Highly oxidized diiron complexes: generation, spectroscopy, and stabilities†
Julia Bernhardette Hildegard
Strautmann
a,
Carl-Georg
Freiherr von Richthofen
a,
Serena
DeBeer George
b,
Eberhard
Bothe
c,
Eckhard
Bill
c and
Thorsten
Glaser
*a
aFakultät für Chemie, Universität Bielefeld, Universitätsstrasse 25, 33615, Bielefeld, Germany. E-mail: thorsten.glaser@uni-bielefeld.de; Fax: +49 (0)521 106 6003; Tel: +49 (0)521 106 6105
bStanford Synchrotron Radiation Lightsource, SLAC, Stanford University, Stanford, CA 94309, USA
cMax-Planck-Institut für Bioanorganische Chemie, Stiftstrasse 34-36, 45470, Mülheim, Germany
First published on 7th April 2009
Abstract
Oxidation of the diferric complex [LFeIIIOFeIIIL] to the monoradical complex [LFeIIIOFeIIIL˙]+ and the diradical complex [L˙FeIIIOFeIIIL˙]2+ is followed by a decay into monomeric complexes including a highly reactive putative [LFeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] complex.
O] complex.
Methane monooxygenase (MMO) and ribonucleotide reductase (RNR) are metalloenzymes that activate dioxygen at nonheme diiron active sites.1 The active intermediates accumulate oxidation equivalents higher than FeIIIFeIII. While the active species in MMO, intermediate Q, is proposed to consist of a high-valent FeIV(μ-O)2FeIV core,2 intermediate X of RNR was first designated a FeIIIFeIII radical species by Mössbauer and EPR spectra (isotropic EPR signal at g = 2.00).3 But now, the formulation as a FeIIIFeIV species is commonly accepted.4
There have been numerous efforts to synthesize model complexes for Q and X.5,6 Our strategy for stabilizing highly oxidized diiron units involves increasing the electron density at the iron atoms by using strongly electron-donating ligands7 such as phenolates and tert-amines. Hence, we synthesized the binuclear ferric complex [LFe(μ2-O)FeL] (1, Fig. 1A)8 which exhibits two oxidations at redox potentials that are uncommonly low for an oxo-bridged binuclear ferric complex (E1 = 0.27 V, E2 = 0.44 V vs. Fc+/Fc). Herein we present the generation and spectroscopic characterization of the one- and the two-electron oxidized species 1+ and 12+, respectively.‡
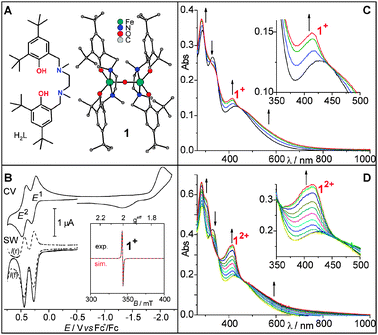 | ||
| Fig. 1 (A) The ligand H2L and the molecular structure of 1;8 (B) electrochemistry of 1 in DCM at −40 °C, scan rate: 100 mV s−1, frequency: 50 Hz. Inset: 10 K X-band EPR spectrum of electrochemically generated 1+; (C) UV/Vis spectra recorded during scanning the oxidative wave of the first oxidation E1 in a CV of 1 at 3 mV s−1 in DCM in an OTTLE cell at −20 °C. This experiment is on the timescale of 2 min. (D) UV/Vis spectra recorded during chronoamperometry of 1 at 0.8 V vs. Fc+/Fc in DCM in an OTTLE cell at −20 °C. This experiment is on the timescale of 80 s. | ||
Preliminary electrochemical studies implied that E1 and E2 are quasi-reversible,8 but extensive CV and SW voltammetry analyses prove E1 and E2 to be reversible (Fig. 1B). The formation of 1+ was followed by UV/Vis spectroscopy during a CV of 1 in an OTTLE cell at −20 °C (Fig. 1C). A new absorption band at 414 nm arises, consistent with the formation of the phenoxyl radical complex [LFeIIIOFeIIIL˙]+.9,10 On the timescale of this experiment (∼2 min), the spectra exhibit isosbestic points. Analogously, the formation of 12+ was followed during chronoamperometry of 1 in DCM at −20 °C in an OTTLE cell (Fig. 1D). The UV/Vis spectra recorded during the two-electron oxidation show a continuous increase in the extinction coefficient at 418 nm. Although the mono- and the dication are formed one after another, the spectra exhibit relatively well-defined isosbestic points implying similar electronic processes for the generation of 1+ and 12+. Re-reduction of 12+ almost restores the spectrum of 1 (Fig. S1, ESI† ), proving the reversibility of this two-electron oxidation on this timescale. A similar increase in the absorption at 419 nm was observed during the chronoamperometry of monomeric [LFeCl] (2), which reveals the presence of the ferric diphenoxyl radical species [L˙˙FeCl]2+ (22+).10 Thus, 12+ is best described as the diferric diphenoxyl radical complex [L˙FeIIIOFeIIIL˙]2+.
