463–5468 CrossRef CAS
- I. C. Kim and A. D. Hamilton, Org. Lett., 2006, 8, 1751–1754 CrossRef CAS
- J. M. Rodriguez and A. D. Hamilton, Angew. Chem., Int. Ed., 2007, 46, 8641–8617
- J. Becerril and A. D. Hamilton, Angew. Chem., Int. Ed., 2007, 46, 4471–4473 CrossRef CAS
- S. M. Biros, L. Moisan, E. Mann, A. Carella, D. Zhai, J. C. Reid and J. Rebek, Bioorg. Med. Chem. Lett., 2007, 17, 4641–4645 CrossRef CAS
- P. Maity and B. König, Org. Lett., 2008, 10, 1473–1476 CrossRef CAS
- J. M. Rodriguez, L. Nevola, N. T. Ross, G.-i. Lee and A. D. Hamilton, ChemBioChem, 2009, 10, 829–833 CrossRef CAS
- C. G. Cummings, N. T. Ross, W. P. Katt and A. D. Hamilton, Org. Lett., 2009, 11, 25–28 CrossRef CAS
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 178–190
- O. M. Stephens, S. Kim, B. D. Welch, M. E. Hodsdon, M. S. Kay and A. Schepartz, J. Am. Chem. Soc., 2005, 127, 13126–13127 CrossRef CAS
- J. D. Sadowsky, W. D. Fairlie, E. B. Hadley, H.-S. Lee, N. Umezawa, Z. Nikolovska-Coleska, S. Wang, D. C. S. Huang, Y. Tomita and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 139–154 CrossRef CAS
- E. A. Harker, D. S. Daniels, D. A. Guarracino and A. Schepartz, Bioorg. Med. Chem., 2009, 17, 2038–2046 CrossRef CAS
- I. Huc, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS
- Z.-T. Li, J.-L. Hou and H.-P. Yi, Chem. Asian J., 2006, 1, 766–778 CrossRef CAS
- J. T. Ernst, J. Becerril, H. S. Park, H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2003, 42, 535–550 CrossRef CAS
- During preparation of this manuscript another group elegantly described libraries of aromatic oligoamides that act as inhibitors of the p53–HDM2 interaction using an ELISA assay, see: A. Shaginian, L. R. Whitby, S. Hong, I. Hwang, B. Farooqi, M. Searcey, J. Chen, P. K. Vogt and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 4904–4916 Search PubMed
- F. Campbell, J. Plante, C. Carruthers, M. J. Hardie, T. J. Prior and A. J. Wilson, Chem. Commun., 2007, 2240–2242 RSC
- J. Plante, F. Campbell, B. Malkova, C. Kilner, S. L. Warriner and A. J. Wilson, Org. Biomol. Chem., 2008, 1, 138–146 Search PubMed
- J.-M. Ahn and S.-Y. Han, Tetrahedron Lett., 2007, 48, 3543–3547 CAS
- I. Saraogi, C. D. Incarvito and A. D. Hamilton, Angew. Chem., Int. Ed., 2008, 47, 9691–9694 CrossRef CAS
- L. Romer, C. Klein, A. Dehner, H. Kessler and J. Buchner, Angew. Chem., Int. Ed., 2006, 45, 6440–6460 CrossRef CAS
- S. Patel and M. R. Player, Expert Opin. Invest. Drugs, 2008, 17, 1865–1882 Search PubMed
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS
- B. L. Grasberger, T. Lu, C. Schubert, D. J. Parks, T. E. Carver, H. K. Koblish, M. D. Cummings, L. V. LaFrance, K. L. Milkiewicz, R. R. Calvo, D. Maguire, J. Lattanze, C. F. Franks, S. Zhao, K. Ramachandren, G. R. Bylebyl, M. Zhang, C. L. Manthey, E. C. Petrella, M. W. Pantoliano, I. C. Deckman, J. C. Spurlino, A. C. Maroney, B. E. Tomczuk, C. J. Molloy and R. F. Bone, J. Med. Chem., 2005, 48, 909–912 CrossRef CAS
- H. Yin, G.-I. Lee, H. S. Park, G. A. Payne, J. M. Rodriguez, S. M. Sebti and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 2704–2707 CrossRef CAS
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS
- J. A. Kritzer, J. D. Lear, M. E. Hodsdon and A. Schepartz, J. Am. Chem. Soc., 2004, 126, 9468–9469 CrossRef CAS
- Z. Nikolovska-Coleska, R. Wang, X. Fang, H. Pan, Y. Tomita, P. Li, P. P. Roller, K. Krajewski, N. G. Saito, J. A. Stuckey and S. Wang, Anal. Biochem., 2004, 332, 261–273 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, protein expression and FP assays. See DOI: 10.1039/b908207g |
| This journal is © The Royal Society of Chemistry 2009 |