Triply-responsive boronic acidblock copolymers : solution self-assembly induced by changes in temperature, pH, or sugar concentration†
Debashish
Roy
,
Jennifer N.
Cambre
and
Brent S.
Sumerlin
*
Department of Chemistry, Southern Methodist University, 3215 Daniel Avenue, Dallas, TX, 75275-0314, USA. E-mail: bsumerlin@smu.edu; Fax: +1 214 768 4089; Tel: +1 214 768 8802
First published on 26th February 2009
Abstract
Boronic acid-containing block copolymers capable of solution self-assembly into micelles and reverse micelles in response to changes in temperature, pH, and sugar concentration were prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization.
Stimuli-responsive or “smart” block copolymers offer promise in drug delivery and medicine because of their ability to undergo self-assembly or dissolution in response to variations in the local environment. Upon the application of an appropriate stimulus, the responsive nature of the block copolymer often leads to selective segmental dehydration and solution self-assembly into micelles, vesicles, etc.1 The most common examples of this type are block copolymers 2 in which one block is hydrophilic and the other stimuli-responsive, giving rise to a copolymer that can be tuned such that it is either double-hydrophilic or amphiphilic, depending on the presence or absence of the stimulus.
Alternatively, another class of adaptive diblock copolymers can be envisioned in which each of the two blocks respond to a different stimulus. Such block copolymers self-assemble into either micelles or reverse micelles, depending on which of the blocks is rendered hydrophobic. Armes and co-workers coined the term “schizophrenic” to describe these systems and pioneered much of the original research regarding their doubly-responsive behavior.3 Various double-hydrophilic or zwitterionic block copolymers based on thermoresponsive4 and pH-responsive4,5 blocks have been used to study doubly-responsive micellization behavior in aqueous media.
Boronic acid-containing macromolecules are a unique class of stimuli-responsive polymers with potential applications as self-healing materials, therapeutic agents, self-regulated drug-delivery systems, and sensors for sugars and glycoproteins.6–8Boronic acid-containing (co)polymers have primarily been synthesized via conventional radical polymerization, which typically results in ill-defined (co)polymers or crosslinked gels. On the other hand, controlled radical polymerization (CRP)9 provides access to well-defined boronic acid (co)polymers, either by atom transfer radical polymerization (ATRP)7,10 or reversible addition–fragmentation chain transfer (RAFT) polymerization.11–13 Jäkle et al. reported the synthesis of boron-containing (co)polymers via ATRP of organoboron monomers or silylated precursors that were borylated through postpolymerization modification.10 We demonstrated the polymerization of a styrenic boronic estervia RAFT polymerization and subsequent postpolymerization deprotection to yield well-defined boronic acid homo- and block copolymers .11 Subsequently, we reported a more straight-forward route to block copolymers via direct RAFT polymerization of unprotected boronic acid acrylamido monomers.12 Because of the well-known ability of boronic acids to undergo a reduction in pKa upon esterification with diols (e.g., glucose), these block copolymers proved to be sugar-responsive, with micellar aggregates undergoing dissociation in aqueous media when exposed to glucose.
Herein, we report the RAFT block copolymerization of a boronic acid acrylamido monomer with N-isopropylacrylamide (NIPAM) and describe the preliminary solution characterization of the resulting block copolymers . RAFT polymerization was selected due to its tolerance of a wide range of functional groups and its proven success for the synthesis of well-defined, water-soluble acrylamido polymers.14 Combining the thermoresponsive nature of PNIPAM with the pH- and diol-responsive solubility of polymeric boronic acids leads to triply-responsive block copolymers capable of forming micelles or reverse micelles depending on the environmental conditions.
