A model study for the concise construction of the oxapentacyclic core of cortistatins through intramolecular Diels–Alder and oxidative dearomatization–cyclization reactions†
Lianzhu
Liu
a,
Yingxiang
Gao
a,
Chao
Che
a,
Na
Wu
a,
David Zhigang
Wang
a,
Chuang-Chuang
Li
*a and
Zhen
Yang
*ab
aLaboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen 518055, China. E-mail: zyang@pku.edu.cn
bKey Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education and Beijing National Laboratory for Molecular Science (BNLMS), College of Chemistry and the State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Science, Peking University, Beijing 100871, China
First published on 9th December 2008
Abstract
A unified strategy towards the facile construction of the [6.7.6.5] oxapentacyclic skeleton of cortistatins is reported, featuring intramolecular Diels–Alder (IMDA) and oxidative dearomatization–cyclization reactions as key steps.
Cortistatins, isolated in 2006 by Kobayashi and co-workers from the marine sponge Corticium simplex, represent a novel family of steroidal alkaloids, with unique structures and prominent biological activities (Fig. 1).1 Of particular interest among these natural products is cortistatin A (1), which was reported to be an extremely potent inhibitor of the migration and proliferation of human umbilical vein endothelial cells (HUVECs) at concentrations as low as 100 pM, and with a therapeutic index of over 3300.1a
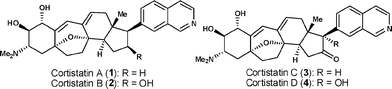 | ||
| Fig. 1 The structures of cortistatins A–D. | ||
Cortistatin A has an unprecedented oxabicyclo[3.2.1]heptene ring system that lies within a complex tetracarbocyclic skeleton, posing a particularly inviting synthetic challenge. Indeed, intensive efforts have been directed towards exploring feasible strategies for the chemical syntheses of these scarce yet pharmacologically significant natural substances, as exemplified by the recent accomplishments of Baran et al. and Nicolaou et al. in the synthesis of (+)-1,2 as well as four elegant model studies.3 Our work with cortistatins initially focused keenly on developing a concise and unified strategy that would allow us to gain rapid access to their oxapentacyclic skeletons, thereby laying the foundation for the total synthesis of these structures and the modular construction of a library of the various analogs necessary for further medicinal chemistry studies.
Retrosynthetically, we envisioned that the oxabicyclo[3.2.1]heptane core, A, endowed with suitable functionalities for further elaboration of the entire structure of cortistatin A, could be derived either from intermediate C, through a Lewis acid-mediated ring opening of the oxabridge,4 or from intermediate D, through the nucleophilic addition of a C-8 hydroxyl group to its phenol ring in the course of a hypervalent iodine5-triggered oxidative dearomatization event.6 Intermediate D could be prepared through a Lewis acid-mediated aromatization of oxabicyclic structure C, which in turn could be constructed via a furan–alkyne intramolecular Diels–Alder reaction (IMDA),7 promoted by a pre-organized favorable conformation of substrate E (see Scheme 1).8 Precursor E could be conveniently assembled from simpler building blocks F, G and H. We believed this strategy would allow for rapid access to the elaborated pentacyclic core of cortistatins in a highly convergent manner. Herein, we report our success in implementing these synthetic designs in the context of cortistatin A.
 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Since the reactivity and selectivity of IMDA reactions are often highly substrate-dependent,9 our initial efforts were focused on evaluating the feasibility of such reactions in substrate 10. The compound was readily prepared through the reaction sequence shown in Scheme 2.