Bulk potentiostatic coulometry of 1 at −40 °C followed by UV/Vis spectroscopy demonstrate a reduced stability of 1+ on the longer timescale of this experiment. The UV/Vis spectra exhibit isosbestic behavior for 10 min and non-isosbestic behavior thereafter (Fig. S2, ESI† ), indicating a slow decay of 1+. The low-temperature EPR spectrum of 1+ taken during the first 10 min exhibits a sharp isotropic signal at g = 2.0050 with 86% spin-concentration (inset Fig. 1B). Despite the similarity to the EPR spectrum of intermediate X, this isotropic signal corroborates the ligand-centered oxidation of 1+.
In order to study the electronic structures and stabilities of 1+ and 12+ further, 1 was also oxidized chemically by one- and two-electrons using the aminyl radical [(C6H4Br)3N](SbCl6) (AR) in DCM. These reactions have been studied by temperature-dependent UV/Vis spectroscopy under Schlenk conditions. Spectra obtained a few seconds after addition of one equivalent of the oxidant solution closely resemble those obtained for electrochemically generated 1+ in Fig. 1C. The extinction coefficient for the radical band at 414 nm decreases with time while a new maximum at ∼580 nm forms (Fig. 2A). This latter maximum is a characteristic feature of mononuclear ferric complexes of the ligand L2−, indicating a cleavage of a Fe–oxo bond.10 The absence of a band around 414 nm indicates that there are no phenoxyl radicals produced in this decay. Longer measurements, especially at slightly higher temperature, reveal a second follow-up reaction (Fig. S3, ESI† ). The kinetic analysis of the first follow-up reaction at −40 °C reveals first-order dependence with k = (4.2 ± 0.1) × 10−4 s−1 (Fig. S4, ESI† ) corresponding to a half-life at −40 °C of ∼27 min. Temperature-dependent measurements (between −30 and −60 °C) allowed the determination of the activation parameters ΔH‡ = (51 ± 1) kJ mol−1 and ΔS‡ = −(90 ± 6) J (mol K)−1 (Fig. S5, ESI† ). The negative value for ΔS‡ is indicative of a more complex reaction scheme than a simple bond cleavage.
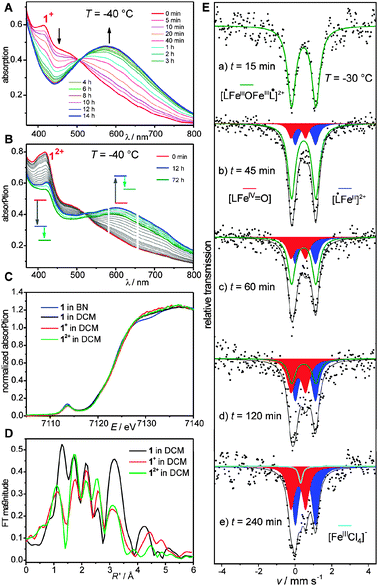 | ||
| Fig. 2 (A) UV/Vis spectra of the decay of 1+, recorded at −40 °C. (B) UV/Vis spectra of the decay of 12+, recorded at −40 °C. (C) Fe K-edge XAS of 1, 1+, and 12+. (D) Fourier transforms of the Fe K-edge EXAFS of 1, 1+, and 12+. (E) Mössbauer spectra recorded at 80 K after the oxidation of 1 to 12+ at −30 °C in DCM using two equivalents of AR recorded on samples quenched at individual times. | ||
Analogously, addition of two equivalents of the oxidant leads to the direct formation of 12+ (red spectrum in Fig. 2B). The strong radical band at 418 nm decreases slowly over ∼12 h at −40 °C while a new band at ∼600 nm with an isosbestic point around 521 nm forms (blue spectrum in Fig. 2B). This decay process of 12+ thus produces two mononuclear iron species of the ligand L2−. The strong decrease of the radical band at 418 nm strongly suggests that not both mononuclear complexes possess phenoxyl radicals as in 12+. However, the feature at ∼418 nm is still strong and therefore implies the presence of a phenoxyl radical in one of the two mononuclear decay products. With ongoing time, the band at 600 nm decreases with a concomitant decrease of the radical band at 418 nm. This is indicative of further follow-up reactions. We refrain from a complete kinetic analysis due to its complexity, but we have examined a first-order analysis of the first reaction step. This reveals k = (3.8 ± 0.5) × 10−5 s−1 at −40 °C and the temperature-dependence of k provides ΔH‡ = 68 (±4) kJ mol−1 and ΔS‡ = −(36 ± 10) J (mol K)−1 as estimates. Interestingly, the more oxidized species 12+ with a higher reactivity for electron transfer exhibits a higher stability for Fe–O bond breaking (τ1/2≈ 6 h).
