
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc selenide stabilized in a quadrilateral network characterized with optical emissions†
Kuan-Lin
Wang
a,
Cai-Fei
Lin
a,
Chin
Cheng
b,
Tzu-Chi
Huang
c,
Bi-Hsuan
Lin
c,
Yu-Lin
Xie
a,
Bo-Yuan
Wang
a,
Jennifer
Kung
d,
Kuang-I
Lin
 e and
Kuei-Fang
Hsu
*a
e and
Kuei-Fang
Hsu
*a
aDepartment of Chemistry, National Cheng Kung University, Tainan 70101, Taiwan. E-mail: hsukf@mail.ncku.edu.tw
bDepartment of Physics, National Cheng Kung University, Tainan 70101, Taiwan
cNational Synchrotron Radiation Research Center, Hsinchu 300092, Taiwan
dDepartment of Earth Science, National Cheng Kung University, Tainan 70101, Taiwan
eCore Facility Center, National Cheng Kung University, Tainan 70101, Taiwan
First published on 11th March 2025
Abstract
A polymorph of K2Zn3Se4 (1) adopting a quadrilateral network induced by the presence of deficient zinc sites is discovered in the ThCr2Si2-type lattice. Upon quenching the synthetic reaction at 630 °C, this lattice crystallizes into a higher symmetry and reduced cell in 1·T. DFT calculations explain a coexistence of two pseudo-enantiomeric forms with an equal energy that favors such a flexible network stabilized in 1. With the partial substitution of Zn2+ ions by Mg2+ ions, one form is then trapped in K2Mg0.5Zn2.5Se4 (2). For the two ZnSe layered wide bandgap semiconductors, photoluminescence (PL) and X-ray excited optical emission (XEOL) measurements reveal a prominent broad defect emission centered at ∼610 nm relative to the emission position observed in the benchmark ZnSe crystal.
Introduction
The semiconductor ZnSe has recently attracted significant attention due to its defect emissions, providing a potential platform for quantum applications.1–4 Its bandgap of 2.6 eV is essential to enhance the stability of these luminescent centers for persistent performance. Materials engineering through the incorporation of alkali chalcogenides to widen the bandgap is a well-established strategy,5,6 which has been applied to discover a series of advanced ZnSe-based semiconductors.7,8 Materials such as K2ZnSe2 and K2Zn3Se4 typically exhibit wider bandgaps exceeding 3.0 eV.The A2Zn3Se4 (A = K, Rb, Cs) family features a two-dimensional (2D) structure originating from the ordered absence of one-quarter of Zn2+ ions within the ThCr2Si2-type lattice. In the noted AxFe1−x/2Se2 (A = K, Cs) materials, the deficiency located around Fe2+ sites in the same type of lattice facilitates structural relaxation into a supercell.7 In this research, a quadrilateral network influenced by deficient Zn2+ sites is discovered in a polymorph of K2Zn3Se4 (1). Beyond their promising application as transparent conductors,8–10 the optical luminescence of these two-dimensional ZnSe-based semiconductors remains unexplored. In this study, we present detailed analyses of crystal structures, DFT calculations, thermal analysis, and defect emissions of K2Zn3Se4 (1) and Mg-substituted K2Mg0.5Zn2.5Se4 (2).
Experimental
Synthesis
Chemicals used in the reaction were potassium (99.5%, Alfa Aesar), zinc (99.8%, Sigma-Aldrich), magnesium turnings (99+%, Alfa Aesar), selenium (99.9%, Sigma-Aldrich), and potassium bromide (99.9%, J. T. Baker). The synthetic preparation was conducted under a dry N2 atmosphere in an OMNI-LAB glove box. A mixture of chemicals was loaded in a fused silica tube (diameter: 8 mm) treated with carbon coating and sealed under vacuum (<10−4 torr).Compound 1 was prepared from K (0.1151 g), Zn (0.2624 g), Se (0.4426 g), and KBr (1.4646 g) in a molar ratio of 1.10/1.50/2.00/4.60. Compound 2 was prepared from K (0.1303 g), Mg (0.0230 g), Zn (0.2107 g), Se (0.4387 g), and KBr (1.5536 g) in a molar ratio of 1.20/0.34/1.16/2.00/4.70. The two compounds were synthesized using identical heating profiles. The reactions were heated to 250 °C and held for 1 h, then the temperature was raised to 800 °C and held for 48 h, followed by cooling to 700 °C in 10 h. Cooling was continued to 400 °C in 60 h, and finally the furnace was turned off. Deionized water was used to eliminate KBr flux from the product. Transparent brown crystals were observed for 1, whereas 2 exhibited transparent yellow crystals.
Compound 1·T was synthesized using the same chemical proportions as compound 1. The reaction tube was heated at 800 °C for 48 h, then cooled to 630 °C in 17 h, and finally quenched in water. After removing the flux, transparent brown crystals were observed.
