 Open Access Article
Open Access ArticleOptimisation of chemical protein cleavage for erythropoietin semi-synthesis using native chemical ligation†
Jonathan P.
Richardson
and
Derek
Macmillan
*
Department of Chemistry, University College London, 20 Gordon Street, London, UK WC1H 0AJ. E-mail: d.macmillan@ucl.ac.uk; Tel: 020-7679 4684
First published on 5th September 2008
Abstract
Selective protein cleavage at methionine residues is a useful method for the production of bacterially derived protein fragments containing an N-terminal cysteine residue required for native chemical ligation. Here we describe an optimised procedure for cyanogen bromide-mediated protein cleavage, and ligation of the resulting fragments to afford biologically active proteins.
Introduction
Chemical ligation is a popular and rapidly expanding area of research impacting numerous aspects of organic chemistry, biomolecular and materials science.1 Native chemical ligation (NCL) in particular, has found numerous applications in such areas.2 This is likely because NCL is a chemoselective coupling reaction between two, normally peptide, components, one bearing a thioester and the other an N-terminal cysteine functionality that reliably takes place in aqueous solution and in the absence of protecting groups. Additionally NCL has interfaced itself completely with biology in that the required thioester and cysteine bearing components can be obtained through chemical synthesis or genetic manipulation making NCL widely accessible to chemists and biologists alike.3 We reported the first use of CNBr-cleaved protein fragments for production of N-terminal cysteine containing components used in native chemical ligation from N-terminally His10- tagged precursors (Fig. 1a).4 Expression of proteins in bacteria with an N-terminal cysteine residue is not a trivial procedure. While some proteins may be amenable to direct expression of a free N-terminal cys residue, more commonly a pre-cysteine sequence, which relies on the availability of a selective protease to remove this sequence and unmask the free cysteine prior to NCL, is employed.5 In our case we investigated the CNBr cleavage reaction because we could neither express the protein directly with an N-terminal cysteine nor cleave a pre-cysteine sequence enzymatically owing to the insolubility of the protein fragment. This is not an uncommon phenomenon, indeed it is remarkable that so many bacterially expressed protein fragments, that are often the reagents for NCL, are soluble to begin with owing to their lack of a complete primary structure. The fact that our protein fragments were soluble only in denaturing reagents or high concentrations of detergent made them incompatible with commonly employed proteases (e.g. Factor Xa). Herein we describe the optimisation of the CNBr cleavage protocol highlighting important features of the process and demonstrate how proteins that have been subjected to this treatment retain biological activity and how inactive protein fragments can regain bioactivity after NCL reactions with synthetic peptide thioesters and refolding. Furthermore we demonstrate that NCL allows for site-specific introduction of a non-canonical acetylenic functional group into the protein erythropoietin (EPO) which is tolerated and may provide a useful handle for further protein modification.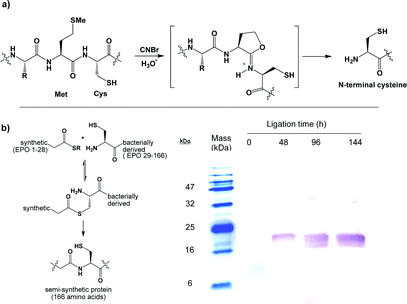 | ||
| Fig. 1 (a) CNBr-mediated protein cleavage yielding an N-terminal cysteine. (b) NCL reaction between synthetic EPO (1–28)-SBn thioester and a bacterially-derived fragment visualised using an anti-EPO monoclonal antibody blot which recognises the synthetic fragment, once appended to the bacterial fragment (product mass = 18.5 kDa). | ||
Optimisation of CNBr-mediated protein cleavage
Initially we investigated the cleavage reaction in a variety of solvents and buffers (5% aqueous TFA; 6 M guanidine hydrochloride + 0.3 M HCl; 6 M urea + 0.3 M HCl; 80% aqueous HCO2H) though quickly settled on 80% formic acid as the best solvent for the reaction as it could most readily dissolve our protein samples. However, we were troubled by the temperamental nature of the reaction, occasionally leading to a lack of efficiency in the subsequent NCL reaction. In some cases the ligation appeared to proceed extremely slowly and with such low efficiency (<5%) that the product, though observable by gel-electrophoresis, could not be isolated (Fig. 1b) leading us to suspect that the integrity of the protein was compromised. We decided to investigate this more thoroughly in the hope that we might