Preparation of a “sliding graft copolymer”, an organic solvent-soluble polyrotaxane containing mobile side chains, and its application for a crosslinked elastomeric supramolecular film†‡
Jun
Araki§
*ab,
Toshiyuki
Kataoka¶
a and
Kohzo
Ito
*ab
aDepartment of Advanced Material Science, Graduate School of Frontier Sciences, University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-City, Chiba 277-8562, Japan. E-mail: jun@shinshu-u.ac.jp; kohzo@molle.k.u-tokyo.ac.jp; Fax: +81-4-7133-0322; Tel: +81-4-7135-6656
bCREST, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 27th November 2007
Abstract
A novel “sliding graft copolymer” (SGC), in which many linear poly-ε-caprolactone (PCL) side chains are bound to cyclodextrin rings of a polyrotaxane, was prepared by ring-opening polymerization of ε-caprolactone initiated by hydroxyl groups of the polyrotaxane. An amorphous, flexible, and sufficiently tough elastomer film was prepared by crosslinking the obtained SGC—a supramolecule possessing a number of mobile side chains—with hexamethylene diisocyanate (HMDI).
Introduction
Polyrotaxanes are the most famous and highly investigated supramolecular materials. They have a necklace-like molecular structure in which many cyclic molecules are threaded onto a linear molecule which has two bulky end groups such that the dissociation of the cyclic molecules is avoided. With their curious characteristics of free sliding and/or rotating of the threaded cyclic molecules, polyrotaxanes have recently attracted increasing attention as raw materials for the production of various substances, in addition to their application in the fundamental research of supramolecular structures.1–3 The recent discovery of inclusion complexation between cyclodextrins (CDs) and linear polymers,4 and subsequent investigation of polyrotaxane preparation from the obtained inclusion complexes (pseudopolyrotaxanes) by using a wide variety of methods, have enabled the facile and efficient production of polyrotaxanes.3b,5,6 In particular, polyrotaxanes prepared from α-CDs, poly(ethylene glycol) (PEG), and bulky end groups have nowadays become one of the most significant and leading candidates in this field of study.Polyrotaxanes have provided various opportunities for material preparation, from nanoscale materials such as “molecular tubes” prepared by the crosslinking of adjacent CDs in a single polyrotaxane followed by dissociation of the included polymer7 and “molecular wires” prepared by the inclusion of a conducting polymer in the molecular tube,8 to large-scale bulky ones such as soft materials having supramolecular polymer networks.9 In addition to the above studies in which polyrotaxanes were used as a single or dominant component, other recent studies have employed polyrotaxanes in combination with other components, particularly other polymers. For example, Wang et al.10 have reported the interesting relaxation and reinforcement effect of polyrotaxanes when incorporated into an epoxy resin matrix. Our recent study11 has also presented the preparation of polyrotaxane–cellulose hybrid fibers and their surprising tensile properties such as elongation up to 190% of the original length. In the future, it is highly expected that the combination of polyrotaxanes and other raw materials will result in new functionalities, in addition to the original characteristics possessed by the associated components. Because the above-mentioned surprising results are thought to be achieved by the sliding motion of the cyclic molecules in polyrotaxanes, it is proposed that these novel materials that have properties that are improved by the addition of polyrotaxanes should be named as “slide-ring materials”.3
To achieve the combination of polyrotaxanes with a wider variety of components, extensive and strict control of polyrotaxane properties such as their solubility in solvents is required. For example, PEG/CD polyrotaxane is soluble only in a limited number of solvents such as DMSO,4 aqueous sodium hydroxide solution,4DMAc–lithium salt system,12 ionic liquids,13 aqueous calcium thiocyanate solution,14 and a specific amine oxide.14 Recently, these difficulties in solubility have been resolved by modifying polyrotaxanes using various methods,12,15 such as the introduction of various types of functional groups onto the CD moiety in polyrotaxanes. These studies have provided various polyrotaxane derivatives that are soluble in general solvents such as water, acetone, THF, chloroform, DMF, pyridine, and so on;12 they have also led to the discovery of the novel thermoreversible gelation of the aqueous solution of methylated polyrotaxane.15
In the course of these continued investigations on the modification of polyrotaxanes, we attempted to prepare a novel type of polyrotaxane having polymeric side chains attached to the cyclic molecules, as represented in Scheme 1. Polyrotaxanes carrying mobile side chains, which are hereafter designated as “sliding graft copolymers” (SGCs), are attractive from both theoretical and practical viewpoints. For example, a theoretical simulation has predicted different conformation states of SGCs, which are dependent on the hydrophobicities/hydrophilicities of the side chains and the polyrotaxane main chain, the lengths of the side chains and the polyrotaxane main chain, or the numbers of side chains.16 The SGC is also expected to be soluble in solvents that are favorable for the side chains due to the solvation of the side chains and steric hindrance, as observed in the polymer brush of colloids.17 These ideas suggest the possibility of controlling the polyrotaxane properties by polymer grafting. The crosslinking of the terminals of the side chains enables the formation of a novel three-dimensional supramolecular network, as shown in Scheme 1.