Single-crystal X-ray diffraction structural analysis
Single crystals were selected for indexing and data collection on a Bruker D8 Venture PHOTON III diffractometer irradiated with graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å). APEX 4 software was utilized for data integration and empirical absorption corrections, while crystal structures were solved and refined using the SHELXTL program.11 Crystal data and atomic coordinates of 1, 2 and 1·T are listed in Tables 1 and 2, respectively.| Compound | 1 | 2 | 1·T |
|---|---|---|---|
| a R 1 = (∑‖Fo| − |Fc‖)/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | |||
| Formula | K2Zn3Se4 | K2Mg0.5Zn2.5Se4 | K2Zn3Se4 |
| f. w. | 590.3 | 572.7 | 590.3 |
| Space group | P2/c | P2/c | C2/m |
| a (Å) | 7.4586(6) | 7.4620(2) | 14.272(19) |
| b (Å) | 5.8956(5) | 5.96870(10) | 4.087(6) |
| c (Å) | 11.3644(9) | 11.3465(2) | 4.093(6) |
| β (°) | 112.375(3) | 112.251(1) | 106.34(3) |
| V (Å3), Z | 462.10(7), 2 | 467.72(2), 2 | 229.1(5), 1 |
| ρ calcd (g cm−3) | 4.255 | 4.065 | 4.277 |
| T (K) | 296 | 296 | 296 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα |
| μ (mm−1) | 24.364 | 22.919 | 24.494 |
| Total refls. | 8353 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 088 088 |
948 |
| Uniq. refls. (Rint) | 1149 (0.0412) | 1155 (0.0236) | 312 (0.0432) |
| Param./restr. | 56/1 | 48/2 | 17/0 |
| Goodness of fit | 1.086 | 1.144 | 1.284 |
| R 1 [I > 2σ(I)] | 0.0611 | 0.0116 | 0.0581 |
| wR 2 [I > 2σ(I)] | 0.1337 | 0.0245 | 0.1592 |
| Δρmax, Δρmin (e Å−3) | 1.541, −1.176 | 0.424, −0.524 | 0.000, 0.000 |
| Atom | Site | x | y | z | U eq. | Occ. |
|---|---|---|---|---|---|---|
| a U eq. is defined as one third of the trace of the orthogonalized Uij tensor. | ||||||
| 1 | ||||||
| K(1) | 2a | 0 | 0 | 0.5 | 0.0340(8) | 1 |
| K(2) | 2e | 0 | 0.4949(6) | 0.5 | 0.0335(8) | 1 |
| Zn(1) | 2f | 0.5 | 0.7495(10) | 0.25 | 0.0525(19) | 0.497(10) |
| Zn(2) | 4g | 0.5002(3) | 0.2497(4) | 0.5213(4) | 0.0206(6) | 0.669(9) |
| Zn(2A) | 4g | 0.4991(10) | 0.2517(14) | 0.4740(9) | 0.0206(6) | 0.258(7) |
| Zn(3) | 2f | 0.5 | 0.2501(5) | 0.25 | 0.0197(8) | 0.678(8) |
| Se(1) | 4g | 0.7114(1) | 0.4906(2) | 0.4325(1) | 0.0231(3) | 1 |
| Se(2) | 4g | 0.2886(1) | 0.0088(2) | 0.3268(1) | 0.0235(3) | 1 |
| 2 | ||||||
| K(1) | 2a | 0 | 0 | 0.5 | 0.0335(1) | 1 |
| K(2) | 2e | 0 | 0.4848(1) | 0.25 | 0.0323(1) | 1 |
| Zn(1) | 2f | 0.5 | 0.2497(1) | 0.75 | 0.0185(1) | 0.906(10) |
| Mg(1) | 2f | 0.5 | 0.2497(1) | 0.75 | 0.0185(1) | 0.100(26) |
| Zn(2) | 4g | 0.4993(1) | 0.7502(1) | 0.5238(1) | 0.0184(1) | 0.825(5) |
| Mg(2) | 4g | 0.4993(1) | 0.7502(1) | 0.5238(1) | 0.0184(1) | 0.182(14) |
| Se(1) | 4g | 0.2878(1) | 0.4749(1) | 0.5664(1) | 0.0181(1) | 1 |
| Se(2) | 4f | 0.2877(1) | 0.9758(1) | 0.3279(1) | 0.0181(1) | 1 |
| 1·T | ||||||
| K(1) | 2b | 0.5 | 0 | 0 | 0.0570(14) | 1 |
| Zn(1) | 4i | 0.2500(2) | 0.5 | 0.7505(7) | 0.0555(9) | 0.75 |
| Se(1) | 4i | 0.3559(1) | 0.5 | 0.3558(4) | 0.0498(7) | 1 |
For 1, two K, three Zn, and two Se atoms were located from the electron density maps. The Zn1 and Zn(3) sites, positioned on the twofold axes, were refined with occupancies of 49.7(10)% and 67.8(8)%, respectively. The Zn(2)/Zn(2A) sites exhibited disorder in the general positions, with a distribution ratio of 66.9(9)%/25.8(7)%. The formula of 1 was determined to be K2Zn3.03(5)Se4 and the stoichiometric K2Zn3Se4 is expressed for clarity. In the final cycle of refinement, all the atoms were anisotropically refined, resulting in R1 and wR2 converging to 0.0611 and 0.1222, respectively.