3-Acrylamidophenylboronic acid (APBA, 1) was polymerized by RAFT at 70 °C with 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid15 (2) as the chain transfer agent (CTA) and 2,2′-azobis(isobutyronitrile) (AIBN) as the initiator with [APBA] : [2] : [AIBN] = 100 : 1 : 0.1 (Scheme 1). The polymerization was conducted in 95% DMF–5% water to prevent crosslinking via boroxine anhydride formation. The RAFT polymerization of APBA was well controlled, as demonstrated by the pseudo-first-order kinetics and linear evolution of Mn with conversion shown in Fig. 1. Importantly, while the boronic acid monomer could be polymerized directly by RAFT, molecular weight analysis by size exclusion chromatography (SEC) via the triple detection method required conversion of the boronic acid groups to pinacolato boronic esters by reacting with pinacol .12
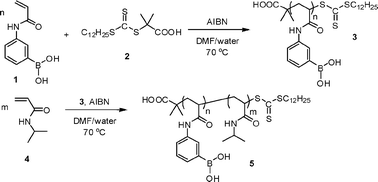 | ||
| Scheme 1 Synthesis of poly(3-acrylamidophenylboronic acid) (PAPBA) and PAPBA-b-poly(N-isopropylacrylamide) (PAPBA-b-PNIPAM) via RAFT. | ||
![(A) Pseudo first-order kinetic plot with selected molar ratios of monomer (APBA)–chain transfer agent (CTA)–initiator (AIBN) and (B) Mnversus monomer conversion ([APBA]–[CTA]–[AIBN] = 100 : 1 : 0.1) for RAFT homopolymerizations of 3-acrylamidophenylboronic acid (APBA, 1) at 70 °C in 95% DMF–5% water.](/image/article/2009/CC/b900374f/b900374f-f1.gif) | ||
| Fig. 1 (A) Pseudo first-order kinetic plot with selected molar ratios of monomer (APBA)–chain transfer agent (CTA)–initiator (AIBN) and (B) Mnversus monomer conversion ([APBA]–[CTA]–[AIBN] = 100 : 1 : 0.1) for RAFT homopolymerizations of 3-acrylamidophenylboronic acid (APBA, 1) at 70 °C in 95% DMF–5% water. | ||
The resulting APBAhomopolymer (3) was used as a macroCTA for block copolymerization with N-isopropylacrylamide (NIPAM, 4) ([NIPAM] : [3] : [AIBN] = 100 : 1 : 0.2; 70 °C; 95% DMF–5% water. While slight low molecular weight tailing was observed in the SEC, likely due to either the presence of a small amount of dead chain ends in the macroCTA or inefficient pinacol protection prior to analysis, copolymerization with NIPAM resulted in an increase in Mn from 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1 for the PAPBA macroCTA to 28100 g mol−1 (Mn,theory = 26000 g mol−1) and a low polydispersity index of 1.15 for PAPBA-b-poly(N-isopropylacrylamide) (PAPBA81-b-PNIPAM109). 1H NMR spectroscopy of the block copolymer confirmed the presence of the peaks associated with each segment. Block copolymer compositions calculated with data from both 1H NMR and SEC were in reasonable agreement (ESI† ).
800 g mol−1 for the PAPBA macroCTA to 28100 g mol−1 (Mn,theory = 26000 g mol−1) and a low polydispersity index of 1.15 for PAPBA-b-poly(N-isopropylacrylamide) (PAPBA81-b-PNIPAM109). 1H NMR spectroscopy of the block copolymer confirmed the presence of the peaks associated with each segment. Block copolymer compositions calculated with data from both 1H NMR and SEC were in reasonable agreement (ESI† ).
The PAPBA-b-PNIPAM block copolymers were expected to be triply-responsive and to self-assemble in response to changes in temperature, pH, and diol concentration (Fig. 2A). PNIPAM was expected to impart thermoresponsive behavior. It is well known that PNIPAM exhibits a sharp phase transition around 32 °C. Below this lower critical solution temperature (LCST), the polymer exists as molecularly dissolved unimers, but heating leads to dehydration and interchain aggregation. Boronic acid-containing (co)polymers are uniquely stimuli-responsive because their water solubility is dictated by pH and solution diol concentration, the latter of which has led to boronic acids being employed as saccharidereceptors.8 The PAPBA block should be hydrophilic at high pH and hydrophobic under acidic conditions.
