DOI:
10.1039/B711248N
(Paper)
Soft Matter, 2008,
4, 145-155
Synthesis of natural–synthetic hybrid materials from cellulose via the RAFT process†
Received
23rd July 2007
, Accepted 18th October 2007
First published on 5th November 2007
Abstract
The synthesis and characterization of a novel natural–synthetic hybrid material based on cellulose is reported. The reversible addition–fragmentation chain-transfer (RAFT) process was used to graft poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) onto a cellulosic substrate. The weight ratio was increased with an increase in monomer concentration, polymerization time and degree of polymerization (DP). We found that the addition of free chain-transfer agent has a pronounced effect on the weight ratio, chain length of grafted polymer, monomer conversion and homopolymer formation in solution. The cellulose-graft-poly(2-(dimethylamino)ethyl methacrylate) copolymers were characterized by gravimetry, elemental analysis, attenuated total reflectance Fourier transform infrared spectroscopy, scanning electron microscopy, thermal analysis and atomic force microscopy. The dithioester end-group present at the chain end of PDMAEMA was removed viaaminolysis. The livingness of the process was utilized to block-copolymerize styrene from the grafted PDMAEMA chains. The hydrophilic/hydrophobic properties of the novel cellulose-g-(PDMAEMA-b-polystyrene) material were illustrated by contact-angle measurements.
Introduction
Poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) is a well-studied water-soluble cationic polymer, which has been used in a variety of applications. For instance, it was proved to be an efficient gene-transfer agent due to its ability in binding DNA by electrostatic interactions.1 In recent years, considerable interest has grown in the grafting of PDMAEMA and its quaternized derivative from various polymeric surfaces, in order to make functional materials. Indeed, PDMAEMA was grafted onto poly(ether–urethane) membranes2 to favor the immobilization of living cells, onto polytetrafluoroethylene (PTFE), to produce positively charged ultrafiltration membranes for the separation of macromolecular solutes,3 for applications as proton-exchange membranes for fuel cells,4 or to be used as materials for the discoloration of wastewaters.5 Grafting of PDMAEMA from styrene–butadiene–styrene (SBS) triblock copolymer membrane was also undertaken, and led to improved oxygen-permeation properties,6 and increased the blood compatibility of the SBS membrane when treated with heparin.7 Other applications include grafting from natural rubber8 to modify its properties, from polymer microspheres to make permanent antibacterial surface,9 and grafting from poly(vinyl alcohol) for a variety of applications (e.g. flocculating agents in water treatment, retention aids in the manufacture of mineral-filled paper, effective clay or shale hydration inhibitors in oilfield treatment).10
Graft polymerization of PDMAEMA onto nontoxic, biodegradable and biocompatible polysaccharides such as chitosan and cellulose has also been reported in the literature.11,12 The chitosan-g-PDMAEMA copolymer has potential applications as a DNA delivery vector in biological and biomedical fields and as an antibacterial agent in textile industry.12 Kim et al. reported the superior flocculation power of chitosan–PDMAEMA graft copolymer compared with chitosan.13 Waly et al.14 reported the preparation of cellulose ion exchanger by grafting cotton fabric with PDMAEMA , followed by quaternization, in order to remove different dyes and heavy metals from aqueous solutions. Sokker et al.15 also reported the DMAEMA grafting onto a cellulosic fabric waste, which could be used as adsorbent for water pollutants (such as dyes) to solve one of the most important environmental problems of the textile industry.
As a support for polymerization, cellulose is one of the most sought after substrates, due to its availability and biocompatibility. To date, the graft polymerizations of DMAEMA onto cellulose that have been reported in the literature are mainly based on conventional free-radical14,16 or irradiation techniques15,17, which usually lead to uncontrolled molecular weight and broad molecular weight distribution of the grafted chains. However, if the grafted molecule is amenable to living radical polymerization, a fine degree of control over the final properties can be obtained. Recently, nitroxide-mediated polymerization (NMP) and atom-transfer living-radical polymerization (ATRP) method have been used to synthesize well-defined cellulosic graft copolymers with specific properties.18–25 In all of these approaches, the cellulose-g-PDMAEMA was well characterized as a material, but very little information was given on the grafted PDMAEMA chains, mainly because the chains were difficult to cleave from the substrate. For instance, molecular weight and PDI data were not measured, but only estimated via the use of a ‘sacrificial initiator’ added to the polymerization system. Reversible addition–fragmentation chain-transfer (RAFT) polymerization is another versatile method that allows the synthesis of polymers with complex molecular architectures, low polydispersity and functionalizable end groups.26 Recent research into RAFT polymerization is covered by two reviews.27,28 The controlled surface modification of different solid substrates such as poly(propylene) lanterns,29–31 core–shell microspheres,32,33silica particles,34–36gold nanoparticles,37,38 poly(vinylidiene fluoride) films,39–42carbon nanotubes,43,44 Merrifield resins45 and cellulose46–49via RAFT graft polymerization has been reported. Furthermore, as far as we are aware, there are only a few papers available in the literature regarding the RAFT homo and block copolymerization of DMAEMA.26,50–52 This work presents a comprehensive study to produce a cellulose–PDMAEMA hybrid material using RAFT polymerization. The chemical process of PDMAEMA-grafting from cellulose substrate is clarified, via the study of the effect of monomer concentration, polymerization time, degree of polymerization (DP), solvent amount, and free chain-transfer agent (free CTA) in solution on the graft ratio of PDMAEMA attached to cellulose fibers. Furthermore, for the first time, the molecular weight and molecular weight distribution of the grafted chains were measured. The final material was characterized in terms of surface properties and thermal properties. The livingness of the polymerization system was then exploited to produce block copolymers of PDMAEMA and polystyrene, and the effect of the block on the hydrophilicity of the cellulose substrate was demonstrated. We show that novel biocompatible materials based on natural cellulose and synthetic PDMAEMA can be produced via the RAFT system, with an excellent control over grafted chains molecular weight.