better understand any additional factors that would contribute towards the development of a more robust procedure.We next set out to investigate the effects of cyanogen bromide concentration and reaction temperature on the cleavage reaction. There was significant precedent for conducting the reaction at elevated temperature (in protein sequencing experiments) so the reaction was performed at 25 and 44 °C. The effect of small changes in cyanogen bromide concentration was generally not significant since incomplete protein cleavage is observed using vast excesses (>100 equivs) of CNBr at room temperature. Furthermore, cleavage efficiency did not greatly change upon increasing the reagent concentration or addition of further solid CNBr to the reaction mixture following 24 h reaction time. We anticipated that increasing the reaction temperature and conducting the reaction in the presence of denaturants would be more beneficial and consequently examined cleavage of the poly-histidine tag at 44 °C. However, an undesirable protein cleavage reaction (observed by SDS-PAGE and LC-MS) appeared to occur at 44 °C, to the extent of approximately 10% in 80% aqueous formic acid and near quantitatively in 6 M guanidine·HCl containing 0.3 M HCl. Based on the size of the resulting fragments (data not shown), this deleterious side-reaction was possibly a result of protein fragmentation following bromination of tryptophan residues in the EPO sequence, a well documented though poorly understood reaction.6 Subsequently it was concluded that CNBr cleavage reactions should not be conducted above room temperature.
To analyse further the reaction in 80% aqueous formic acid at ambient temperature, the mass spectra of the initial His10-tagged protein fragment comprising erythropoietin residues 29–166 (Fig. 2a) and the crude isolated product from the CNBr cleavage reaction were compared. We observed that although the cleavage had been successful, in that little starting material was observed, the product appeared to be a minimum of 111 Da larger than anticipated (Fig. 2b) and was heterogeneous with the spectrum displaying several deconvoluted molecular ions with mass differences of approximately 27.5 Da (Fig. 2c), presumably arising from significant formylation of the cleaved protein. This was surprising as the formation of formyl amides/esters was not considered to be a stable modification under the reaction conditions and the extent of formylation can vary from one cleavage to the next, indeed in many cases it is not observed at all. We estimated the addition of 5–10 formyl groups to the protein backbone and suspected that formyl modification of the α-NH2group and/or side-chain thiol may be responsible for the poor performance of the fragment in the subsequent NCL reaction shown in Fig. 1b. The crude cleaved products were then redissolved in 6 M guanidine hydrochloride at either pH 1 or pH 12 (containing 0.1 M HCl and 0.1 M NaOH respectively) at 37 °C for 1–16 h. After exposure to 0.1 M HCl for 16 h the sample was homogeneous and had the correct observed mass (Fig. 2d). In contrast, while after 0.5 h the NaOH treated material had almost entirely reverted to the deformylated state, after 16 h the sample appeared heterogeneous (Fig. 2e) and to have a lower observed mass than that calculated indicating that several elimination reactions had occurred.
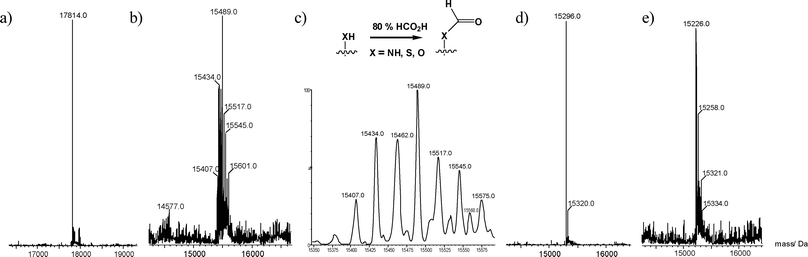 | ||
| Fig. 2 Optimisation of CNBr protein cleavage. (a) The His10-fusion protein prior to cleavage; calculated average mass = 17813.3 Da. (b) Isolated protein after CNBr cleavage in 80% formic acid. (c) Expansion of the molecular ion region shows multiple formylation. (d) The same protein sample after treatment with 0.1 M HCl in 6 M guanidine·HCl for 16 h, calculated average mass = 15292.7 Da (an identical spectrum is obtained upon exposure of the sample to 0.1 M NaOH in 6 M guanidine·HCl for 30 min). (e) Protein sample after treatment with 0.1 M NaOH in 6 M guanidine·HCl for 16 h. | ||
From these experiments we concluded that the CNBr reaction is best conducted in aqueous formic acid (particularly suitable for proteins and fragments of low solubility) but the reaction should not be conducted above room temperature due to undesirable side-reactions. Incomplete protein cleavage at methionine is not problematic since any unreacted His10-tagged starting material can be removed by passage through a short Ni2+-NTA column. Additionally the cleaved protein may be formylated though formylation can be reliably reversed upon exposure to dilute (0.1 M) aqueous HCl.