 | ||
| Scheme 1 Schematic illustration of the preparation of a sliding graft copolymer (SGC) and the crosslinked SGC film. | ||
Although the preparation of the SGC, i.e., the polyrotaxane with sliding side chains, is highly interesting and relevant as stated above, it is our understanding that no instances of this have been reported. To prepare the SGC, two different strategies should be examined, i.e., post-grafting of preformed polymers18 and living polymerization of monomers initiated by the functional groups on the cyclic molecules.19 The latter has been employed for the preparation of polysaccharides with hydrophobic side chains, such as copolymers of dextran,20 cellulose and its derivatives,21pullulan,22 and starch or its analogues.23 In the present study, we report the successful preparation of an SGC using many hydrophobic poly(ε-caprolactone) (PCL) side chains by the ring-opening polymerization of the ε-caprolactone (ε-CL) monomer that is initiated by the hydroxyl groups on the CD moieties in the polyrotaxane, according to the previously reported method for the preparation of hydroxypropyl cellulose grafted with PCL.21b Subsequent crosslinking between the terminals of the PCL side chains provides an elastomer-like SGC film with a supramolecular inner structure.
Materials and methods
Hydroxypropylated polyrotaxane was supplied by Advanced Softmaterials, Inc. (Tokyo, Japan). Based on information provided by the supplier, polyrotaxane was prepared by the hydroxypropylation of PEG/CD polyrotaxane consisting of PEG 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 90–100 hydroxypropylated α-CDs and terminal adamantane moieties—a method very similar to that used in our previous study.12b The properties of hydroxypropylated polyrotaxane, such as molecular weight and molecular substitution of cyclodextrin moiety, are supplied as supplementary information.‡ All other chemicals were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). In addition, ε-CL was stored over molecular sieves 4 Å, and other chemicals were used as received unless otherwise noted.
000, 90–100 hydroxypropylated α-CDs and terminal adamantane moieties—a method very similar to that used in our previous study.12b The properties of hydroxypropylated polyrotaxane, such as molecular weight and molecular substitution of cyclodextrin moiety, are supplied as supplementary information.‡ All other chemicals were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). In addition, ε-CL was stored over molecular sieves 4 Å, and other chemicals were used as received unless otherwise noted.
The SGC was prepared by the so-called “bulk polymerization” of ε-CL monomers, which was initiated by the hydroxyl groups of the hydroxypropylated CDs and catalyzed by tin 2-ethylhexanoate (stannous octoate, Sn(Oct)2). In this method, ε-CL (10 ml) was added to hydroxypropylated polyrotaxane (1 g) that had been exhaustively dried in vacuo; this was followed by 30 min of gentle magnetic stirring under an argon atmosphere. After polyrotaxane was dispersed homogeneously in ε-CL, Sn(Oct)2 (0.2 ml) was added and the mixture was allowed to react in an oil bath at 100 °C for 4 h under a continuous argon flow. The obtained viscous liquid was dissolved in toluene (40 ml) at room temperature and poured with vigorous stirring into hexane (450 ml) to precipitate the SGC and remove the unreacted monomers and the catalyst. After discarding the supernatant and drying in vacuo, the SGC polymer was obtained as an opaque solid with a varied yield (8.5–10.0 g).