For 2, the electron density maps revealed the presence of two K, two M (M = Zn/Mg), and two Se atoms. The M(1) site on the twofold axis was occupied by both Zn and Mg atoms with a distribution ratio of 91(1)%/10(3)%. The M(2) site in the general position was refined with a ratio of 83(1)%/18(1)% for the disordered Zn/Mg atoms. The formula of 2 was then determined to be K2Mg0.46(5)Zn2.56(2)Se4 and the stoichiometric K2Mg0.5Zn2.5Se4 is expressed for clarity. In the final cycle of refinement, all the atoms were anisotropically refined, resulting in R1 and wR2 values converging to 0.0116 and 0.0245, respectively.
For 1·T, the electron density maps revealed the presence of one K, one Zn, and one Se atoms. The crystal of 1·T quenched at 630 °C crystallized a unit cell close to a tetragonal lattice with a = 14.27(2) Å, b = 4.087(6) Å, and c = 4.093(6) Å. Examination of the (hk0) reflections, as shown in Fig. S1(a),† revealed a lattice with C-centered symmetry. However, reflections such as (110), (200) and (310) at low 2θ angles indicated a lattice exhibiting a degree of disordering. An alert B of a tetragonal system was then suggested by the checkCIF report due to the model being constructed by lower C2/m symmetry. The Zn(1) site is 25% deficient on the mirror position that derived the formula K2Zn3Se4. In the final cycle of refinement, all the atoms were anisotropically refined, resulting in R1 and wR2 values converging to 0.0581 and 0.1592, respectively.
Electron microscopy
Semiquantitative analysis was conducted using a Hitachi SU8000 scanning electron microscope (SEM) equipped with a Bruker XFlash 5010 energy-dispersive spectrometer (EDS). The data were acquired at an accelerating voltage of 15 keV and analyzed using the EMAX Suite version 1.90 software. The EDS analyses in Fig. S2a and 2b† revealed that the averaged compositions of crystals 1 and 2 are K1.00(1)Zn1.46(6)Se2.07(7) and K1.00(1)Mg0.17(1)Zn1.26(6) Se1.95(5), respectively.Powder X-ray diffraction analysis
The pure phase was characterized by powder X-ray diffraction (PXRD) patterns using a Bruker AXS D2 PHASER equipped with a Cu Kα tube (1.5406 Å) operating at 30 kV and 10 mA. A one-dimensional LYNXEYE detector, designed with more than 150 integrated slits, recorded 2θ positions with an angular accuracy of less than 0.02°. The measured PXRD patterns agreed well with the calculated patterns derived from single-crystal X-ray structural models, as demonstrated in Fig. S3.†12Theoretical calculations
In order to delve further into these observed experimental results, first-principles density functional theory (DFT)13 calculations were carried out using the CASTEP14 module implemented in Materials Studio software. The number of electrons treated as valence electrons are 9, 12, 16 and 2 for K, Zn, Se and Mg, respectively, and the OTFG norm-conserving pseudopotentials were used for the interactions between the valence electrons and the ions. Plane waves with kinetic energy up to 1350 eV were included in the basis expansion and the cutoff energy was increased to 1450eV for convergence tests when necessary. The PBEsol15 scheme of the generalized gradient approximation (GGA)16 functionals was employed for the exchange-correlation energy functional. It is well known that the local density (LDA)17 functional predicts overbinding lattice constants and the revised PBE scheme of the GGA functionals results in underbinding ones.18 The PBEsol functional was proposed to improve the description of the PBE scheme.19DFT calculations were applied to two pseudo-enantiopure forms in K2Zn3Se4 (1) with space group symmetry P2/c to start with (Fig. 2). The primitive unit cell consists of twice the compound formula, i.e. 4K, 6Zn, and 8Se atoms. Four of the six Zn atoms in the unit cell occupy the Zn(2) sites, while the other two Zn atoms occupy either the Zn(1) or the Zn(3) sites to generate the two enantiopure forms, which were found to be identical in energy and lattice parameters. In these calculations, the Monkhorst–Pack20k-point set of 4 × 4 × 3 was used for sampling the integration of the physical quantities over the first Brillouin zone. The k-point sets with similar density in reciprocal space were used for the other structures examined in the present study. The lattice parameters of the relaxed structure were found to be a = 7.36 Å (−1.3%), b = 5.86 Å (−0.6%), c = 11.34 Å (−0.2%), α = γ = 90°, and β = 112.58° (+0.2%), where the corresponding deviations from the experimental data are presented in parentheses.
To study the possible origin of the coexisting Zn(1) and Zn(3) sites as observed experimentally, periodic structures generated involving building blocks of both enantiomers were investigated in the DFT calculations. The crystal structure with alternate presence of the two enantiopure forms along the a axis exhibits the same P2/c symmetry as the experimental observations. The completely relaxed structure turned out to have about the same energy as twice the energy of one enantiopure form, i.e. displaying a miniscule energy cost (up to the accuracy of the calculations, i.e. 0.1 meV/2fu) in creating the alternate presence of these two enantiopure forms. In addition, the supercells with alternate building blocks of the two enantiomers along the b axis resulted in an energy higher than that of the enantiopure form by more than 0.6 eV/2fu and a modified space group symmetry of C2/m, i.e. it is no longer in possession of the P2/c symmetry. The relaxation along the c axis retains the P2/c symmetry but with an energy of 0.2 eV/2fu higher than that of the enantiopure form.