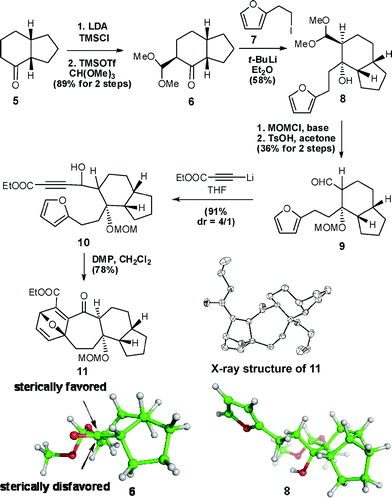 | ||
| Scheme 2 The synthesis of key intermediate 11. | ||
Ketone 6 was synthesized in an 89% yield from 510via the conversion of its carbonyl group to a silyl enol ether, followed by reaction with CH(OMe)311 in the presence of TMSOTf. 6 then underwent a coupling reaction with a furan-based alkyl lithium reagent, generated in situ by the treatment of iodide 7 with t-BuLi, to give hydroxyketal 8 in a 58% yield. Its relative stereochemistry was established to be that shown in Scheme 2 on the basis of nuclear Overhauser effect analysis (see the ESI† ). Next, protection of the hydroxyl group as its MOM ether and acetal deprotection in the presence of TsOH converted 8 into aldehyde9 in a 36% yield. 9 was then reacted with 3-ethoxy-3-oxoprop-1-ynyl lithium to give propargyl alcohol 10 in a 91% yield as a pair of diastereoisomers. The IMDA reaction of the unactivated alkyne moiety in 10 proved to be unsuccessful under a variety of conditions screened. To our delight, treating 10 with Dess–Martin periodinane (DMP) at 25 °C smoothly promoted the desired IMDA reaction to yield the easily crystallized pentacyclic product, 11, in a 78% yield. The structure of 11 was determined unambiguously by X-ray crystallography.†
The seemingly trivial step of MOM deprotection in 11, however, proved to be problematic. We therefore explored the direct coupling reaction between aldehyde12 and 3-ethoxy-3-oxoprop-1-ynyl lithium. To this end, 8 was first treated with TsOH in the presence of acetone to afford hydroxyaldehyde12 in an 87% yield, which, upon treatment with AlMe3 in THF and subsequent reaction with 3-ethoxy-3-oxoprop-1-ynyl lithium (that might direct the attack of the lithium reagent onto the aldehyde from the less hindered face), gave the expected product, 14, in a 71% yield as a single diastereoisomer. The excellent diastereoselectivity observed in this reaction may be attributed to the formation of tetracoordinate organoaluminum complex 13,12 which might direct the attack of the lithium reagent on the aldehyde to the less hindered face (Scheme 3). The IMDA reaction of substrate 14 was next investigated, and its reactivity profile was found to be similar to that of 10. Although 14 itself did not cyclize to 15, it did under DMP oxidation conditions, yielding epimeric products 16a and 16b in a 3 : 1 ratio.
 | ||
| Scheme 3 The syntheses of compounds 16a and 16b. | ||
Having assembled the [6.7.6.5] pentacyclic core structure, we next turned our attention to investigating the feasibility of forming the oxabicyclo[3.2.1]heptane ring system of cortistatins by direct nucleophilic addition of the hydroxyl group at C-8 in the presence of activating agents, such as protic or Lewis acids.
We speculated that due to the coordination of Lewis acids with substrates 16a and 16b, the C–O bond of the oxabridge in the substrate would be polarized, and thereby suited for an SN2 displacement by the C-8 hydroxyl group to afford the desired product, 17. A range of Lewis acids were screened, such as Me3Al, ZnCl2 and MgBr2, yet none of them were successful. This failure might be attributed to both the steric hindrance surrounding the C-8 tertiary alcohol13 and the ring strain of the pentacyclic oxabicyclo[3.2.1]heptane.14
Gratifyingly, it was found that BF3·Et2O was capable of effecting aromatization of the oxabicyclo[3.2.1]heptane ring in 16a and 16b, leading to phenol 18 in a 63% yield (Scheme 4).15 The stereochemistry of 18 was confirmed by X-ray crystallography.† These findings suggested new opportunities to install the oxabicycloheptene ring via a hypervalent iodine-mediated oxidative dearomatization–cyclization sequence (18 → 19), as illustrated in Scheme 5.16
 | ||
| Scheme 4 The synthesis of compound 18. | ||
![The synthesis of oxabicyclo[3.2.1]heptane 19.](/image/article/2009/CC/b817376a/b817376a-s5.gif) | ||
| Scheme 5 The synthesis of oxabicyclo[3.2.1]heptane 19. | ||
Phenol 18 was then subjected to the action of several known oxidative dearomatization–cyclization protocols, as shown in Scheme 5, resulting, unfortunately, in either decomposition or recovery of the starting material. Presumably, the significant electron deficiency of the aromatic ring in 18 demands a more reactive oxidant in a more polar medium that would facilitate the formation and stabilization of the putative carbocation intermediate. In this context, the PhI(OOCCF3)2 reagent employed earlier by Dai and Danishefsky in their cortistatin A model study17 caught our attention.