Experimental
Materials
All solvents, monomer, and other chemicals were purchased from Aldrich (Gillingham, Dorset, UK) at the highest purity available unless otherwise stated. 2-(Dimethylamino)ethyl methacrylate (DMAEMA) (98%) and styrene were filtered before use through an activated basic alumina (Brockmann I) column. 2,2′-Azobis-(isobutyronitrile) (AIBN) (Fluka, 98%) was purified by recrystallization from methanol and dried at room temperature in a vacuum oven. Hexylamine (Aldrich, 99%) was used as received. The S-methoxycarbonylphenylmethyl dithiobenzoate (MCPDB) RAFT agent was synthesized following a procedure available in the literature.46 A filter paper, Whatman no. 1, was used as the cellulose substrate due to its high cellulose content (>98% alpha cellulose). The synthesis procedure for the cellulose-s-methoxycarbonylphenylmethyl dithiobenzoate RAFT agent (cellulose CTA; DS = 0.63) is described elsewhere.47All other chemicals and solvents were used as received.
The cellulose samples before and after graft polymerization were analyzed for C, H, S and N contents at the Laboratory of Microanalysis, Chemistry Department, the University of Leeds, UK. The C, H and N contents were determined by combustion followed by chromatographic separation and thermal conductivity detection using a Carlo Erba EA 1108 elemental analyzer. The method for sulfur analysis is described elsewhere.47–53
ATR FT-IR spectra of the grafted and ungrafted cellulose samples were obtained on a Perkin-Elmer Spectrum One FT-IR spectrometer using a single reflection horizontal ATR accessory. Each spectrum was collected in the range of 4000–400 cm−1 by cumulating 100 scans at a resolution of 4 cm−1. The scan speed was set at 0.5 cm s−1. Baselines were corrected for all spectra using the Perkin-Elmer Spectrum software.
Raman spectra of cellulose samples, before and after the block extension and aminolysis reaction, were recorded using a Perkin-Elmer system 2000 NIR FT Raman spectrometer. The laser source was a Nd:YAG with diode pumping. The scan speed was set at 0.2 cm s−1 for 100 scans. Baseline was corrected for all spectra using the Perkin-Elmer Spectrum software.
Thermogravimetric analyses on cellulose filter paper, cellulose CTA, PDMAEMA, and cellulose-g-PDMAEMA copolymers were carried out using a TA Instrument TGA 2050 thermogravimetric analyzer under a nitrogen atmosphere. The samples (2–3 mg) were heated from room temperature to 500 °C at a rate of 10 °C min−1. The TA Instruments Thermal Advantage Universal Analysis software was used for calculating the onset and end decomposition temperature.
The surface morphology of the virgin filter paper, cellulose CTA and cellulose-g-PDMAEMA copolymers was observed by SEM using a JEOL JSM-820 scanning microscope operated at an accelerating voltage of 10 Kev. The dried cellulose samples were coated with a 30 nm gold layer using a Bio-Rad diode sputter coating unit. Electron micrographs of each sample were recorded at a magnification of ×2000.
Atomic force microscopy was performed in tapping mode using the Veeco Multomode AFM with a Nanoscope IV controller. A standard flow-through liquid cell was used (volume approximately 200 μl). The tips used for the experiments were sharpened NPS-10 silicon levers with spring constant 2.0 N m−1 (as determined by the thermal method) 54 and resonance frequencies about 164 KHz.
The number-average molecular weight (Mn) and polydispersity index (Mw/Mn)) of free and cleaved poly(2-(dimethylamino)ethyl methacrylate) grafts were performed at Polymer Laboratories, UK, using a SEC system equipped with a differential refractive index (DRI) detector (PL-GPC 50), and two PLgel 5.0 μm MIXED-C columns (300 × 7.5 mm) in series (Polymer Laboratories, UK). THF with 5% triethylamine (TEA) was used as the eluent at a flow rate of 1.0 mL min−1 at ambient temperature. All samples were prepared accurately at nominally 2 mg mL−1. The SEC system was calibrated with polymethylmethacrylate (PMMA) EasiVial narrow standards with molecular weights ranging from 690 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 944000 g mol−1.
1H (400 MHz)) nuclear magnetic resonance (NMR) spectra of free polymer formed in graft polymerization reactions were recorded on a Bruker 400 UltraShield spectrometer at 25°C. Methylene chloride-d2 and d-chloroform were used as solvents. DMAEMA monomer conversion was assessed by measuring the disappearance of the vinyl protons (2 H per mol of monomer) of the monomer by respect to aromatic protons (5 H per mol of toluene) of toluene, which was used as polymerization solvent. Although, there is overlapping of chloroform protons with toluene protons, the contribution of the formers can be neglected. Indeed, the results were also verified using methylene chloride-d2 in place of d-chloroform, and we found a variation of conversion by only 1–2%.
944000 g mol−1.
1H (400 MHz)) nuclear magnetic resonance (NMR) spectra of free polymer formed in graft polymerization reactions were recorded on a Bruker 400 UltraShield spectrometer at 25°C. Methylene chloride-d2 and d-chloroform were used as solvents. DMAEMA monomer conversion was assessed by measuring the disappearance of the vinyl protons (2 H per mol of monomer) of the monomer by respect to aromatic protons (5 H per mol of toluene) of toluene, which was used as polymerization solvent. Although, there is overlapping of chloroform protons with toluene protons, the contribution of the formers can be neglected. Indeed, the results were also verified using methylene chloride-d2 in place of d-chloroform, and we found a variation of conversion by only 1–2%.
General procedure for the RAFT graft polymerization of DMAEMA onto cellulose
Two series of experiments were conducted keeping the initial monomer concentration at 0.5 M and 2.0 M, respectively. In all cases, the molar ratio of monomer, cellulose CTA and initiator (AIBN) was 100 : 1 : 0.1. All polymerizations were performed at 60 °C in septa-sealed round bottom flasks after purging with nitrogen for 10 min. The reactions were stopped after pre-determined time, by cooling the reaction flasks in an ice–water bath, and the polymerization flasks opened to air. The crude cellulose-g-PDMAEMA solid samples were thereafter repeatedly washed with toluene (5 × 150 mL) and THF (5 × 150 mL) to remove surface contaminations such as unreacted monomer, non-attached (free) homopolymer and initiator. The samples were then extracted in THF (2 × 250 mL) using a Soxhlet apparatus for 36 h, to ensure the complete removal of loosely attached (not covalently bound) free PDMAEMA. An aliquot of Soxhlet-extracted solution (50 mL) was also analyzed by SEC (after complete evaporating of solvent) to confirm that there was no more extractable PDMAEMA present in the grafted sample. Finally, the sample was dried to a constant weight under vacuum at 60 °C. The mass gain of the cellulose CTA due to PDMAEMAgraft was determined viagravimetry.