The observed Mg-doped system of K2Mg0.5Zn2.5Se4 (2) was simulated by stoichiometric compounds of the formula K4MgZn5Se8 in the DFT calculations. To preserve the P2/c symmetry, the following four structures were considered. The first two structures have Mg replacing the Zn(1) site with double the primitive cell of the K2Zn3Se4 cell in the a and b axis, respectively, i.e. with the formula K8Mg2Zn10Se16 in a primitive unit cell. The other two structures have the Mg replacing the Zn(2) site with either quadruple cells in the a direction or double cells in both the a and b directions, i.e. with the formula K16Mg4Zn20Se32 in a primitive unit cell. Both the structures with Mg replacing the Zn(2) site are lower in energy than those with Mg replacing the Zn(1) site. The lowest energy one is the one with quadruple cells along the a direction.
Differential thermal analysis
Differential thermal analysis (DTA) was performed using a computer-controlled Shimadzu DTA-50 thermal analyzer. The sample and Al2O3 as a reference were loaded with the same weight of about 25 mg and were sealed in fused silica ampoules under a vacuum. The heating and cooling program of the DTA chamber was run at a constant rate of 10 °C min−1. Thermal measurements were monitored across multiple melting and recrystallization cycles for 1 and 2, as shown in Fig. S4.†UV-vis–NIR spectroscopy
Diffuse reflectance spectra were obtained using a Hitachi U-4100 UV-vis–NIR spectrophotometer covering the wavelength range of 200–2600 nm. The system was equipped with a single monochromator, a photomultiplier detector for the UV/vis region, and a PbS-based detector for the NIR region. Measurements of the powder sample were performed using BaSO4 powder as a reference. The absorption data F(R) were calculated using the Kubelka–Munk equation: F(R) = (1 − R)2/2R, where R is the reflectance at a given energy. The absorption data were further fitted using the Tauc equation [hν·F(R)]2 = A(hν − Eg), where h is Planck's constant, ν is the frequency of light, F(R) is the absorption data, A is a proportionality constant, and Eg is the bandgap.21 The Eg value was then determined as the point of intersection between the baseline and the line extrapolated from the linear portion of the absorption edge. The bandgap values of 1 and 2 are about 2.97 eV and 3.06 eV, respectively (see Fig. S5†).Photoluminescence measurement
Photoluminescence (PL) measurement was conducted using a custom-built laser scanning confocal system employing excitation from a 458 nm pulsed laser (Coherent Genesis MX 460-500) with a repetition rate of 8 MHz. The laser excitation light was focused onto the sample by a 50× objective (JAPAN, LMPlan FL, N 0.5) with an on-sample power tuned at 30 mW, 50 mW and 70 mW. PL spectra were acquired through a fiber-coupled spectrometer (Photonic Workshop, model HORIBA iHR 550) equipped with a 1800 grooves per mm grating and a thermoelectric-cooled CCD (Newton DU920P-BVF).X-ray excited optical luminescence and X-ray absorption measurement