In order to obtain independent spectroscopic signatures for the electronic structures of 1, 1+, and 12+, we used Mössbauer (on 57Fe enriched samples) and X-ray absorption spectroscopy (XAS). Both methods reveal slight differences of 1 as a solid and in DCM solution indicative of some structural rearrangements in solution as observed for the mononuclear complexes.10 It should be mentioned that even for the solid state data, the separation of 3 Fe–O components (2 Fe–OPh at 1.92 Å and 1 Fe–Ooxo at 1.81 Å) is beyond the resolution of the EXAFS data (Table S2, ESI† ). Interestingly, the EXAFS fitting requires a decrease in the Fe–N distances from 2.25 Å in the solid to 2.14 Å in solution. The pre-edge and edge features of all four spectra are essentially identical (Fig. 2C), indicating that 1, 1+, and 12+ are ferric complexes. However, the EXAFS spectra exhibit significant differences (Fig. S6, ESI† ). The Fourier transforms show that the Fe–oxo distances of 1+ and 12+ are decreased as compared to 1 (Fig. 2D, Table S2 (ESI† )), indicating a shortening of the Fe–oxo bonds by oxidation.
These findings are corroborated by Mössbauer spectra recorded from specially prepared samples in frozen DCM (Fig. 2E).‡ The sample of 12+, taken 15 min after addition of two equivalents AR at −30 °C, shows a slightly larger isomer shift (0.47 mm s−1) than in 1 (0.44 mm s−1), indicating less covalent bonding. Samples taken after longer reaction times exhibit an intensity increase of the low velocity line along with some intensity arising between the two former absorption lines. The signal-to-noise ratio in the spectra‡ does not allow meaningful simulations of individual spectra. However, taking into account the information from the temperature-dependent UV/Vis spectra, a self-consistent simulation of all spectra could be performed with two evolving new subspectra with δ = 0.17, |ΔEQ| = 0.80 mm s−1 (red spectrum) and δ = 0.56, |ΔEQ| = 1.10 mm s−1 (blue spectrum). The isomer shift of the red component is consistent with the formulation as a FeIV species, whereas the blue component is ascribed to a ferric species of lower covalency.
All experimental results are consistent with the following reaction schemes:
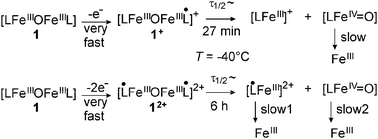
One- and two-electron oxidation of 1 results in the fast generation of the monoradical complex 1+ and the diradical complex 12+, respectively. Coordinated phenoxyl radicals donate less charge to the FeIII ions than a coordinating phenolate ligand. This reduced electron density is compensated by a stronger charge donation from the bridging oxo ligand.11 This results in an unsymmetrical bonding situation for the two FeIII–oxo bonds in 1+, while in the diradical complex 12+, the oxo ligand has to donate more charge to both FeIII ions in comparison to the precursor 1. The unsymmetrical Fe–O–Fe bond situation in 1+ already phases into the unsymmetrical decay of the dimer into two mononuclear complexes. The former bridging oxo ligand remains on the iron ion with the stronger Fe–oxo bond in 1+, i.e. that with the coordinated phenoxyl ligand. This would result in the hypothetical [L˙FeIIIO] species, but the absence of a phenoxyl radical band in the UV/Vis spectrum and the Mössbauer data indicates that it relaxes by intramolecular electron transfer to the ferryl species [LFeIVO]. In contrast, both FeIII–oxo bonds in 12+ are strengthened as evidenced by the higher activation energy for FeIII–oxo bond cleavage (ΔH‡ = (68 ± 4) kJ mol−1 in 12+vs.ΔH‡ = (51 ± 1) kJ mol−1 in 1+). Cleavage of one FeIII–oxo bond results formally in the two species [L˙FeIIIO] and [L˙FeIII]2+. While the former relaxes as in the case of 1+ to the ferryl species [LFeIVO], the radical ferric species [L˙FeIII]2+ exhibits no electronic structure relaxation consistent with the persistence of a radical band in the UV/Vis spectrum10 due to the absence of a stabilizing oxo ligand. The higher isomer shift of 0.56 mm s−1 for this species indicates less covalent bonding of this ferric complex consistent with the presence of a coordinated phenoxyl radical and the absence of an oxo ligand. The oxidized mononuclear species [L˙FeIII]2+ and [LFeIVO] undergo further decay reactions as evidenced by the time-dependence of the UV/Vis spectra. The high reactivity of the [LFeIVO] despite the stabilization by the strongly electron-donating ligand set and the weak-field nature of this ligand set in comparison to ligand environments used for more stable FeIVO units12 might indicate the presence of a high-spin FeIV (S = 2) ion.13
In summary, we have generated and characterized two highly reactive dinuclear iron complexes, which accumulate oxidation equivalents as in X and Q. Further studies on the final products and on the reactivity of the highly oxidized intermediates with properly chosen substrates are currently being pursued in our laboratories. In addition, the identification of the two main drawbacks of the current ligand, i.e. the ligand-centered oxidation and dissociation into mononuclear species, is being used for optimization of the ligand design.