Theoretical number average molecular mass
The theoretical number average molecular masses, Mn,theo were calculated using following the equation. | | | Mn,theo = MCTA + (Mmon× [M]0× Convmon (%)/[CTA]0) | (1) |
Where [M]0, [CTA]0, Convmon (%), Mmon and MCTA are the initial concentration of the monomer and chain-transfer agent, monomer fraction conversion (determined using 1H NMR spectroscopy), molar mass of the monomer and chain-transfer agent, respectively. The contribution of the molar mass of the chains initiated by the thermal initiator (AIBN) was neglected.
Weight ratio
The weight ratio (wt%) of each PDMAEMA-grafted filter paper was calculated using the following equation. | |  | (2) |
Where “weightgraft” is the dry weight of each cellulose CTA sample after grafting with PDMAEMA and Soxhlet extraction with tetrahydrofuran and “weightcell–CTA” is the initial weight of each cellulose CTA sample.
General procedure for the raft graft polymerization DMAEMA onto cellulose with the presence of free chain-transfer agent
All polymerizations were performed at 60 °C in toluene with a thermal initiator (AIBN). The molar ratios of cellulose CTA to free CTA (MCPDB) ([cell–CTA]0/[free CTA]0) varied from 1 : 0.5 to 1 : 2.5. A series of experiments was conducted keeping the initial monomer-to-cellulose–CTA ratio constant at 100 : 1 and initial monomer concentration at 2.0 M. Another batch of reactions was performed using different monomer to cellulose–CTA ratio ([M]0–[cell–CTA]0 of 50 : 1 and 25 : 1) using the same initial monomer concentration (2.0 M). Another batch was also carried out using different monomer-to-cellulose CTA ratio ([M]0–[cell–CTA]0 of 200 : 1, 100 : 1 and 50 : 1) keeping the amount of solvent constant. In all cases, the ratio cellulose–CTA-to-initiator ([cell–CTA]0–[AIBN]0) was 1 : 0.1. The percentage monomer conversion in solution was measured by 1H NMR spectroscopy. The polymer solution was precipitated into a large amount of cold hexane. The supernatant was removed and the polymer re-dissolved in dichloromethane and re-precipitated into cold hexane. Finally, the viscous polymer was dried overnight under reduced pressure. Molecular weight and polydispersity index of free PDMAEMA were determined by SEC. The weight ratio of each PDMAEMA-grafted filter paper was calculated using eqn (2).
In a typical reaction (Entry C2, Table 1), cellulose–CTA sample (0.167 g, RAFT agent loading = 0.317 mmol) was immersed in a round bottom flask containing toluene (9.0 g) as the solvent. 2-(Dimethylamino)ethylmethacrylate (31.7 mmols, 5.0 g), free CTA (MCPDB) (0.317 mmols, 0.096 g) and 2,2′-azobis(isobutyronitrile) (0.0317 mmol, 0.005 g) were then added in the flask in ratios of 100 : 1 : 0.1 The molar ratios of cellulose CTA to free CTA ([cell–CTA]0–[free CTA]0) was set at 1 : 1. The reaction flask was deoxygenated with nitrogen for approximately 15 min and then placed in a preheated oil bath at 60 °C. The initial monomer concentration was kept at 2.0 M. The reaction was stopped after 20 h by cooling the flask in an ice–water bath and exposed to air. The solid product was first washed with toluene, DCM, THF, water, ethanol followed by Soxhlet extraction with THF for 24 h. The percentage monomer conversion was measured by 1H NMR spectroscopy (conversion ∼66%). The weight ratio was calculated using eqn (2) after drying the sample at 40 °C under vacuum for 24 h. The weight ratio obtained = 17 wt%.
| Samples |
Monomer : cellulose–CTA : free CTA : initiator/mol |
Monomer conversion in solution (%) |
Weight ratio (wt%) |
N-content (wt%) |
|
Reaction time = 20 hours; monomer concentration = 2.0 M
|
| B3 |
100 : 1 : 0 : 0.1 |
78 |
10.0 |
1.3 |
| C1 |
100 : 1 : 0.5 : 0.1 |
67 |
15.0 |
1.7 |
| C2 |
100 : 1 : 1.0 : 0.1 |
66 |
17.0 |
2.0 |
| C3 |
100 : 1 : 1.5 : 0.1 |
63 |
21.0 |
2.2 |
| C4 |
100 : 1 : 2.0 : 0.1 |
55 |
20.0 |
2.1 |
| C5 |
100 : 1 : 2.5 : 0.1 |
51 |
19.0 |
2.0 |
General procedure for the RAFT graft polymerization of DMAEMA onto cellulose without free chain-transfer agent
The same procedure as described above was followed. In a typical reaction, cellulose–CTA sample (0.167 g, RAFT agent loading = 0.317 mmol) was immersed in a round bottom flask containing toluene (9.0 g) as the solvent. 2-(Dimethylamino)ethylmethacrylate (31.7 mmols, 5.0 g) and 2,2′-azobis(isobutyronitrile) (0.0317 mmols, 0.005 g) were then added to the flask in a ratio of 100 : 0.1. The reaction flask was deoxygenated with nitrogen for approximately 15 min and then placed in a preheated oil bath at 60 °C. The initial monomer concentration was kept at 2.0 M. The reaction was stopped after 20 h by cooling the flask in an ice–water bath and exposed to air. The solid product was first washed with toluene, DCM, THF, water, ethanol followed by Soxhlet extraction with THF for 24 h. The percentage monomer conversion was measured by 1H NMR spectroscopy (conversion ∼78%). The weight ratio was calculated using eqn (2) after drying the sample at 40 °C under vacuum for 24 h. The weight ratio obtained = 10 wt%.
Removal of RAFT end group from the cellulose-g-PDMAEMAviaaminolysis
A typical aminolysis experiment was carried out as follows. The cellulose-g-PDMAEMA sample with 15% weight ratio (0.09 g, loading of immobilized RAFT agent = 0.17 mmol) (Entry C1, Table 1) was immersed into a round bottom flask containing tetrahydrofuran (10 mL) for 4 h. The septa-sealed flask was deoxygenated with nitrogen for approximately 10 min. An excess amount of hexylamine (1.9 mmol, 0.19 g,) was then added into solution under nitrogen. The reaction was left at room temperature for 24 h. The sample was repeatedly washed with THF and Soxhlet extracted with THF for 16 h, and finally dried under vacuum at 60 °C to constant weight.