X-ray excited optical luminescence (XEOL) and X-ray absorption (XAS) experiments were conducted at the Taiwan Photon Source (TPS) 23A X-ray nanoprobe beamline located at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The capabilities of the TPS 23A X-ray nanoprobe beamline have previously been described in detail by Wu et al.22 This beamline can deliver an X-ray beam spot size of less than 100 nm. By using an optical fiber (with a core diameter of 400 μm) attached to a spectrometer (iHR550, HORIBA), we collected XEOL spectra and XEOL maps by using a deep thermoelectric cooling charge-coupled device (Syncerity BI UV-vis with 2048 × 70 pixels) and a photomultiplier tube (PMT), respectively. The XAS spectra were measured using a silicon drift detector (SDD; Vortex-ME4, Hitachi).Results and discussion
Structure
A quadrilateral network of ZnSe is unprecedentedly stabilized in a polymorph of K2Zn3Se4 (1), whose lattice is extended by the half-filled Zn(1) and Zn(3) sites on a twofold axis along with the split Zn(2)/Zn(2A) sites at the general position (see Fig. 1(a)). How the network related to the topology identified in the first phase7 can be elucidated from the following structural description. The spatial arrangement of Zn(1) and Zn(3) sites in the network of 1 is present in an enantiomeric symmetry along the b axis, as their atomic coordinates are closely related by (1/2, y, 1/4) and (1/2, 1 − y, 1/4) (see Table 2). Therefore, the network extended by the full occupation of 2Zn1 + 4Zn2 or 2Zn3 + 4Zn2 sites in the unit cell then corresponds to the previous phase quenched at 850 °C followed by furnace cooling (see Fig. 2). Why the present quadrilateral lattice stabilized in 1 will be further explained from the aspect of DFT calculations and thermal analysis.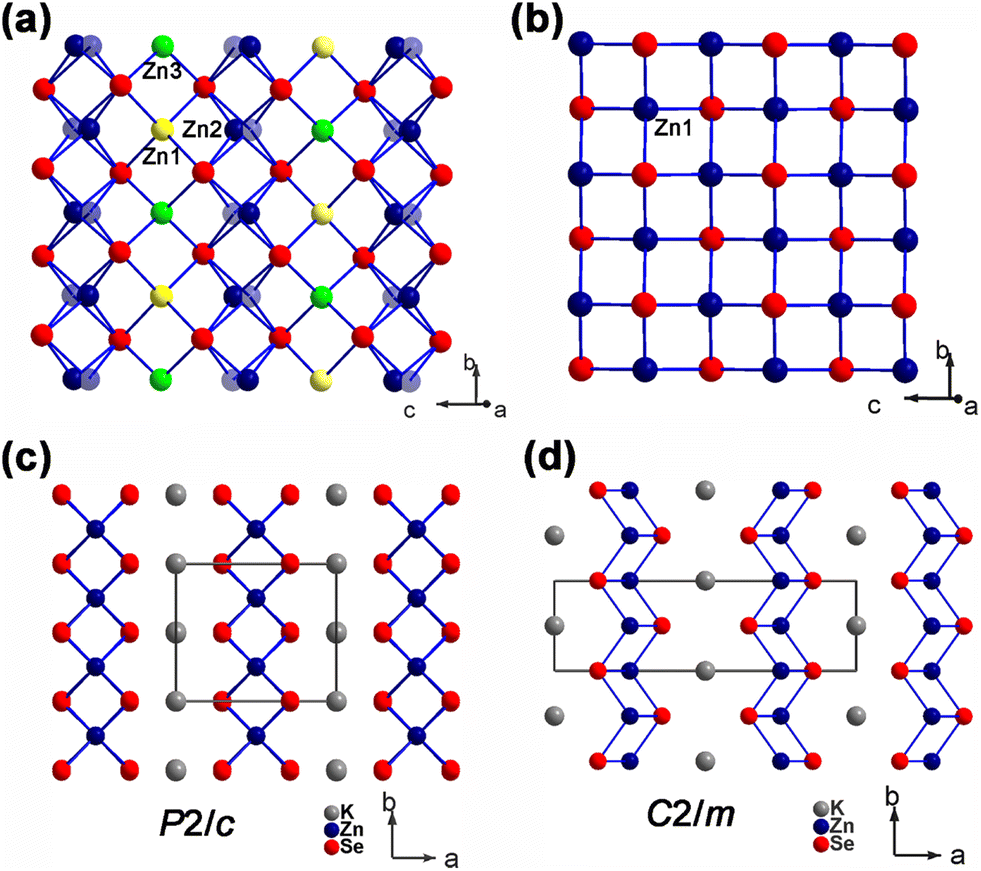 | ||
| Fig. 1 (a) A quadrilateral ZnSe layer composed of the half-filled Zn(1) and Zn(3) sites and the disordered Zn(2) site in 1. (b) A square ZnSe layer composed of the Zn(1) sites with 25% deficiency in 1·T. (c) The stacking of the quadrilateral layer in 1 with the symmetry P2/c. (d) The stacking of the square layer in 1·T with the symmetry C2/m. | ||
 | ||
| Fig. 2 The two pseudo-enantiomeric forms based on 4Zn(2) + 2Zn(1) or 4Zn(2) + 2Zn(3) in the unit cell of K2Zn3Se4 (1). | ||
DFT calculations
DFT calculations confirmed the equal energy and lattice parameters of the two pseudo-enantiopure forms as expected (Fig. 2). However, the coexistence of these two forms would not have been observed if the boundary energy between them is prominently positive, i.e. indicating a repulsive interaction between the two forms. The DFT calculations suggested a negligible energy cost for the phase featuring two alternating forms along the a axis when compared to the energy of the single form. As a result, the two pseudo-enantiopure forms can coexist with random distribution along the a axis, which maximizes the system's entropy and thereby minimizes the free energy of the system. A supercell structure consisting of alternative stacking of the two pseudo-enantiopure forms has been evident in the sulfide analogue.8 Instead of relaxing into a supercell, the two forms are constrained within a single network in the present zinc selenide 1.The advantage of the minute energy cost of the alternate presence of the two enantiopure forms is disrupted by the partial substitution of Zn2+ ions by Mg2+ ions, causing one form to be trapped and crystallized in K2Mg0.5Zn2.5)Se4 (2). The DFT studies of the stoichiometric compounds of K4MgZn5Se8 deliver the same conclusion as that of the experimental observations, i.e. the preferred substitution of the Zn(2) atoms by the doped Mg atoms. In addition, the Mg formation energy of K4MgZn5Se8 with respect to K4Zn6Se8 was evaluated as −1.078 eV per Mg atom.