Semi-synthesis of an erythropoietin analogue by NCL
Having demonstrated that the bacterial fragment could be reliably produced, we were encouraged to proceed towards the assembly of EPO itself, comprising 166 amino acids. First we assembled EPO residues 1–28 on 0.1 mmol scale employing pre-loaded glycine-sulfamylbutyryl resin (Scheme 1).7 The peptide synthesis was conducted in automated fashion on an Applied Biosystems 433A automated peptide synthesiser using the FastMoc protocol. EPO is a glycoprotein so we wanted the potential to site-selectively modify the semi-synthetic protein with glycosylation or polyethylene glycol (PEG) using chemistry developed in our group8 and consequently introduced the novel unnatural amino acid 1 at position 24 (normally a glycosylation site).9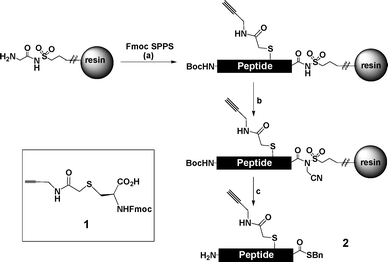 | ||
| Scheme 1 Synthesis of EPO residues 1–28 as a C-terminal benzyl thioester. Amino acid 1 was introduced at position 24. Reagents and conditions: (a) HBTU–HOBt–DIPEA (b) ICH2CN, DIPEA, DMF, 24 h (c) BnSH, NaSPh, DMF, 16 h then 95 : 2.5 : 2.5 v/v/v TFA–EDT–H2O, 5 h. | ||
Upon exposure to iodoacetonitrile, then benzylmercaptan following established protocols7 the thioester 2 was isolated following precipitation from cold diethyl ether, and purification by semi-preparative HPLC to obtain the product in an unoptimised yield of 5% (based on resin loading).
With the required fragments in hand, NCL reactions between the bacterially derived fragment and synthetic thioester were performed in 6 M guanidine hydrochloride containing 300 mM sodium phosphate buffer (pH 7.5), 50 mM 4-mercaptophenylacetic acid (MPAA),10 and 20 mM tris-carboxyethylphosphine (TCEP). The ligation reaction could be conveniently monitored by LC-MS (Fig. 3), was judged to be complete (trace bacterial fragment remaining) by LC-MS after 4 h, and could be driven to completion by addition of excess synthetic thioester. Remarkably, when 2-mercaptoethanesulfonic acid (MESNa) was employed as the thiol additive, in place of MPAA, the reaction proceeded with no more than 50% conversion after 3 days. The product protein, full length EPO containing an acetylene functionality at residue 24, was obtained after dialysis against water at 4 °C and centrifugation to obtain the precipitated protein, which was redissolved in 6 M guanidine·HCl, reduced with dithiothreitol, and then refolded by overnight dialysis against 2% w/v N-lauroylsarcosine, 50 mM Tris-HCl; pH 8.0, 40 μM CuSO4.11
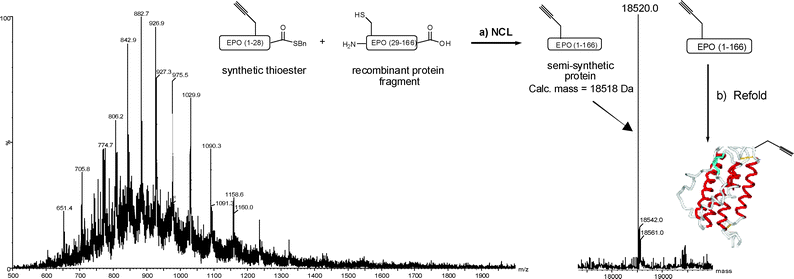 | ||
| Fig. 3 NCL between synthetic thioester 2 and recombinant EPO (residues 29–166). The mass spectrum (left) is observed for the ligation product after 4 h and corresponds to the desired mass (right). Reagents and conditions: (a) 6 M guanidine·HCl, 300 mM Na phosphate buffer; pH 7.5, 50 mM MPAA, 20 mM TCEP, 4 h. (b) 2% w/v N-lauroylsarcosine, 50 mM Tris·HCl; pH 8.0, 40 μM CuSO4. The schematic EPO structure is modified from PDB entry 1BUY. | ||
Biological analysis
An important prerequirement is that CNBr treatment has no deleterious effect on the biological activity of the protein. Additionally, while no biological activity should be attributable to the protein fragments, the biological activity of the protein must be re-established following the NCL reaction. To first assess the benign nature of the CNBr treatment, and subsequent deformylation reaction, EPO[M54L] (full length EPO bearing a single M54L mutation) was expressed and purified from E. coli.12 The M54L mutation was incorporated into the wild-type protein to render it compatible with CNBr mediated removal of the poly-histidine tag. The CNBr reaction was performed exactly as described for the protein fragment then the purified protein was refolded by