Initially, implementation of the PhI(OOCCF3)2-promoted dearomatization–cyclization in our more complex setting proved to be highly solvent-dependent, and did not initially yield the desired product, 19, in CH2Cl2, THF, CH3CN or CF3CH2OH (see the ESI for details† ). It was found, however, that the reaction in CH3NO2 gave 19 in a 30% yield, the major by-product being generated through the capture of the carbocation intermediate by an external hydroxyl source, such as water, rather than by the intramolecular C-8 hydroxyl group. Indeed, the employment of highly anhydrous CH3NO2 in the presence of pre-activated molecular sieves significantly improved the yield of 19 to 60%. The structure of 19 was established via NMR and HRMS studies.
In summary, we have explored a furan-based IMDA reaction to construct the [6.7.6.5] pentacyclic core of cortistatin A and efficiently installed its unique oxabicyclo[3.2.1]heptene ring system through a concise oxidative dearomatization–cyclization sequence. The work described herein demonstrates a synthetically robust strategy for the rapid construction of the framework of cortistatins and their various analogues. A total synthesis of cortistatin A based upon these key findings is currently under way in our laboratory.
Notes and references
- (a) S. Aoki, Y. Watanabe, M. Sanagawa, A. Setiawan, N. Kotoku and M. Kobayashi, J. Am. Chem. Soc., 2006, 128, 3148 CrossRef CAS; (b) Y. Watanabe, S. Aoki, D. Tanabe, A. Setiawan and M. Kobayashi, Tetrahedron, 2007, 63, 4074 CrossRef CAS; (c) S. Aoki, Y. Watanabe, D. Tanabe, A. Setiawan, M. Arai and M. Kobayashi, Tetrahedron Lett., 2007, 48, 4485 CrossRef CAS; (d) S. Aoki, Y. Watanabe, D. Tanabe, M. Arai, H. Suna, K. Miyamoto, H. Tsujibo, K. Tsujikawa, H. Yamamoto and M. Kobayashi, Bioorg. Med. Chem., 2007, 15, 6758 CrossRef CAS.
- (a) R. A. Shenvi, C. A. Guerrero, J. Shi, C.-C. Li and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7241 CrossRef CAS; (b) K. C. Nicolaou, Y.-P. Sun, X.-S. Peng, D. Ploet and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2008, 47, 7310 CrossRef CAS.
- (a) S. Yamashita, K. Iso and M. Hirama, Org. Lett., 2008, 10, 3413 CrossRef CAS; (b) E. M. Simmons, A. R. Hardin, X. Guo and R. Sarpong, Angew. Chem., Int. Ed., 2008, 47, 6650 CrossRef CAS; (c) M. Dai and S. J. Danishefsky, Tetrahedron Lett., 2008, 49, 6610 CrossRef CAS; (d) M. Dai, Z. Wang and S. J. Danishefsky, Tetrahedron Lett., 2008, 49, 6613 CrossRef CAS; (e) D. D. T. Craft and B. W. Gung, Tetrahedron Lett., 2008, 49, 5931 CrossRef CAS.
- For information regarding the metal-mediated ring opening of oxabridges, see: M. Lautens, K. Fagnou and S. Hiebert, Acc. Chem. Res., 2003, 36, 48 Search PubMed.
- (a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS; (b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS.