Thermal analysis
The DTA curve of K2Zn3Se4 (1), shown in Fig. S4(a),† displays consistent but weak endothermic and exothermic peaks at ∼562 °C and ∼538 °C, respectively, across two consecutive cycles. Rather than being cooled to 400 °C, the crystal 1·T was synthesized by quenching the reaction above the crystallization temperature of 1 at 630 °C. Despite the limitation of crystal quality, a reduced cell with higher C-centered symmetry is characterized for the structure 1·T (Table 1). The square network stabilized in 1·T is extended by a single site of ZnSe4 tetrahedra, where a 25% deficiency localized at the Zn(1) site (see Fig. 1(b)). Consequently, the quadrilateral lattice in 1, which exhibits nearly 50% deficiencies at both Zn(1) and Zn(3) sites, may become increasingly mobile at elevated temperature and lead to the periodic lattice appearing in a higher symmetric manner.On the other hand, the enantiomers described previously in the structure may act as the fundamental topology for the formation of the three polymorphs of K2Zn3Se4. The first phase quenched at 850 °C and resolved by powder X-ray diffraction may reveal an in situ structure with one enantiomeric form present. As the temperature decreases to 630 °C, the two enantiomeric forms may become slightly steady and disordered in the higher symmetry 1·T phase. Finally, in the 1 phase observed at 400 °C, both enantiomeric forms are trapped but constrained in the quadrilateral lattice with lower symmetry.
PL and XEOL
The photoluminescence behavior of crystal 1 is excited using a 458 nm laser. As shown in Fig. 3(a), a broadband emission with a maximum at 610 nm is observed. The ZnSe crystal, prepared for comparative purposes, displays a broadband emission centered at 630 nm, as shown in Fig. 3(c). This emission is attributed to the recombination of deep defect centers.23,24 The luminescence intensity on the crystal 1 increases as the laser power increased from 30 mW to 70 mW, which indicates that this layered ZnSe semiconductor retains a considerable density of defect recombination. Furthermore, substituting Zn2+ with Mg2+ in crystal 2 finely modulates the energy bandgap to 3.06 eV. The disordered Zn/Mg sites in the network of 2 may create more complex energy states of defects within the wide bandgap,25 leading to a broad emission centered at 640 nm under a higher energy of laser power (see Fig. 3(b)).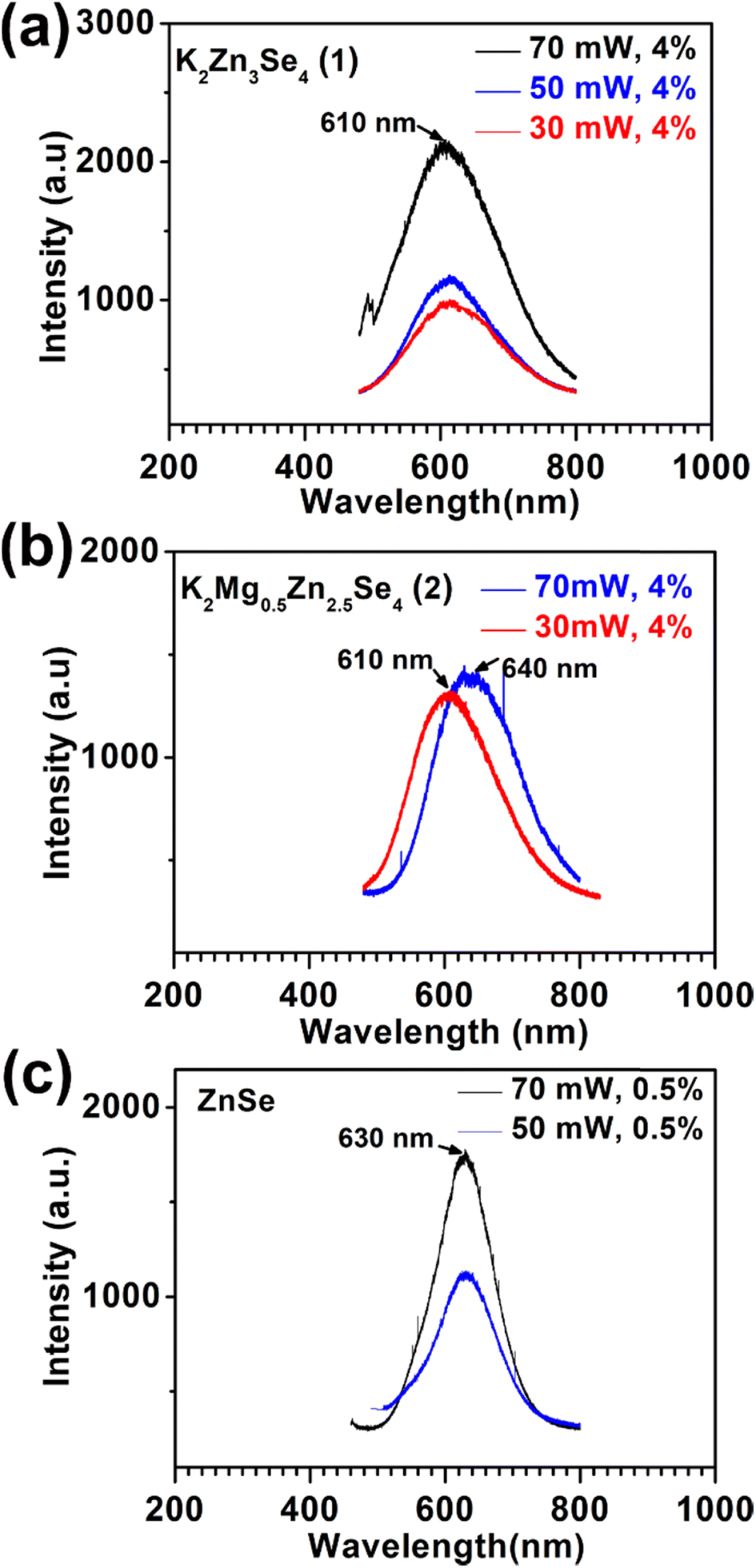 | ||
| Fig. 3 The PL spectrum excited by a 458 nm laser with variable laser power of 30 mW, 50 mW, and 70 mW and tunable laser transmittance of 0.5% and 4% on the crystals of (a) K2Zn3Se4 (1), (b) K2Mg0.5Zn2.5Se4 (2), and (c) ZnSe. | ||
Since the advantages of the X-ray synchrotron source are continuous and tunable, the emission properties of 1 can be further studied by using XEOL spectra with tuning X-ray energy across the Zn and Se K-edge. The XAS spectra of the Zn and Se K-edge of 1 are shown in Fig. 4(a) and (b), respectively. Five different X-ray energies across the Zn and Se K-edge have been selected to study their XEOL emission spectra, which are shown in Fig. 4(c) and (d), respectively. The emission evolution behaviors of 1 with X-ray energy across the Zn and Se K-edge are shown in Fig. 4(e) and (f), respectively. The results of the XEOL spectra exhibit positive edge jump behavior with X-ray energy across both the Zn and the Se K-edge. The emission behaviors suggest that the emission intensity of 1 will be proportional to its absorption coefficient. Similar positive edge jump behavior had also been observed by using soft X-ray to study the ZnO nanoneedle sample26 or using hard X-ray to study the ZnO wafer.27