Cleaving of PDMAEMA chains from cellulose backbone for molecular weight analyses
Poly(DMAEMA) chains were cleaved from the cellulose backbone under acidic conditions. In a typical acid hydrolysis experiment, 0.2 g cellulose-g-poly(DMAEMA) sample were immersed into a round bottom flask containing 12 mL of 6 M HCl aqueous solution. The flask was stirred at 100 °C for 72 h. The reaction mixture was filtered to separate the solid cellulose particles, and the HCl aqueous solution was removed by evaporation. The remaining polymer was analyzed by NMR and IR (see ESI† for typical spectra). It was found that some DMAEMA repeating units were hydrolyzed to acrylic acid, but the degree of hydrolysis was so low that it could be neglected in the molecular weight determination by SEC analyses. The polymer was dissolved in organic GPC eluent (THF + 5 v/v% triethylamine + 0.5 v/v% toluene as internal standard) and the molecular weight (Mn) and molecular weight distribution (Mw/Mn) were analyzed using PMMA calibration.
Block extension of styrene from the PDMAEMA-grafted cellulose fibers (synthesis of cellulose-g-PDMAEMA-b-PS)
In a typical chain-extension reaction, the cellulose-g-PDMAEMA sample (0.43 g, RAFT agent loading = 0.65 mmol) was immersed in a round bottom flask containing toluene (12.4 g) as the solvent. Styrene (65.0 mmols, 6.8 g), free CTA (MCPDB) (0.65 mmols, 0.2 g) and 2,2′-azobis(isobutyronitrile) (0.06 mmol, 0.01 g) were then added to the flask in ratios of 100 : 1 : 0.1 The molar ratios of cellulose CTA to free CTA ([cell–CTA]0–[free CTA]0) was set at 1 : 1. The reaction flask was deoxygenated with nitrogen for approximately 15 min and then placed in a preheated oil bath at 60 °C. The initial monomer concentration was kept at 3.0 M. The reaction was stopped after 36 h by cooling the flask in an ice–water bath and exposed to air. The solid product was washed thoroughly with toluene and THF, followed by Soxhlet extraction with THF for 24 h. The percentage monomer conversion was measured by 1H NMR spectroscopy (conversion ∼25%). The weight ratio was calculated using eqn (2) after drying the sample at 60 °C under vacuum for 24 h. The weight ratio obtained = 20 wt%.
Results and discussion
Cellulose chain-transfer agent
RAFT graft polymerization from a solid support can be performed using both the R-group approach (the RAFT agent is attached to the support via the leaving and re-initiating R group), and the Z-group approach (the RAFT agent is attached to the support via the stabilizing Z group). The R-group approach allows for higher graft densities at the surface by comparison to those achieved using the Z-group approach, which suffers from hindrance problems.45,49 In this study, the RAFT agent (MCPDB) was attached to the cellulose fiber surface via its R group, in order to form a cellulose-supported chain-transfer agent (Scheme 1).
The kinetics of DMAEMA polymerization from cellulose was investigated for two monomer concentrations (0.5 and 2.0 M). Weight ratios were determined from elemental analyses and gravimetrically. Gravimetric analyses were performed following Soxhlet extraction of non-covalently bound homopolymer with a suitable solvent, and evaporation of solvent from the cellulose samples. Both techniques agree remarkably, as we observed that the increase in %N content (from the pendant amine group of DMAEMA) follow the increase in grafting determined by gravimetry (Table 2). The presence of PDMAEMA chains grafted on cellulose was confirmed by ATR FT-IR spectroscopy. Prior to analyses, the modified cellulose substrate was submitted to Soxhlet extraction in the presence of tetrahydrofuran in order to remove the unreacted DMAEMA and homo-PDMAEMA that may be physically adsorbed in the cellulose matrix. Fig. 1 shows the ATR FT-IR spectrum of a homo-PDMAEMA prepared by RAFT polymerization, a cellulose-g-PDMAEMA and a cellulose CTA sample. The IR Spectrum (Fig. 1b) of the cellulose-g-PDMAEMA shows the appearance of new bands at 2941 cm−1 (for C–H stretching of methyl and –CH2– groups), 2819 cm−1, 2767 cm−1 (for C–H stretching of –N(CH3)2group), 1454 cm−1 (for –CH2– bending) and 1144 cm−1 (for C–N stretching) by comparison to that of cellulose CTA, and these bands are characteristic peaks of PDMAEMA (Fig. 1a). Furthermore, there is no intense band appearing in these spectral regions from the cellulose–CTA sample (Fig. 1c). The peak for ester group at 1747 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) in the cellulose CTA sample is also moved to about 1723 cm−1, characteristic of the CO stretching of PDMAEMA.
O stretching) in the cellulose CTA sample is also moved to about 1723 cm−1, characteristic of the CO stretching of PDMAEMA.
| Samples |
Monomer : cellulose–CTA : initiator/mol |
Time/h |
Monomer conversion in solution (%) |
Weight ratio (wt%) |
N-content (wt%) |
|
Monomer concentration = 0.5 M.
Monomer concentration = 2.0 M.
|
| A1a |
100 : 1 : 0.1 |
4 |
13 |
1.0 |
— |
| A2a |
100 : 1 : 0.1 |
8 |
32 |
3.0 |
— |
| A3a |
100 : 1 : 0.1 |
20 |
60 |
6.0 |
— |
| A4a |
100 : 1 : 0.1 |
48 |
75 |
10.0 |
— |
| B1b |
100 : 1 : 0.1 |
4 |
29 |
7.0 |
0.80 |
| B2b |
100 : 1 : 0.1 |
8 |
48 |
8.0 |
0.92 |
| B3b |
100 : 1 : 0.1 |
20 |
78 |
10.0 |
1.30 |
| B4b |
100 : 1 : 0.1 |
48 |
94 |
20.0 |
2.30 |
The kinetics of PDMAEMA grafting was also followed viaATR FT-IR spectroscopy. Fig. 2 shows an overlap of ATR FT-IR spectra of the cellulose-g-PDMAEMA copolymers recorded at various time intervals, and it is clear that the intensity of the characteristic peaks of the PDMAEMA at around 2941, 2819, 2767, 1723, 1454 and 1144 cm−1 increase with polymerization time. Furthermore, when comparing polymerization using the monomer concentrations 0.5 and 2.0 M, it is clear that the same peaks increase with monomer concentration.