A crosslinked SGC film was prepared by dissolving the obtained SGC (250 mg) in anhydrous toluene (5 ml), which was followed by the addition of dibutyltin dilaurate (DBTDL, 50 µl) and hexamethylene diisocyanate (HMDI, 32 µl), casting onto a Teflon petri dish (50 mm in diameter), and drying overnight at room temperature. Crosslinking with HMDI was developed during the evaporation of toluene, yielding a semi-transparent thin film, which was further annealed at 105 °C for 2 h. The obtained film was cooled to room temperature and washed with a sufficient amount of toluene or acetone, which was followed by air drying on a teflon sheet. An uncrosslinked SGC film was prepared simply by casting and drying the SGC–toluene solution without DBTDL and HDMI because the uncrosslinked SGC was melted at an elevated temperature, as described in the following Results and discussion section.
1H NMR spectra were recorded in CDCl3 at 400 MHz on a JEOL JNM-AL400 spectrometer at room temperature, and chemical shifts were referenced by the solvent (7.24 ppm). Gel permeation chromatography (GPC) measurements were conducted using a Shimadzu CLASS-VP chromatograph equipped with a Shodex K-G guard column, Shodex K-800D, and two Shodex K-806L columns, with chloroform as the eluent (40 °C, 1.0 ml min−1); an RI detector and PEG standards were also used. Fourier transform-infrared (FT-IR) spectra were recorded on a Nicolet 4700 FT-IR spectrometer (Thermo Electron Co., Ltd.) equipped with a diamond attenuated total reflection (ATR) accessory (SensIR technologies, DurasamplIR II). Samples were pressed onto a diamond window, and the spectra obtained by 128 scans under air at a resolution of 4 cm−1 were analyzed using the attached OMNIC software. Polarized optical micrographs of the samples were observed with an Olympus BX51 microscope. The mechanical properties of the crosslinked SGC films were measured by tensile measurements with TA Instruments RSA III. The specimens with span lengths and widths of 10 mm and 5 mm, respectively, were measured at the drawing rate of 0.1 mm s−1 under ambient conditions.
Results and discussion
Preparation of the sliding graft copolymer (SGC)
A novel SGC comprising a hydroxypropylated polyrotaxane as the main chain and PCL as the mobile side chains was successfully prepared by the ring-opening polymerization of ε-CL, which was initiated by the hydroxyl groups on the hydroxypropylated CD moiety in the polyrotaxane and catalyzed by Sn(Oct)2. This method has been employed for the preparation of similar types of graft copolymers in which the PCL or poly(lactic acid) side chains are grafted onto the main chain of dextran,20 cellulose derivatives,21pullulan,22 and starch.23 The strategies employed in the previous studies are broadly classified into two parts: a reaction in a solution comprising solvents such as toluene or THF20 and a so-called bulk polymerization in which the parent polymer is dispersed (or dissolved) in a large amount of monomer without any other solvent.21b The unmodified polyrotaxane is known to be soluble only in a limited number of solvents.4,12–14 Our preliminary experiments conducted on the ring-opening polymerization of ε-CL onto unmodified polyrotaxane solution in dry DMSO were unsuccessful, yielding a sample insoluble in solvents of PCL such as toluene or THF even under various reaction conditions (temperature or reaction time). Moreover, bulk polymerization of ε-CL onto unmodified polyrotaxane did not yield a toluene-soluble product even after a sufficient reaction period of 2 h. By using hydroxypropylated polyrotaxane as a starting material, the same reaction showed rapid dissolution of polyrotaxane in monomer ε-CL and the reaction mixture became transparent within the initial 15 min of the reaction. The sample collected by precipitation with hexane after 2 h of reaction at 100 °C showed excellent solubility in toluene and THF, suggesting the successful grafting of a sufficient amount of PCL side chains onto hydroxypropylated polyrotaxane. Considering the above preliminary results, we selected hydroxypropylated polyrotaxane as the starting main chain and bulk polymerization as the reaction scheme. It seems that this difference in reactivity between unmodified and hydroxypropylated polyrotaxane is caused because the inter- and intramolecular hydrogen bonding in the former12–14 prevent the grafting of the monomer, whereas the hydroxyl groups in the latter exhibit a higher accessibility due to a decreased tendency toward aggregation by hydrogen