overnight dialysis against 2% w/v N-lauroylsarcosine. The protein solution obtained was concentrated to approximately 2.0 mL in a centrifugal protein concentrator (Millipore, MWCO = 10 kDa) and was determined to have a concentration of 55 μM. Since unglycosylated EPO is reported to be approximately as active as glycosylated EPOin vitro, this protein was analysed, as a positive control, in both a receptor binding and cell proliferation assay. A difficulty in conducting the assay is the insolubility of the unglycosylated erythropoietin protein as the protein could not be solubilised in the required solvent (100% DMSO). The N-lauroylsarcosine required to solubilise unglycosylated EPO samples is known to interfere with receptor binding even at low concentrations, though we reasoned that the protein should be sufficiently active at low concentrations such that meaningful data could be obtained. Consequently we proceeded with the receptor binding (Fig. 4a) and cell proliferation assays (Fig. 4b–c) hoping that the effect of the presence of N-lauroyl sarcosine would be more limited in the TF-1 cell proliferation assay, and while analysis of our molecules at 1 μM resulted in death of the cell culture during the assay due to the presence of the detergent, more positive results were obtained below this concentration.![(a) Receptor binding assay comparing the glycosylated rhEPO standard (squares) and the bacterially-derived EPO[M54L] (circles). (b) TF-1 cell proliferation assay for bacterially-derived EPO[M54L]. (c) TF-1 cell proliferation assay for semi-synthetic EPO prepared by NCL.](/image/article/2008/OB/b811501j/b811501j-f4.gif) | ||
| Fig. 4 (a) Receptor binding assay comparing the glycosylated rhEPO standard (squares) and the bacterially-derived EPO[M54L] (circles). (b) TF-1 cell proliferation assay for bacterially-derived EPO[M54L]. (c) TF-1 cell proliferation assay for semi-synthetic EPO prepared by NCL. | ||
The biological assays13 were conducted by MDS-Pharma Services (Taipei, Taiwan) so shall only be discussed briefly here. The receptor binding assay was an [125I]erythropoietin radioligand-EPO receptor (derived from human recombinant NSO cells) inhibition assay where residual radioactivity is measured after a 2 h incubation with the test molecule at 25 °C. Initially the results were disappointing with the EPO[M54L] protein, though still considerably active with an IC50 = 26.5 nM, demonstrating 100 fold less activity than the recombinant human EPO standard. The IC50 of the semi-synthetic protein could not be determined under identical conditions. However, the recombinant EPO reference sample had been spiked with only 0.02% N-lauroylsarcosine and this may have contributed to its unusually large IC50 of 320 pM (Lit. = 100 pM).14 In the cell proliferation assay, an EPO dependent cell line TF-1 was incubated for 3 days at 37 °C with standard, bacterially expressed EPO[M54L] and semi-synthetic protein. Cell proliferation was determined by quantification of [3H]thymidine uptake. As suspected, this assay seemed a little less sensitive to the presence of N-lauroylsarcosine and EC50's of 0.73 nM and 11.4 nM could be obtained for the bacterially expressed EPO[M54L] and semi-synthetic protein respectively. No activity could be measured for the bacterial fragment (EPO residues 29–166) used in the NCL reactions in either the receptor binding or the cell proliferation assay. While considerably less active than the therapeutic glycoprotein rhEPO (Lit. EC50 = 10–30 pM)14 our results are highly encouraging, though need to be interpreted with caution owing to the unavoidable presence of N-lauroylsarcosine which inhibits the EPO–EPOR interaction by 20% at concentrations as low as 0.002% w/v (data not shown).