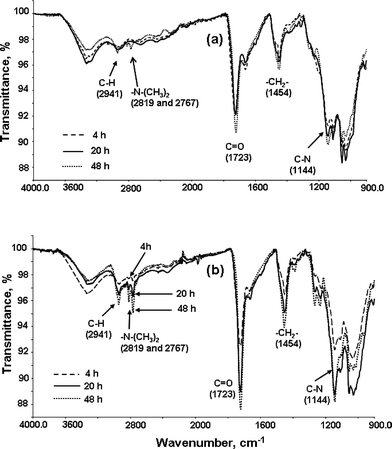 |
| | Fig. 2 Overlay of the ATR FT-IR spectra of cellulose-g-PDMAEMA copolymers (a) synthesized using 0.5 M monomer concentration; (b) synthesized using 2.0 M monomer concentration. | |
We observed a near to linear increase of the weight ratio with time and monomer conversion for both monomer concentrations, suggesting that the weight ratio could be predicted depending on the time of reaction (Fig. 3). A higher weight ratio was obtained for the highest initial monomer concentration, with a pronounced increase being observed in the very early stage of the polymerization. It is likely that at high monomer concentration, we observe a loss of control over the polymerization early in the reaction, leading to uncontrolled polymeric chains growing from the surface, before the RAFT equilibrium is established, and control over polymer growth is obtained. The polymerization for higher monomer concentration was faster, with 94% conversion obtained in 48 hours for a monomer concentration of 2.0 M, whilst 75% conversion was reached in the same time for a monomer concentration of 0.5 M.
 |
| | Fig. 3 Weight ratio versuspolymerization time for an initial monomer concentration of 0.5 M (■) and 2.0 M (▲). The dashed line is a line of best fit. | |
Table 3 Molecular weight and PDI value of homo, free and grafted PDMAEMA
| Samples |
Monomer : cellulose–CTAa: freeCTAa : initiator |
Monomer conversion (%)d |
M
n,theo
d /g mol−1 |
M
n
e /g mol−1 |
M
w
e /g mol−1 |
PDIe |
|
CTA: chain-transfer agent.
PDMAEMA in solution obtained from RAFT graft polymerization from cellulose.
Hydrolyzed PDMAEMA grafted from cellulose viaRAFT polymerization.
Determined using 1H NMR spectroscopy.
Measured by SEC.
|
|
Poly(DMAEMA)freeb |
100 : 1 : 0 : 0.1 |
78 |
12500 |
60100 |
116900 |
1.94 |
|
Poly(DMAEMA)graftc |
100 : 1 : 0 : 0.1 |
78 |
12500 |
4100 |
5600 |
1.36 |
|
Poly(DMAEMA)freeb |
100 : 1 : 1.5 : 0.1 |
63 |
4300 |
16600 |
21400 |
1.29 |
|
Poly(DMAEMA)graftc |
100 : 1 : 1.5 : 0.1 |
63 |
4300 |
3000 |
3400 |
1.14 |
|
Poly(DMAEMA)freeb |
100 : 1 : 2.5 : 0.1 |
51 |
2600 |
12500 |
15850 |
1.27 |
| poly(DMAEMA)graftc |
100 : 1 : 2.5 : 0.1 |
51 |
2,600 |
2700 |
3100 |
1.15 |
A series of graft polymerization was carried out with the addition of free chain-transfer agent (MCPDB) to control the growth of the grafted polymeric chains on cellulose. We observed that the addition of free chain-transfer agent not only permit to control the molecular weight and molecular weight distribution of the free polymer in solution, but also allows for an increase in weight ratio (Table 2) by comparison to a system without free CTA. As expected, an increase in the concentration in CTA in solution leads to a decrease in Mn, for both grafted and free PDMAEMA. We observe (Table 2) that an increase in free CTA leads to lower monomer conversions for a given time (20 hours). This can be explained by the effect of retardation, usually observed in RAFT polymerization mediated by dithiobenzoates. However, as a general trend, we see that the weight ratio either increases or remains constant with decreasing conversion (and increasing concentration in free CTA). This suggests that the overall weight ratio increases with increase in free CTA, and this is conflicting with the information provided by table 3, which shows that an increase in CTA seems to lead to a decrease in molecular weight of the grafted chains. In this system, the number of grafted chains remains constant, as they are initiated from the surface by the R group of the CTA. Therefore, an increase in weight ratio should only originate from grafted chains extending their molecular weight. However, it is possible that the free CTA adsorbs at the surface of the substrate viahydrogen bonding between its carbonyl group and the cellulose OH groups, thus increasing the number of chains attached to the surface, whilst lowering their molecular weight. Another potential explanation is related to the RAFT process itself. It has been shown that the localized high concentration in CTA at the surface of a substrate can be at the origin of a phenomenon of ‘radical hopping’, which can lead to termination reactions between surface-immobilized chains.34 These terminations can occur early in the reaction, thus reducing the number of growing polymeric chains. Addition of free CTA can effectively prevent radical hopping (and therefore termination reactions), thus increasing the number of living chains by comparison to polymerization with a lower concentration in free CTA. It is remarkable that the addition of free chain-transfer agent has a direct effect on the molecular weight of the grafted chains, with their molecular weight getting very close to that predicted, and their PDI being lowered to values as low as 1.15, whilst the PDIs of free chains remain unaffected (∼1.3).
Finally, we observed that the monomer conversion decreases slightly with the addition of free CTA in solution (Table 2). For instance, 63% conversion was reached in 20 hours for a system using an optimal ratio of free CTA–cellulose CTA (1.5 : 1) whilst 78% was reached when no free chain-transfer agent is used. Once again, the PDMAEMA grafting was assessed by both gravimetric and %N content analyses, and both techniques agreed well.