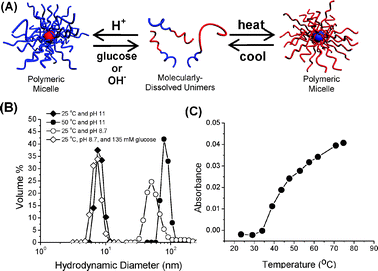 | ||
| Fig. 2 (A) Block copolymer self-assembly–dissociation in response to changes in pH or temperature. (B) Hydrodynamic size distributions of poly(3-acrylamidophenylboronic acid)-b-poly(N-isopropylacrylamide) (PAPBA-b-PNIPAM) at 25 °C and pH 11, at 50 °C and pH 11, at 25 °C and pH 8.7, and at 25 °C and pH 8.7 in the presence of 135 mM glucose. (C) Turbidimetry results at λ = 600 nm for the determination of lower critical solution temperature for PAPBA-b-PNIPAM. | ||
Above the pKa of the boronic acid, PAPBA exists as a boronate polyanion; acidification to pH < pKa should result in the boronate moieties being converted to neutral/hydrophobic boronic acid groups and subsequent chain dehydration. The diol-responsive nature of PAPBA arises from its ability to reversibly form cyclicboronate esters with 1,2- and 1,3-diols, which effectively lowers the pKa of the boronic acid. The net result of this process leads to neutral/hydrophobic boronic acidpolymers at pH < pKa and anionic/hydrophilic boronate esterpolymers in the presence of a sufficient concentration of diol. The combination of these three stimuli should lead to a copolymer with a PNIPAM block that is sensitive to changes in temperature and a PAPBA block that is sensitive to changes in either pH or diol concentration. Therefore, it was anticipated that heating a solution of molecularly dissolved unimers above the LCST of the PNIPAM block would lead to micelles with PAPBA coronas and PNIPAM cores. On the other hand, lowering the pH of a solution of PAPBA–b-PNIPAM molecularly dissolved unimers below the pKa of the boronic acid block should lead to dehydration of the organoboron segment and subsequent self-assembly to form micelles with a hydrophilic PNIPAM corona and a hydrophobic PAPBA core. These aggregates should be capable of dissociation to unimers when exposed to either an increase in pH or the addition of low molecular weight diol.
Solutions of PAPBA–b-PNIPAM at 25 °C and pH ≈ 11 yielded unimers with an approximate hydrodynamic diameter (Dh) of 8 nm, as determined by DLS, which is consistent with both blocks being water soluble. Upon lowering the solution pH to 8.7, below the pKa of the PABPA units (pKa ≈ 9), aggregates of approximately 55 nm were observed (Fig. 2B). Returning the solution to pH ≈ 11 by adding NaOH resulted in dissociation of the aggregates back to unimers. Similarly, the diol-responsive nature of the block copolymers was investigated by DLS. Addition of glucose to a solution of PAPBA-b-PNIPAM at pH 8.7 and 25 °C also caused aggregate dissociation to unimers of approximately 7 nm (Fig. 2B).
The temperature-responsive nature of the PAPBA-b-PNIPAM block copolymers was investigated by turbidimetry, DLS, and NMR spectroscopy. Because the LCST of PNIPAM can be affected by the nature of adjacent comonomers or block segments, it was necessary to determine the critical aggregation temperature of the PNIPAM block in the PAPBA81-b-PNIPAM109block copolymer . Turbidimetry measurements revealed a rather broad transition around 42 °C (Fig. 2C). The thermoresponsive nature resulted in aggregate formation when a solution of the block copolymer was heated above this temperature at pH ≈ 11, consistent with the formation of polymeric micelles with a PNIPAM core and a PAPBA corona. Aggregates with a hydrodynamic diameter of 78 nm were observed by DLS (Fig. 2B). The slightly larger aggregate size in this case, as compared to the reverse micelle structure, could be the result of the highly hydrated and extended nature of the anionic coronal chains.