In conclusion we have optimised a procedure for the production of bacterially derived protein fragments containing an N-terminal cysteine residue for use in NCL reactions. The protein fragments are homogenous by SDS-PAGE and LC-MS after a reaction sequence involving CNBr treatment, followed by a 0.1 M HCl deformylation step, and Ni2+ mediated removal of unreacted starting material. The biologically inactive fragments so produced then efficiently participate in NCL reactions with synthetic thioesters, in the presence of 4-mercaptophenylacetic acid (MPAA) as the thiol additive, and the resulting reconstituted proteins can be refolded to yield bioactive molecules. Though solubility was a concern for erythropoietin analysis, the acetylenic handle introduced at position 24 of EPO will facilitate site-selective modification with polyethyleneglycol (PEG) or oligosaccharides8 improving both solubility and hence biological activity in future studies.
Experimental details
General experimental details
1H NMR spectra were recorded at 350 and 300 MHz, 13C NMR spectra were recorded at 63 and 75 MHz respectively on a Bruker 250Y instrument. Electrospray mass spectroscopy was carried out on a Waters Acquity UPLC-SQD MS system with an applied voltage of 50 V. Semi-preparative HPLC was performed using a Phenomenex LUNA C18 column and a gradient of 5–60% acetonitrile containing 0.1% TFA over 45 minutes (flow rate of 4.0 mL min−1). All reagents and solvents were standard laboratory grade and used as supplied unless otherwise stated. Where a solvent was described as dry, it was purchased as anhydrous grade. All organic extracts were dried over anhydrous magnesium sulfate prior to evaporation under reduced pressure. All resins and Fmoc amino acids for peptide synthesis were purchased from Novabiochem.Synthesis of amino acid 1
L-Cysteine hydrochloride (268 mg, 1.70 mmol) was dissolved in water (2.0 mL) and 2-bromoacetyl propargylamide (300 mg, 1.70 mmol) was added. Solid sodium hydrogen carbonate was added in small portions (with evolution of CO2) until pH 8.0 was established. The reaction mixture was then stirred at room temperature for a further 2 h. The reaction mixture was then frozen in liquid nitrogen and lyophilised to afford the crude product as a pale brown solid and was used without further purification, 1H-NMR (300 MHz, D2O) δ (ppm): 3.99 (2H, d, J = 2.5 Hz, CH2N), 3.93 (1H, q, αCH, J = 7.7 Hz, J = 4.3 Hz), 3.37 (2H, s, C(O)CH2S), 3.17 (1H, dd, J = 14.8 Hz, J = 4.3 Hz, CH2S), 3.06 (1H, dd, J = 14.8 Hz, J = 7.7 Hz, CH2S), 2.61 (1H, t, J = 2.5 Hz, CH). ESI-MS calculated for C8H13N2O3S 217.0641 [MH]+, found: 217.0642.The crude propargylamide was then re-dissolved in water (2.0 mL) and Et3N (146 μl) was added. Fmoc-succinimide (550 mg, 1.63 mmol) was dissolved in acetonitrile (2.0 mL) and this solution was added in one portion to the aqueous amino acid solution and stirring was continued at room temperature for a further 1.5 h. The pH of the reaction was monitored throughout the reaction to ensure it remained approximately 9, adding further Et3N as required. After 1.5 h, the reaction mixture was evaporated to dryness and the residue was partitioned between dichloromethane (30.0 mL) and 2 M HCl (30.0 mL). The organic phase was separated and the aqueous phase was extracted with dichloromethane (1 × 30.0 mL). The combined organic extracts were washed with 2 M HCl (25.0 mL) and sat. aq. NaCl (25.0 mL), dried with MgSO4, filtered and the solvent was removed under vacuum to afford the crude product as an off-white solid. The crude product was purified by flash column chromatography over silica (a short column: 5 cm silica, eluent 100% EtOAc then 20% MeOH in EtOAc) to afford the pure product (370 mg, 47%) as a white foam. Rf = 0.05 (4 : 1 EtOAc–MeOH), νmax (cm−1) 3283 (OH, v, acid), 2359, 2123 (w, alkyne), 1709 (CO, v, acid) 1650 (CO, v, amide). 1H-NMR (400 MHz, CD3OD) δ (ppm): 7.77–7.26 (8H, m, ArH), 4.41–4.27 (3H, m, CHCH2O and αCH), 4.20 (1H, t, J = 7.1 Hz, Fmoc,CH), 3.96 (2H, d, J = 2.5 Hz, CH2N), 3.23 (2H, s, COCH2S), 3.11 (1H, dd, J = 13.9 Hz, J = 4.5 Hz, CH2S), 2.93 (1H, dd, J = 13.3 Hz, J = 8.1 Hz, CH2S), 2.56 (1H, t, J = 2.5 Hz, CH). 13C-NMR (75 MHz, CDCl3) δ (ppm): 128.8 (CH, Ar-CH), 128.2 (CH, Ar-CH), 126.3 (CH, Ar-CH), 120.9 (CH, Ar-CH), 80.0 (CH), 68.0 (CH2), 56.1 (CH), 48.4 (CH), 36.1 (CH), 29.8 (CH2). ESI-MS calculated for C23H23N2O5S 439.1322 [MH]+, found: 439.1317.