The effect of free chain-transfer agent addition onto weight ratio strongly suggests that the polymerization occurring at the cellulose surface is indeed controlled by the RAFT process. In order to confirm this observation, three degrees of polymerization (DPs) were targeted by changing the ratio monomer : cellulose–CTA, whilst keeping the ratio cellulose–CTA : free CTA = 1 : 1.5 (table 4). We observe that a decrease in monomer concentration leads to slower polymerizations (for the same period of time, conversions decrease with decreasing monomer concentration) and this also affects the grafting ratio, which is lowered with conversion. This decrease in rate when decreasing monomer concentration is also observed in conventional free-radical polymerization, and in this specific case, could also be explained by the rate-retardation effect discussed above. Fig. 4 shows overlay of ATR-FTIR spectra (in the range from 2850 to 2700 cm−1) of cellulose-g-PDMAEMA copolymers with different DP (targeted). As expected, the intensity of the peaks of PDMAEMA at 2819 and 2767 cm−1 due to –N(CH3)2group increase with the DPtargeted values.
 |
| | Fig. 4 Overlay of the ATR FT-IR spectra of cellulose-g-PDMAEMA copolymers with different targeted DP values. | |
Table 4
Graft polymerization of DMAEMA varying the monomer unit (monomer concentration constant, 2.0 M)
| Samples |
Monomer : cellulose–CTA : free CTA : initiator /mol |
Monomer concentration/M |
Monomer conversion in solution (%) |
Weight ratio (wt%) |
N-content (wt%) |
|
Reaction time = 24 hours.
|
| D1 |
200 : 1 : 1.5 : 0.1 |
1.9 |
51 |
22.0 |
2.4 |
| D2 |
100 : 1 : 1.5 : 0.1 |
1.1 |
42 |
11.0 |
1.3 |
| D3 |
50 : 1 : 1.5 : 0.1 |
6.0 |
38 |
2.8 |
0.8 |
Thermal analysis
Thermogravimetric analyses (TGA) were carried out in order to evaluate the effect of the polymer grafting on the thermal stability of cellulose fiber at different PDMAEMA weight contents. Fig. 5 shows the thermograms of unmodified filter paper, cellulose CTA, PDMAEMA and cellulose-g-PDMAEMA copolymers with different weight ratios. We observe that unmodified filter paper is more thermally stable than cellulose CTA, grafted fibers and PDMAEMA, and that the degradation temperature of grafted cellulose samples decreased as PDMAEMA content increased. Furthermore, unmodified filter paper and cellulose CTA show a single, well-defined decomposition curve with an extrapolated onset temperature at ∼347 and ∼248 °C, respectively, whereas we observe a two-step degradation profile for cellulose-g-PDMAEMA samples. For instance, a typical graft copolymer sample (cellulose-g-PDMAEMA, 48 h polymerization, and 20 wt% weight ratio) exhibited two thermal decomposition peaks at ∼197 and ∼310 °C (Fig. 5). A similar TGA profile was also observed for grafted samples with different targeted DP (Fig. 6). In both cases, the decomposition temperature is lowered when the molecular weight of PDMAEMA is increased. It is also noteworthy that PDMAEMA synthesized via RAFT has a three-step degradation profile where the first degradation step at ∼167 °C, the second step at ∼306 °C and the third step at ∼403 °C were observed. The first degradation step may be associated with the decomposition of the dithioester end-group of the polymeric chain. Previous work by our group has shown that dithiobenzoates decompose in this range of temperatures.55 We explain the decomposition of cellulose CTA and cellulose-g-PDMAEMA at lower temperature than cellulose by the presence of the dithioester functional group, which is cleaved at lower temperatures and triggers the decomposition process. It should be noted that the occurrence of degradation on a cellulosic compound due to graft polymerization of methyl methacrylate has been previously reported before by Román-Aguirre et al.56 The authors observed that degradation temperature of grafted fiber samples decreased as PMMA content increased.
Surface morphology
The surface morphology of the cellulose-g-PDMAEMA fibers were examined with SEM (Fig. 7). The apparent surface morphology of unmodified cellulose fiber and CTA-immobilized cellulose fiber show no apparent modification. On the other hand, we observed an increase in polymer present at the surface of the cellulose fiber when increasing weight ratio, as noticed by Karlsson and Gatenholm.57,58
 |
| | Fig. 7
SEM
photomicrographs of (a) virgin cellulose filter paper; (b) cellulose CTA; (c) cellulose-g-PDMAEMA (7% weight ratio, 4 h polymerization); (d) cellulose-g-PDMAEMA (10% weight ratio, 20 h polymerization); (e) cellulose-g-PDMAEMA) (20% weight ratio, 48 h polymerization). | |
The surface morphology of PDMAEMA-grafted filter papers were also investigated by AFM. Fig. 8 shows the height and phase images of unmodified and grafted cellulose surface. AFM reveals the fibrils of unmodified cellulose as shown in Fig. 8(a). The morphology of the cellulose-g-PDMAEMA (Fig. 8(b)) sample appears modified, which confirms the coverage of the surface by the grafted polymer. The phase image of the grafted sample also showed that the there is some material variations from spot to spot, and grafting does not completely cover the substrate. The fine structure on the right-hand side of phase image (Fig. 8(b)) suggested that the coating is comparatively thin in this region. In other areas, the coating appeared to be thick enough to hide the fibril. This observation, coupled with the narrow PDIs observed for the grafted polymeric chains, show that polymer growth does not occur regularly at the surface of the cellulose substrate, certainly due to the irregular presence of CTA on the fibers. A similar AFM surface morphology of grafted cellulose fiber was observed by Karlsson and Gatenholm,57 and Carlmark et al.20
 |
| | Fig. 8
AFM images of (a) virgin cellulose filter paper and (b) paper grafted with PDMAEMA (10% weight ratio, 20 h polymerization). | |
Removal of color from the cellulose-g-PDMAEMA copolymersviaaminolysis