- (a) A. Pelter and S. M. A. Elgendy, J. Chem. Soc., Perkin Trans. 1, 1993, 1891 RSC; (b) P. Wipf, Y. Kim and H. Jahn, Synthesis, 1995, 154; (c) S. Quideau, L. Pouységu and D. Deffieux, Synlett, 2008, 467 CrossRef CAS.
- (a) C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179 CrossRef CAS; (b) D. D. Sternbach, D. M. Rossana and K. D. Onon, J. Org. Chem., 1984, 49, 3427 CrossRef CAS; (c) M. E. Jung and J. Gervay, J. Am. Chem. Soc., 1989, 111, 5469 CrossRef CAS.
- (a) G. Brieger and J. N. Bennett, Chem. Rev., 1980, 80, 63 CrossRef CAS; (b) S. M. Weinreb, Acc. Chem. Res., 1985, 18, 16 CrossRef CAS; (c) M. K. Diedrich, F.-G. Klarner, B. R. Beno, K. N. Houk, H. Senderowitz and W. C. Still, J. Am. Chem. Soc., 1997, 119, 10255 CrossRef.
- (a) X.-H. Du, H. V. Chu and O. Kwon, Org. Lett., 2003, 5, 1923 CrossRef CAS; (b) W. S. Jen, Development of new asymmetric organocatalytic methods and progress towards the total synthesis of guanacastepene A, PhD Thesis, California Institute of Technology, Pasadena, California, 2004 (http://etd.caltech.edu/etd/available/etd-10282003-135857/) Search PubMed; (c) C.-C. Li, S. Liang, X.-H. Zhang, Z.-X. Xie, J. Chen, Y. Wu and Z. Yang, Org. Lett., 2005, 7, 3709 CrossRef CAS.
- S. Boßhammer and H. Gais, Synthesis, 1998, 919 CrossRef.
- S. Murata, M. Suzuki and R. Noyori, Tetrahedron, 1988, 44, 4259 CrossRef CAS.
- T. Ooi, N. Kagoshima, H. Ichikawa and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 3328 CrossRef CAS.
- (a) A. Pelter, A. Hussain, G. Smith and R. S. Ward, Tetrahedron, 1997, 53, 3879 CrossRef CAS; (b) R. Venkateswarlu, C. Kamakshi, S. G. A. Moinuddin, P. V. Subhash, R. S. Ward, A. Pelter, S. J. Coles, M. B. Hursthouse and M. E. Light, Tetrahedron, 2001, 57, 5625 CrossRef CAS; (c) S. Quideau, M. Lebon and A.-M. Lamidey, Org. Lett., 2002, 4, 3975 CrossRef CAS.
- G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95 CrossRef CAS.
- J. T. Repine, D. S. Johnson, A. D. White, D. A. Favor, M. A. Stier, J. Yip, T. Rankin, Q. Ding and S. N. Maiti, Tetrahedron Lett., 2007, 48, 5539 CrossRef CAS.
- For selected examples of oxidative dearomatization in synthesis, see: (a) N. T. Vo, R. D. M. Pace, F. O’Hara and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 404 CrossRef CAS; (b) J. Gagnepain, F. Castet and S. Quideau, Angew. Chem., Int. Ed., 2007, 46, 1533 CrossRef CAS; (c) S. P. Cook, A. Polara and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 16440 CrossRef CAS; (d) L. H. Mejorado and T. R. R. Pettus, J. Am. Chem. Soc., 2006, 128, 15625 CrossRef CAS; (e) A. Berube, I. Drutu and J. L. Wood, Org. Lett., 2006, 8, 5421 CrossRef CAS; (f) J. Zhu and J. A. Porco, Jr, Org. Lett., 2006, 8, 5169 CrossRef CAS; (g) Y. Hayashi, H. Gotoh, T. Tamura, H. Yamaguchi, R. Masui and M. Shoji, J. Am. Chem. Soc., 2005, 127, 16028 CrossRef CAS.
- M. J. Dai and S. J. Danishefsky, Heterocycles, 2008 Search PubMed , COM-08-S(F)6.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation data. CCDC 699943 and 700771. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b817376a |
| This journal is © The Royal Society of Chemistry 2009 |