 | ||
| Fig. 4 The XAS spectra of K2Zn3Se4 (1) with (a) the Zn K-edge and (b) the Se K-edge. The XEOL spectra of K2Zn3Se4 (1) with X-ray energy across (c) the Zn K-edge and (d) the Se K-edge. The emission evolution behaviors of 1 with X-ray energy across (e) the Zn K-edge and (f) the Se K-edge. | ||
Fig. 5(a) shows the emission intensity distribution of the ZnSe crystal with wavelength λem = 610 nm.28,29 Based on the excellent spatial resolution using the nano-focused beam, the different local areas P1–P5 marked in Fig. 5(a) are selected to investigate their XEOL emission spectra as shown in Fig. 5(b). The emission intensity distribution of the Mg-substituted crystal 2 with wavelength λem = 600 nm is shown in Fig. 5(c). The XEOL spectra of the compound with different local areas P1–P3 marked in Fig. 5(c) are used to study their emission properties as shown in Fig. 5(d). As compared with zinc blend, both ZnSe-based layered semiconductors 1 and 2 exhibit weak and slightly narrow emission bands under X-ray excitation at room temperature.
 | ||
| Fig. 5 (a) Emission intensity distribution of ZnSe at λem = 610 nm. XEOL emission spectra of the local areas P1–P5 from (a) are shown in (b). (c) Emission intensity distribution of K2Mg0.5Zn2.5Se4 (2) at λem = 600 nm. XEOL emission spectra of the local areas P1–P3 from (c) are shown in (d). | ||
Conclusions
A flexible layered structure is discovered in the wide bandgap semiconductors of K2MgxZn3−xSe4. The occurrence of deficiencies or substitution around the zinc sites affects the two-dimensional lattice crystallizing in various layered topology. Instead of near-band-edge emission, defect emission as characterized by PL and XEOL demonstrates a consistent and stable performance to allow these layered ZnSe materials to be suitable as host lattices30 for further electron implantation in potential quantum research.Data availability
The data supporting this article have been included as part of the ESI.† Crystal structures in Fig. 1 and 2 were plotted using the software Diamond 3.0. Optical spectra in Fig. 3–5 were plotted using Origin 8.5Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. F. H. acknowledges the National Science and Technology of Taiwan for the support (NSTC 112-2113-M-006-005) and the use of XRD003100 and EM003600 belonging to the Core Facility Center of National Cheng Kung University. L. B. H. acknowledges NSTC 112-2112-M-213-011 and NSTC 113-2112-M-213-013.References
- Y. Wu, K. J. Mirrielees and D. L. Irving, Appl. Phys. Lett., 2022, 120, 202102 CrossRef.
- Y. Wu, K. J. Mirrielees and D. L. Irving, J. Phys. Chem. Lett., 2022, 13, 8380–8385 CAS.
- E. Kirstein, E. A. Zhukov, D. S. Smirnov, V. Nedelea, P. Greve, I. V. Kalitukha, V. F. Sapega, A. Pawlis, D. R. Yakovlev, M. Bayer and A. Greilich, Commun. Mater., 2021, 2, 91 CrossRef CAS.