The cellulose CTA and grafted cellulose showed a strong red color due to the presence of dithioester end group. In order to remove the colour, whilst introducing functional end-groups at the end of the grafted PDMAEMA, we performed an aminolysis reaction, as previously reported by Rizzardo et al.59 and Lima et al.60 We observed that an excess amount of hexylamine removed the red color from the cellulose, to give an orange color, close to the color of the esterified cellulose used as an intermediate for the synthesis of the cellulose CTA.47 The removal of the dithioester group was confirmed by elemental analyses, ATR FT-IR and Raman spectroscopy (see ESI†). The thermal analyses (Fig. 9) of cellulose-g-PDMAEMA before and after aminolysis showed that the thermal stability of the grafted sample was increased after aminolysis due to the removal of the labile dithioester functionality. The increased thermal stability of polymer due to the absence of dithioester function was also observed by Lima et al.60
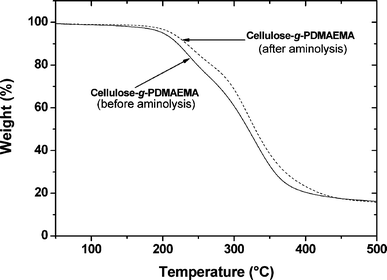 |
| | Fig. 9
TGA curves of PDMAEMA-grafted cellulose before and after aminolysis. | |
Formation of a block-copolymer on the cellulose-g-PDMAEMA surface
The livingness of the PDMAEMA-grafted chains was tested by block extension with styrene. An increase in mass was observed after styrene polymerization, with a weight ratio of cellulose-g-PDMAEMA-b-PS at 20 wt-%. ATR FT-IR spectroscopy of cellulose-g-PDMAEMA-b-PS (Fig. 10) also confirmed the formation of the polystyrene block by showing the characteristic peaks of PS at 3082, 3061, 3027 (aromatic C–H vibration); 1601, 1493 (aromatic ring vibration); 758, 696 cm−1 (deformation vibration of the –CH– group of the monosubstituted benzene ring). The cellulose-g-PDMAEMA-b-PS surface was also characterized by FT-Raman spectroscopy (see ESI†) and the increase of intensities of peaks due to monosubstituted aromatic ring of polystyrene was observed. The cellulose-g-PDMAEMA-b-PS surface was also characterized by TGA (Fig. 11). It was observed that the decomposition temperature was increased after chain extension with polystyrene, confirming successful block formation.61
 |
| | Fig. 11 Overlay of TGA curves of cellulose-g-PDMAEMA and cellulose-g-PDMAEMA-b-PS | |
The molecular weight of the PS in solution was determined at 5000 g mol−1 with a PDI of 1.07. The molecular weight of the newly formed block copolymers could not be determined, as the presence of PS chains increased dramatically the hydrophobicity of the cellulose substrate, making its degradation difficult. The degrees of hydrophobicity of the cellulose-g-PDMAEMA and cellulose-g-PDMAEMA-b-PS were tested via contact-angle measurements. The cellulose-g-PDMAEMA surface was found to adsorb the water droplets very quickly, due to the hydrophilic nature of PDMAEMA. However, the cellulose-g-PDMAEMA-b-PS showed an enhanced hydrophobicity, as illustrated in Fig. 12. The results suggest that the hydrophilic nature of PDMAEMA-grafted cellulose surface has been altered due to the hydrophobic nature of the polystyrene layer. The surface morphology of cellulose-g-PDMAEMA-b-PS was also observed by SEM (see ESI†). It was observed that the surface morphology of graft block copolymer is quite different from that of the graft copolymer. The surface of graft block copolymer looked smoother and this may be due to the formation of a second layer of polystyrene over the first layer of cellulose-g-PDMAMEA.
 |
| | Fig. 12 Photographs showing the behavior of a water droplet at the surface of (a) cellulose-g-PDMAEMA and (b) cellulose-g-PDMAEMA-b-PS. | |
Conclusions
In this work, we have demonstrated an easy technique to control the grafting of poly(2-(dimethylamino)ethyl methacrylate) from cellulose fibers surface via the RAFT process. The graft ratio was increased when increasing monomer concentration, polymerization time and degree of polymerization. SEM photomicrographs, AFM images and thermogravimetric decomposition patterns of cellulosic graft copolymer confirmed the successful grafting of poly(2-(dimethylamino)ethyl methacrylate) onto cellulose, and illustrated the properties of this novel hybrid natural–synthetic material. Grafted block copolymers of PDMAEMA and polystyrene were also produced, and the hydrophilic/hydrophobic properties of the resultant material were demonstrated. Finally, aminolysis reaction was shown to effectively remove the characteristic red color of the cellulosic graft copolymer formed by RAFT polymerization.
Acknowledgements
D.R. acknowledges the Department of Color and Polymer Chemistry, the University of Leeds, for financial support. The authors are grateful to Mr Kazlauciunas for assistance with the thermogravimetric analysis and SEM photographs, Dr Ian Willoughby, Polymer Laboratories, for GPC data, and Professor Simon Biggs and Dr Chris Hodges, Institute of Particle Science and Engineering, at the University of Leeds, for AFM images.
References
- P. Van de Wetering, J.-Y. Cherng, H. Talsma, D. J. A. Crommelin and W. E. Hennink, J. Controlled Release, 1998, 53, 145–153 CrossRef.
- J. Guan, C. Gao, L. Feng and J. Shen, Eur. Polym. J., 2000, 36, 2707–2713 CrossRef CAS.
- K. Yamada, T. Gondo and M. Hirata, J. Appl. Polym. Sci., 2001, 81, 1595–1604 CrossRef CAS.
- Y. Yan, M. Yi, M. Zhai and H. Ha, Gaofenzi Xuebao, 2004, 6, 871–876 Search PubMed.
- K. Yamada, C. Takagi and M. Hirata, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2003, 44, 926–927 CAS.
- J. M. Yang, C. P. C. Chian and K. Y. Hsu, J. Membr. Sci., 1999, 153, 175–182 CrossRef CAS.
- J. M. Yang, Y. J. Jong, K.Y. Hsu and C. H. Chang, J. Biomed. Mater. Res., 1998, 39, 86–91 CrossRef CAS.
- P. C. Oliveira, A. Guimaraes, J.-Y. Cavaille, L. Chazeau, R. G. Gilbert and A. M. Santos, Polymer, 2005, 46, 1105–1111 CrossRef CAS.
- Z. Cheng, X. Zhu, Z. L. Shi, K. G. Neoh and E. T. Kang, Ind. Eng. Chem. Res., 2005, 44, 7098–7104 CrossRef CAS.
- S.Y. Zheng, Z. C. Chen, D. S. Lu, Q. Wu and X. F. Lin, J. Appl. Polym. Sci., 2005, 97, 2186–2191 CrossRef CAS.
- D. K. Singh and A. R. Ray, J. Appl. Polym. Sci., 1997, 66, 869–877 CrossRef CAS.
- J. Liang, P. Ni, M. Zhang and Z. Yu, J. Macromol. Sci., Pure Appl. Chem., 2004, 41, 685–696 CrossRef.
- C.-H. Kim, J.-W. Lee, B.-O. Jung, B.-K. Chang, K.-S. Choi and J.-J. Kim, Kongop Hwahak, 1995, 6, 130–139 Search PubMed.