J.B.H.S. is thankful for a doctoral fellowship of the Fonds der Chemischen Industrie. This publication was made possible by the Fonds der Chemischen Industrie, the DFG and by Grant Number 5P41RR001209 from the NCRR, a component of the NIH. SSRL operations are funded by DOE, BES. The SMB program is supported by the NIH, NCRR and by DOE, BER.
Notes and references
- M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müller and S. L. Lippard, Angew. Chem., Int. Ed., 2001, 40, 2782 CrossRef CAS; E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235 CrossRef CAS.
- L. Shu, J. C. Nesheim, K. Kauffmann, E. Münck, J. D. Lipscomb and L. Que, Jr, Science, 1997, 275, 515 CrossRef CAS.
- N. Ravi, J. M. Bollinger, Jr, B. H. Huynh, D. E. Edmondson and J. Stubbe, J. Am. Chem. Soc., 1994, 116, 8007 CrossRef CAS.
- B. E. Sturgeon, D. Burdi, S. Chen, B.-H. Huynh, D. E. Edmondson, J. Stubbe and B. M. Hoffman, J. Am. Chem. Soc., 1996, 118, 7551 CrossRef CAS.
- H.-F. Hsu, Y. Dong, L. Shu, V. G. J. Young and L. Que, Jr, J. Am. Chem. Soc., 1999, 121, 5230 CrossRef CAS; E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987 CrossRef CAS.
- A. Hazell, K. B. Jensen, C. J. McKenzie and H. Toftlund, Inorg. Chem., 1994, 33, 3127 CrossRef CAS; A. Ghosh, F. Tiago de Oliveira, T. Yano, T. Nishioka, E. S. Beach, I. Kinoshita, E. Münck, A. D. Ryabov, C. P. Horwitz and T. J. Collins, J. Am. Chem. Soc., 2005, 127, 2505 CrossRef CAS; G. Xue, D. Wang, R. De Hont, A. T. Tiedler, X. Shan, E. Münck and L. Que, Jr, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20713 CrossRef CAS.
- A. Chanda, D.-L. Popescu, F. Tiago de Oliveira, E. Bominaar, A. D. Ryabov, E. Münck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606 CrossRef.
- T. Glaser, R. H. Pawelke and M. Heidemeier, Z. Anorg. Allg. Chem., 2003, 629, 2274 CrossRef CAS.
- B. Adam, E. Bill, E. Bothe, B. Goerdt, G. Haselhorst, K. Hildenbrand, A. Sokolowski, S. Steenken, T. Weyhermüller and K. Wieghardt, Chem.–Eur. J., 1997, 3, 308 CrossRef CAS; R. C. Pratt and T. D. P. Stack, Inorg. Chem., 2005, 44, 2367 CrossRef CAS.
- J. B. H. Strautmann, S. DeBeer George, E. Bothe, E. Bill, T. Weyhermüller, A. Stammler, H. Bögge and T. Glaser, Inorg. Chem., 2008, 47, 6804 CrossRef CAS.
- T. Glaser, K. Rose, S. E. Shadle, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 442 CrossRef CAS; T. Glaser, B. Hedman, K. O. Hodgson and E. I. Solomon, Acc. Chem. Res., 2000, 33, 859 CrossRef CAS.
- L. Que, Jr, Acc. Chem. Res., 2007, 40, 493 CrossRef CAS.
- A. E. Anastasi, P. Comba, J. McGrady, A. Linke and H. Rohwer, Inorg. Chem., 2007, 46, 6420 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, details of the physical measurements, UV/Vis spectra recorded during electrochemical experiments, course of the absorption at 580 nm during the decay of 1+, Eyring plot, rate constants, XAS-spectra, and results of the fit of the EXAFS. See DOI: 10.1039/b903500a |
| ‡ The parent complex 1 is only soluble in DCM. Dissolution of 1 in other solvents leads to a color change to blue, indicative of the formation of mononuclear complexes. Thus, all characterizations were performed in DCM although DCM is a problematic solvent for XAS and Mössbauer spectroscopy due to strong absorptions of X-ray and γ radiation. DCM is usually considered to be opaque for Mössbbauer radiation (see ESI† for absorber optimization). |
| This journal is © The Royal Society of Chemistry 2009 |