bonding, similar to other modified polyrotaxanes.15As shown in Fig. 1, the GPC measurements of the obtained SGC sample reveal a fraction having a higher molecular weight (component a, Mw = 2.0–2.5 × 105) than that of the starting hydroxypropylated polyrotaxane (Mw = 1.24 × 105, see Table in ESI‡). Fig. 2 shows the FT-IR spectra of the starting hydroxypropylated polyrotaxane and the SGC. The most striking absorption peak in the spectrum of the SGC appears at 1722 cm−1, which is attributed to carbonyl stretching in the PCL side chains.24 Other absorption peaks that appear at 2941, 2865, 1294, and 1240 cm−1 also show a good correspondence with those reported for bulk PCL;24 however, the absorbance from hydroxyl groups at 3430 cm−1 considerably decreases in relation to that of hydroxypropylated polyrotaxane because the hydroxyl groups are present only on the terminals of the PCL side chain in the SGC. Fig. 3 shows the 1H NMR spectrum of the SGC in CDCl3. While strong absorption peaks from the protons in the PCL side chains25 are observed at 1.39, 1.64, 2.31, and 4.06 ppm, peaks from the protons in the polyrotaxane main chain, i.e., protons of PEG (3.5 ppm) and anomeric protons of CDs (4.46 and 5.09 ppm) also appear, although they are very weak and obscured. Resonances caused by the anomeric protons in polysaccharide graft copolymers are reported to split into two peaks,22 which is very similar to our present result. Although the two peaks at 4.46 and 5.09 ppm are considered to be from the C(1)H protons in the anhydroglucose units with and without the grafted PCL chains, detailed information regarding the attribution is currently unclear.
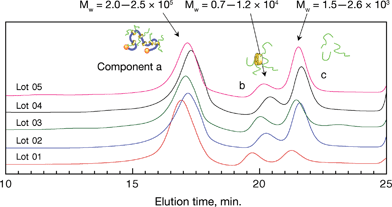 | ||
| Fig. 1 GPC profiles of the as-prepared SGC for different batches. | ||
 | ||
| Fig. 2 FT-IR spectra of (a) the starting hydroxypropylated polyrotaxane, (b) the SGC, and (c) the crosslinked SGC film. | ||
 | ||
| Fig. 3 1H NMR of the SGC (CDCl3, 400 MHz). | ||
From the GPC, FT-IR, and 1H NMR results displayed in Fig. 1–3, successful grafting of the PCL side chains onto hydroxypropylated polyrotaxane is confirmed. However, the GPC profiles for different batches show other fractions with lower molecular weights such as Mw = 0.7–1.2 × 104 and 1.5–2.6 × 103, as shown in Fig. 1 (component b and c). These undesirable fractions with the higher and lower molecular weights are thought to be CDs grafted with PCLs that are dissociated during the reaction and free PCL chains generated by the ring-opening polymerization of CL that is initiated by a trace amount of water in the reaction system, respectively. Similar impurities are also reported in the previous study on hydroxypropyl cellulose grafted with PCL,21b and can be successfully removed by fractional precipitation using a combination of a good and a poor solvent (THF and hexane, respectively). However, our preliminary trial for removal of the low-molecular-weight fractions using fractional precipitation was unsuccessful, and it seems difficult to perform this separation with other methods such as conventional precipitation because the solubility/insolubility of the three fractions including the SGC itself are thought to be almost equal. Considering these difficulties, we have used as-prepared SGC containing the impurities for the following preliminary analysis and investigation of film formation. The removal of fractions with lower molecular weights should be considered in future studies for the investigation of fundamental properties of the SGC as well as for the possibility of changes in the properties of materials prepared from it.
At the present state, it is still difficult to perform a detailed characterization of the SGC, such as the measurement of the number or the length of the side chains, due to problems regarding the removal of the above-mentioned impurities. Although the estimation of the molecular weight of the side chains by means of 1H NMR21b is not applicable, the molecular weight of the side chains is hypothetically estimated to be 1.5–2.6 × 103 if the ring-opening polymerization onto the main polyrotaxane and that of the free PCL are assumed to be initiated simultaneously.
The obtained SGC was soluble in good solvents of PCL, such as toluene, THF, and chloroform. In contrast to unmodified polyrotaxanes or other polyrotaxane derivatives, which do not melt below their thermal decomposition, the SGC melted into a transparent viscous liquid at ca. 60 °C, which is close to the melting point of PCL with a molecular weight of 1.0 × 105.