Variable-temperature NMR spectroscopy was used to monitor the dehydration of each responsive block under conditions of changing pH and temperature (Fig. 3),16 though analysis of the diol-response was complicated by the presence of large peaks from glucose. PAPBA81-b-PNIPAM109 was dissolved in D2O–NaOD at pH ≈ 11, and 1H NMR spectroscopy was conducted below and above the LCST of the PNIPAM block (Fig. 3A). At 25 °C, the peaks for each block were observed. When the temperature was raised to 60 °C, the CH(CH3)2 and –CH(CH3)2 peaks of the PNIPAM block at δ ≈ 3.9 and 1.1 ppm, respectively, were significantly attenuated (Fig. 3B). When the pH was lowered by the addition of DCl and the 1H NMR spectrum was collected at 25 °C, the PNIPAM peaks were fully restored, as determined by integration of the signals before and after heating. Under these conditions, the peaks associated with PAPBA in the aromatic region (δ ≈ 6.5–7.5) and the peaks related to the PAPBA backbone (at e.g., δ ≈ 2.4 ppm) were significantly diminished, indicative of dehydration of the boronic acid-containing block (Fig. 3C).
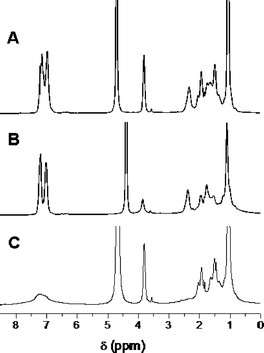 | ||
| Fig. 3 1H NMR spectra of PAPBA81-b-PNIPAM109 at (A) 25 °C and pH 11, (B) at 60 °C and pH 11, and (C) at 25 °C and pH 8. | ||
By exploiting the unique chemistry of boronic acid-containing polymers, we have prepared a novel example of triply-responsive “schizophrenic” block copolymers . The combination of PNIPAM with PAPBA leads to a single system capable of self-assembly in response to changes in temperature, pH, and the concentration of a model diol. The ready availability of boronic acids with a range of easily tuned pKa values indicate this general method can be used to prepare a wide variety of novel triply-responsive block copolymers .
This material is based upon work supported by the National Science Foundation under grant no. DMR-0846792. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. Acknowledgement is made to the donors of the American Chemical Society Petroleum Research Fund (45286-G7). We thank Dr Sudershan R. Gondi for synthesizing DMP.
Notes and references
- J. F. Lutz, Polym. Int., 2006, 55, 979 CrossRef CAS
.
-
(a) S. Hou, E. L. Chaikof, D. Taton and Y. Gnanou, Macromolecules, 2003, 36, 3874 CrossRef CAS
-
(a) V. Bütün, S. Liu, J. V. M. Weaver, X. Bories-Azeau, Y. Cai and S. P. Armes, React. Funct. Polym., 2006, 66, 157 CrossRef
- A. B. Lowe, M. Torres and R. Wang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5864 CrossRef CAS
-
(a) S. Liu, N. C. Billingham and S. P. Armes, Angew. Chem., Int. Ed., 2001, 40, 2328 CrossRef CAS
- W. Niu, C. O’Sullivan, B. M. Rambo, M. D. Smith and J. J. Lavigne, Chem. Commun., 2005, 4342 RSC
- F. Jäkle, J. Inorg. Organomet. Polym. Mater., 2005, 15, 293 CrossRef
- T. D. James and S. Shinkai, Top. Curr. Chem., 2002, 218, 159 CAS
-
(a) W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS
-
(a) Y. Qin, G. Cheng, A. Sundararaman and F. Jäkle, J. Am. Chem. Soc., 2002, 124, 12672 CrossRef CAS
- J. N. Cambre, D. Roy, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2007, 129, 10348 CrossRef CAS
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477 RSC
- Y. Ma, K. Ohno, Y. Tsujii and T. Fukuda, Polym. Prepr., Jpn., 2007, 56, 219 Search PubMed
-
(a) A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754 CrossRef CAS
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and selected H NMR spectra and SEC traces. See DOI: 10.1039/b900374f |
| This journal is © The Royal Society of Chemistry 2009 |