- A. Waly, F. A. Abdel-Mohdy, A. S. Aly and A. Hebeish, J. Appl. Polym. Sci., 1998, 68, 2151–2157 CrossRef CAS.
- H. H. Sokker, E. S. Abdel Halim, A. S. Aly and A. Hashem, Adsorption Sci. Technol., 2004, 22, 679–691 Search PubMed.
- L.-M. Zhang and Y.-B. Tan, Macromol. Mater. Eng., 2000, 280(281), 59–65 CrossRef.
- L. Jun, Y. Min, L. Jiuqiang and H. Hongfei, J. Appl. Polym. Sci., 2001, 81, 3578–3581 CrossRef.
- W. H. Daly, T. S. Evenson, S. T. Iacono and R. Walker Jones, Macromol. Symp., 2001, 174, 155–163 CrossRef CAS.
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS.
- A. Carlmark and E. Malmström, Biomacromolecules, 2003, 4, 1740–1745 CrossRef CAS.
- Q. Zhou, L. Greffe, M. J. Baumann, E. Malmstrom, T. T. Terri and H. Brumer, Macromolecules, 2005, 38, 3547–3549 CrossRef CAS.
- I. Ikeda, T. Higuchi and Y. Maeda, Sen'i Gakkaishi, 2002, 58, 308–313 Search PubMed.
- M. Coskun and M. M. Temuz, Polym. Int., 2005, 54, 342–347 CrossRef CAS.
- S. B. Lee, R. R. Koepsel, S.W. Morley, K. Matyjaszewski, Y. Sun and A. J. Russell, Biomacromolecules, 2004, 5, 877–882 CrossRef CAS.
- Emma Oestmark, Simon Harrisson, L. Wooley Karen and E. Malmstroem Eva, Biomacromolecules, 2007, 8(4), 1138–1148 CrossRef CAS.
- J. Chiefari, B.Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- L. Barner, N. Zwaneveld, S. Perera, Y. Pham and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4180–4192 CrossRef CAS.
- L. Barner, S. Perera, S. Sandanayake and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 857–864 CrossRef CAS.
- L. Barner, J. F. Quinn, C. Barner-kowollik, P. Vana and T. P. Davis, Eur. Polym. J., 2003, 39, 449 CrossRef CAS.
- R. Joso, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and L. Barner, Aust. J. Chem., 2005, 58, 468–471 CrossRef CAS.
- L. Barner, C. Li, X. Hao, M. H. Stenzel, C. Barner-Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5067–5076 CrossRef CAS.
- Y. Tsujii, M. Ejaz, K. Sato, A. Goto and T. Fukuda, Macromolecules, 2001, 34, 8872–8878 CrossRef CAS.
- M. Baum and W. J. Brittain, Macromolecules, 2002, 35, 610–615 CrossRef CAS.
- L. Chunzhao and B. C. Benicewicz, Macromolecules, 2005, 38, 5929–5936 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, P. Zhang, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559–5562 CrossRef CAS.
- A.-S. Duwez, P. Guillet, C. Colard, J.-F. Gohy and C.-A. Fustin, Macromolecules, 2006, 39, 2729–2731 CrossRef.
- Y. Chen, L. Ying, W. H. Yu, E. T. Kang and K. G. Neoh, Macromolecules, 2003, 36, 9451–9457 CrossRef CAS.
- L. Ying, W. H. Yu, E. T. Kang and K. G. Neoh, Langmuir, 2004, 20, 6032–6040 CrossRef CAS.
- Y. Chen, W. Sun, Q. Deng and L. Chen, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3071–3082 CrossRef CAS.
- M. Grasselli and N. Betz, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 236, 201–207 CrossRef CAS.
- C.-Y. Hong, Y.-Z. You and C.-Y. Pan, Chem. Mater., 2005, 17, 2247–2254 CrossRef CAS.
- C.-Y. Hong, Y.-Z. You and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2419–2427 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 6770 CrossRef CAS.
- S. Perrier, P. Takolpuckdee, J. Westwood and D. M. Lewis, Macromolecules, 2004, 37, 2709–2717 CrossRef CAS.
- D. Roy, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 10363 CrossRef CAS.
- D. Roy, Aust. J. Chem., 2006, 59, 229 CrossRef CAS.
- M. Hernández-Guerrero, T. P. Davis, C. Barner-kowollik and M. H. Stenzel, Eur. Polym. J., 2005, 41, 2264 CrossRef.
- Q. Xiong, P. Ni, F. Zhang and Z. Yu, Polym. Bull., 2004, 53, 1–8 CrossRef CAS.
- M. Sahnoun, M.-T. Charreyre, L. Veron, T. Delair and F. D'agosto, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4180–4192.
- X. Xin, Y. Wang and W. Liu, Eur. Polym. J., 2005, 41, 1539–1545 CrossRef CAS.
-
H. F. Walton, Principles & Methods of Chemical Analysis,Prentice Hall, Englewood Cliffs, NJ, USA, 2nd edn, 1964, p. 175 Search PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868 CrossRef CAS.
- M. Legge Thomas, T. Slark Andrew and Sebastien. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(24), 6980–6987 CrossRef.
- M. Román-Aguirre, A. Márquez-Lucero and E. A. Zaragoza-Contreras, Carbohydr. Polym., 2004, 55, 201–210 CrossRef CAS.
- J. O. Karlsson and P. Gatenholm, Polymer, 2000, 41, 1551–1559 CrossRef CAS.
- J. O. Karlsson and P. Gatenholm, Polymer, 1997, 38, 4727–4731 CrossRef CAS.
- E. Rizzardo, J. Chiefari, B. Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad and S. H. Thang, Macromol. Symp., 1999, 143, 291–307 CAS.
- V. Lima, X. Jiang, J. Brokken-Zijp, P. J. Schoenmakers, B. Klumperman and R. V. D. Linde, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 959–973 CrossRef CAS.
- D. Beattie, K. H. Wong, C. Williams, L. A. Poole-Warren, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2006, 7, 1072–1082 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional images, IR and Raman spectra. See DOI: 10.1039/b711248n |
| ‡ Present address: Key Centre for Polymer Colloids, School of Chemistry, Building F11, Eastern Avenue, The University of Sydney, NSW 2006, Australia. E-mail: S.Perrier@chem.usyd.edu.au; Fax: +61 (0)2 9351 3329; Tel: +61 (0)2 9351 3366. |
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here. 