Preparation and characterization of the crosslinked SGC film
As shown in Fig. 4a, a semi-transparent flexible film could be prepared by crosslinking of the SGC obtained with hexamethylene isocyanate (HMDI) in dry toluene as explained above. The crosslinking reaction with HDMI, which was catalyzed by dibutyltin dilaurate (DBTDL), occurred within the cast solution when it was allowed to stand overnight and was further facilitated by annealing at 105 °C for 2 h. The obtained film did not melt even at 105 °C, in contrast to the starting SGC which melted at ca. 60 °C as described above. The casting of the SGC dissolved in dry toluene without the addition of HDMI was also examined, yielding a brittle opaque film as shown in Fig. 4a. | ||
| Fig. 4 (a) Appearances of the crosslinked (left) and uncrosslinked (right) SGC cast films. (b) Polarized optical micrographs of the crosslinked (left) and uncrosslinked (right) SGC cast films. | ||
The FT-IR spectrum of the crosslinked SGC film is also shown in Fig. 2, which reveals a typical absorbance from carbamate linkage formed by isocyanate crosslinking at 1579, 1620, and 3323 cm−1. The absorbance is almost absent at around 3500 cm−1, which is typical of the hydroxyl groups; this is probably due to the complete consumption of the hydroxyl groups by crosslinking. From the polarized optical micrographs of the crosslinked and uncrosslinked SGC films shown in Fig. 4b, it is observed that the former is completely amorphous, whereas the latter shows separated dark and bright domains, representing amorphous polyrotaxane and semi-crystalline PCL side chains, respectively, which are very similar to the case of hydroxypropyl cellulose grafted with PCL.21b This disappearance of crystallinity in the SGC film after crosslinking seems to be a reason for the flexibility of the crosslinked SGC films, although the detailed mechanism is still unknown.
Typical mechanical properties of the obtained film are summarized in Table 1. Repeated drawing of the film resulted in a hysteresis of the stress–strain profile, which was typically observed for the other elastomeric materials26 (typical stress–strain curve of the film is supplied as supplementary information‡). It is our understanding that this is the first time that the elastomeric properties of a crosslinked SGC film as well as the above-mentioned flexibility have been investigated for dry supramolecular materials; the results are surprising in comparison with the previous results for the similar crosslinked polyrotaxane gel that shows resin-like hardening after drying.9b
| Young's modulus/MPa | Tensile strength/MPa | Strain at break (%) |
|---|---|---|
| 8.66 ± 1.42 | 13.2 ± 5.2 | 310 ± 44 |
Our present preliminary study demonstrates the preparation of a novel graft copolymer with mobile side chains as well as the first elastomeric materials prepared from a supramolecular raw material. The preparation of the SGC readily contributes to the examination of the previously established theoretical treatment.16 The results are also expected to provide new functional materials that possess the excellent advantages of the “slide-ring materials”, such as astonishing physical properties achieved by optimum relaxation of the inner stress.9b,27 However, the physical properties of the film are thought to be affected by a wide variety of unidentified parameters such as the length and number of side chains, the amount of added crosslinkers, and the amount of free PCL chains; these parameters should be further elucidated in future investigations. Other properties such as thermodynamics, viscoelasticity, and biodegradability are appealing aspects of the film and should also be further investigated.
Notes and references
- (a) A. Harada, Coord. Chem. Rev., 1996, 148, 115–133 CrossRef CAS; (b) A. Harada, A. Hashidzume and Y. Takashima, Adv. Polym. Sci., 2006, 201, 1–43.
- (a) T. Takata, N. Kihara and Y. Furusho, Adv. Polym. Sci., 2004, 171, 1–76 CAS; (b) T. Takata, Polym. J., 2006, 38, 1–20 CAS.
- (a) K. Ito, Polym. J., 2007, 39, 489 CAS; (b) J. Araki and K. Ito, Soft Matter, 2007, 3, 1456–1473 RSC.
- (a) A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS; (b) A. Harada, J. Li, T. Nakamatsu and M. Kamachi, J. Org. Chem., 1993, 58, 7524–7528 CrossRef CAS.
- N. Kihara, K. Hinoue and T. Takata, Macromolecules, 2005, 38, 223–226 CrossRef CAS.
- J. Araki, C.-M. Zhao and K. Ito, Macromolecules, 2005, 38, 7524–7527 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1993, 364, 516–518 CrossRef CAS.
- (a) M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS; (b) T. Shimomura, T. Akai and K. Ito, J. Chem. Phys., 2002, 116, 1753–1756 CrossRef CAS.