Peptide thioester synthesis (2, EPO residues 1–28)
The peptide C-terminal benzyl thioester was prepared on 0.1 mmol scale using standard procedures. Briefly, commercially available H-Gly-pre-loaded sulfamylbutyryl resin was extended using HBTU–HOBt as coupling reagents in the presence of DIPEA (Fastmoc protocol) in automated fashion using an Applied Biosystems 433A automated peptide synthesiser. The coupling time was 0.5 h. The dry resin-linked target sequence: APPRL![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) CDSR
CDSR![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif)
![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif)
![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif) RY A
RY A![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) C*TTG-resin (underlined residues were double coupled, C* corresponds to amino acid 1) was then transferred to a 5.0 mL vial. The resin was resuspended in anhydrous DMF (4.0 mL) and, following the addition of ICH2CN (0.2 mL) and DIPEA (0.2 mL), the reaction mixture was stirred at room temperature with the exclusion of light for 24 h. The resin was then filtered off and washed exhaustively with DMF, then DCM. The alkylated resin was then resuspended in anhydrous DMF and, following addition of benzylmercaptan (0.3 mL) and NaSPh (6 mg), was stirred at room temperature for 16 h. The resin was then filtered and washed with DMF (5.0 mL) then DCM (5.0 mL). The combined eluents were evaporated to dryness under vacuum and the residue then exposed to a solution comprised of 95% TFA, 2.5% EDT, 2.5% H2O (5.0 mL), with stirring for 5 h. The crude product was then collected by centrifugation at 3000 rpm following precipitation from cold diethyl ether (50.0 mL). The fully deprotected and precipitated product was redissolved in 25% aqueous MeCN and purified by semi-preparative HPLC. The major peak (retention time = 26 min, 6 mg, 5.3%) was analysed by ESI-MS and was found to correspond to the desired product. This fraction was lyophilised and used in subsequent NCL reactions.
C*TTG-resin (underlined residues were double coupled, C* corresponds to amino acid 1) was then transferred to a 5.0 mL vial. The resin was resuspended in anhydrous DMF (4.0 mL) and, following the addition of ICH2CN (0.2 mL) and DIPEA (0.2 mL), the reaction mixture was stirred at room temperature with the exclusion of light for 24 h. The resin was then filtered off and washed exhaustively with DMF, then DCM. The alkylated resin was then resuspended in anhydrous DMF and, following addition of benzylmercaptan (0.3 mL) and NaSPh (6 mg), was stirred at room temperature for 16 h. The resin was then filtered and washed with DMF (5.0 mL) then DCM (5.0 mL). The combined eluents were evaporated to dryness under vacuum and the residue then exposed to a solution comprised of 95% TFA, 2.5% EDT, 2.5% H2O (5.0 mL), with stirring for 5 h. The crude product was then collected by centrifugation at 3000 rpm following precipitation from cold diethyl ether (50.0 mL). The fully deprotected and precipitated product was redissolved in 25% aqueous MeCN and purified by semi-preparative HPLC. The major peak (retention time = 26 min, 6 mg, 5.3%) was analysed by ESI-MS and was found to correspond to the desired product. This fraction was lyophilised and used in subsequent NCL reactions.