- L. S. dos Santos, W. G. Schmidt and E. Rauls, Phys. Rev. B, 2011, 84, 115201 Search PubMed.
- J.-H. Zhang, D. J. Clark, J. A. Brant, K. A. Rosmus, P. Grima, J. W. Lekse, J. I. Jang and J. A. Aitken, Chem. Mater., 2020, 32, 8947–8955 CAS.
- Y.-H. Su, W.-L. Chen, H. R. Byun, Y.-F. Zhang, M.-R. Zhuang, Y.-C. Lin, C.-K. Chang, P.-Y. Wang, C.-C. Lin, K.-I. Lin, H.-K. Liu, M.-K. Lee, J. I. Jang, Y.-M. Chang and K.-F. Hsu, Inorg. Chem., 2023, 62, 1570–1579 CrossRef CAS PubMed.
- A. Bhutani, X. Zhang, P. Behera, R. Thiruvengadam, S. E. Murray, A. Schleife and D. P. Shoemaker, Chem. Mater., 2020, 32, 326–332 CAS.
- J. Lin, M. Li, Y. Wang, Y. Li, F. Sun, X. Chen, Z. Guo, J. Zhao and W. Yuan, J. Alloys Compd., 2022, 927, 167098 CrossRef CAS.
- P. Zavalij, W. Bao, X. F. Wang, J. J. Ying, X. H. Chen, D. M. Wang, J. B. He, X. Q. Wang, G. F. Chen, P. Y. Hsieh, Q. Huang and M. A. Green, Phys. Rev. B, 2011, 83, 132509 CrossRef.
- H. Shi, B. Saparov, D. J. Singh, A. S. Sefat and M.-H. Du, Phys. Rev. B, 2014, 90, 184104 CrossRef.
- APEX 4 version 2021.4-0, Bruker Analytical X-r ay Systems, Madison, WI, 2021 Search PubMed.
- Diffrac plus TOPAS v. 4.2 (Manual), BRUKER AXS GmbH, Karlsruhe, 2009 Search PubMed.
- D. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, John Wiley & Sons, Inc, Hoboken, NJ, 2009 Search PubMed.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.:Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 102, 039902 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671–6687 CrossRef CAS PubMed.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566–569 CrossRef CAS.
- G. X. Zhang, A. M. Reilly, A. Tkatchenko and M. Scheffler, New J. Phys., 2018, 20, 063020 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- S. H. Moon, S. J. Park, Y. J. Hwang, D. K. Lee, Y. Cho, D. W. Kim and B. K. Min, Sci. Rep., 2014, 4, 4408 CrossRef PubMed.
- Y.-H. Wu, Y.-Y. Lin, J.-L. Chen, S.-Y. Fu, S.-C. Huang, C.-Y. Lee, B.-Y. Chen, G.-C. Yin, E.-W. Huang, M.-T. Tang and B.-H. Lin, J. Synchrotron Radiat., 2022, 29, 456–461 CrossRef CAS PubMed.
- M. S. Brodyn, V. Y. Degoda, N. Y. Pavlova, G. P. Podust, Y. P. Kogut, M. Alizadeh and B. V. Kozhushko, Optik, 2020, 208, 164139 CrossRef CAS.
- V. Degoda, I. Doroshenko, Y. Kogut, H. Podust and N. Pavlova, Results Opt., 2022, 9, 100286 CrossRef.
- R. Beyer, H. Burghardt, F. Firszt and D. R. T. Zahn, J. Appl. Phys., 1998, 84, 5345–5347 CrossRef CAS.
- L. Armelao, F. Heigl, S. Brunet, R. Sammynaiken, T. Regier, R. I. R. Blyth, L. Zuin, R. Sankari, J. Vogt and T.-K. Sham, ChemPhysChem, 2010, 11, 3625–3631 CrossRef CAS PubMed.
- B.-H. Lin, H.-Y. Chen, S.-C. Tseng, J.-X. Wu, B.-Y. Chen, C.-Y. Lee, G.-C. Yin, S.-H. Chang, M.-T. Tang and W.-F. Hsieh, Appl. Phys. Lett., 2016, 109, 192104 CrossRef.
- V. Y. Degoda, N. Y. Pavlova, G. P. Podust and A. O. Sofiienko, Phys. B, 2015, 465, 1–6 CrossRef CAS.
- V. Y. Degoda, G. P. Podust and M. Alizadeh, Adv. Condens. Matter Phys., 2019, 2019, 8784109 Search PubMed.
- K. De Greve, S. M. Clark, D. Sleiter, K. Sanaka, T. D. Ladd, M. Panfilova, A. Pawlis, K. Lischka and Y. Yamamoto, Appl. Phys. Lett., 2010, 97, 241913 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: (hk0) reflection patterns, SEM images together with energy dispersive spectra, powder X-ray diffraction patterns, differential thermal analysis, and UV-vis-NIR reflectance spectra. CCDC 2408439, 2408440, and 2408441. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01251h |
| This journal is © The Royal Society of Chemistry 2025 |