- (a) G. Fleury, G. Schlatter, C. Brochon and G. Hadziioannou, Polymer, 2005, 46, 8494–8501 CrossRef CAS; (b) Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- X.-S. Wang, H. K. Kim, Y. Fujita, A. Sudo, H. Nishida and T. Endo, Macromolecules, 2006, 39, 1046–1052 CrossRef CAS.
- J. Araki, T. Kataoka, N. Katsuyama, A. Teramoto, K. Ito and K. Abe, Polymer, 2006, 47, 8241–8246 CrossRef CAS.
- (a) J. Araki and K. Ito, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 532–538 CrossRef CAS; (b) J. Araki and K. Ito, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6312–6323 CrossRef CAS.
- S. Samitsu, J. Araki, T. Kataoka and K. Ito, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1985–1994 CrossRef CAS.
- J. Araki, T. Kataoka and K. Ito, J. Appl. Polym. Sci., 2007, 105, 2265–2270 CrossRef CAS.
- (a) M. Kidowaki, C. Zhao, T. Kataoka and K. Ito, Chem. Commun., 2006, 4102–4103 RSC; (b) T. Kataoka, M. Kidowaki, C. Zhao, H. Minamikawa, T. Shimizu and K. Ito, J. Phys. Chem. B, 2006, 110, 24377–24383 CrossRef CAS.
- K. Watariguchi, Y. Sakai and K. Ito, manuscript in preparation for submission.
- J. Araki, M. Wada and S. Kuga, Langmuir, 2001, 17, 21–27 CrossRef CAS.
- (a) R. Gref, J. Rodrigues and P. Couvreur, Macromolecules, 2002, 35, 9861–9867 CrossRef CAS; (b) H. Feng and C.-M. Dong, Biomacromolecules, 2006, 7, 3069–3075 CrossRef CAS.
- T. Ouchi, T. Kontani and Y. Ohya, Polymer, 2003, 44, 3927 CrossRef CAS.
- I. Ydens, D. Rutot, P. Degée, J.-L. Six, E. Dellacherie and P. Dubois, Macromolecules, 2000, 33, 6713–6721 CrossRef CAS.
- (a) Y. Teramoto and Y. Nishio, Biomacromolecules, 2004, 5, 407–414 CrossRef CAS; (b) R. Shi and H. M. Burt, J. Appl. Polym. Sci., 2003, 89, 718–723 CrossRef CAS.
- D. H. Donabedian and S. P. McCarthy, Macromolecules, 1998, 31, 1032–1039 CrossRef CAS.
- (a) E.-J. Choi, C.-H. Kim and J.-K. Park, Macromolecules, 1999, 32, 7402–7408 CrossRef CAS; (b) P. Dubois, M. Krishnan and R. Narayan, Polymer, 1999, 40, 3091–3100 CrossRef CAS.
- T. Elzein, M. Nasser-Eddine, C. Delaite, S. Bistac and P. Dumas, J. Colloid Interface Sci., 2004, 273, 381–387 CrossRef CAS.
- P. Dubois, R. Jerome and P. Teyssie, Polym. Bull., 1989, 22, 475–482 CrossRef CAS.
- W. Wang, P. Ping, H. Yu, X. Chen and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5505–5512 CrossRef CAS.
- (a) T. Karino, M. Shibayama and K. Ito, Physica B, 2006, 385–386, 692–696 CrossRef CAS; (b) T. Karino, M. Shibayama, Y. Okumura and K. Ito, Physica B, 2006, 385–386, 807–809 CrossRef CAS.
Footnotes |
| † The HTML version of this article has been enhanced with colour images. |
| ‡ Electronic supplementary information (ESI) available: Typical stress–strain curve for the crosslinked SGC film; properties of the starting hydroxypropylated polyrotaxane. See DOI: 10.1039/b715231k |
| § Present address: Young Researchers Empowerment Project, Shinshu University, Tokida 3-5-1, Ueda City, Nagano 386-8567, Japan. Fax: +81-268-21-5587; Tel: +81-268-21-5587 |
| ¶ Present address: Department of Material and Life Chemistry, Faculty of Engineering, Kanagawa University, 3-27-1 Rokkakubashi, Kanagawa-ku, Yokohama 221-8686, Japan |
| This journal is © The Royal Society of Chemistry 2008 |