Cleavage of proteins using CNBr
Following expression and purification of a target protein from E. coli as previously described,4 the protein (3–10 mg) was dissolved in the minimum volume of 80% aqueous formic acid (usually 1.0–2.0 mL). CNBr (5 mg) was added and the reaction mixture stirred for 16 h at room temperature under nitrogen with the exclusion of light. The reaction mixture was then evaporated to dryness and the residue resuspended in 6 M guanidine hydrochloride (1.0 mL).Deformylation of cleaved proteins and removal of uncleaved material using Ni2+ affinity chromatography
The crude product from the CNBr cleavage reaction, redissolved in 6 M guanidine hydrochloride (1.0 mL) was treated with HCl (added from a 3 M stock solution) to obtain a final HCl concentration of 0.1 M and was incubated at 37 °C for 16 h. The deformylation reaction was monitored by LC-MS. Alternatively the protein could be deformylated under basic conditions by exposure to 0.1 M NaOH (added to 6 M guanidine hydrochloride from a 10 M stock solution) for 0.5 h at room temperature. After deformylation, the pH was adjusted to 8.0 with 10 M NaOH or 3 M HCl respectively and diluted to 5.0 mL in 6 M guanidine hydrochloride containing 20 mM Tris-HCl; pH 8.0, 0.5 M NaCl, 5 mM imidazole. The resulting solution was treated with 2-mercaptoethanol to a final concentration of 5 mM and incubated at 37 °C with shaking for 1 h. The reduced protein sample was loaded onto a pre-equilibrated Ni2+-NTA column (Novagen) and washed with 5 mM 2-mercaptoethanol in 6 M guanidine hydrochloride containing 20 mM Tris-HCl; pH 8.0, 0.5 M NaCl, 5 mM imidazole (10.0 mL). The combined eluents were dialysed against water at 4 °C for 16 h to precipitate the protein and the crude precipitate was lyophilised and used directly in native chemical ligation.Native chemical ligation
The cleaved protein (approx 3.0 mg) was dissolved in 6 M guanidine hydrochloride containing 300 mM Na phosphate buffer; pH 7.5 (0.5 mL), containing 20 mM TCEP and 50 mM MPAA. Solid peptide thioester (approx 2.0 mg) was weighed into a 1.5 mL eppendorf tube and the protein solution was added in a single portion. The cap was closed and the reaction was shaken at room temperature for 4 h. A sample (3.0 μL) was taken for LC-MS analysis after which time the reaction appeared complete. Further TCEP (10 mM) was added and the reaction shaken at room temperature for a further 1 h. The reaction could then be purified by loading the reaction mixture directly on a semi-preparative HPLC column. However, owing to the completeness of the reaction and the insolubility of the protein, the crude product was collected as a precipitate after pouring into cold water (6.0 mL), allowing it to stand at 4 °C for 16 h and centrifugation at 3000 rpm for 15 min. This process also serves to remove excess unreacted or hydrolysed thioester, which are highly water soluble. The crude product was taken up in 6 M guanidine·HCl (2.0 mL) and treated with 50 mM DTT at 37 °C for 1 h.Refolding of EPO samples
The reduced EPO samples obtained in 2.0 mL guanidine·HCl were diluted to 20.0 mL with 6 M guanidine·HCl and refolded by dialysis against 2% w/v N-lauroylsarcosine, 50 mM Tris.HCl; pH 8.0, 40 μM CuSO4 (2 × 2 L) then concentrated to approximately 2.0 mL using a centrifugal protein concentrator (Millipore) with a 10 kDa molecular weight cut-off filter membrane. The samples were then snap-frozen in liquid nitrogen and stored at −80 °C prior to use in biological assays.13Acknowledgements
The authors acknowledge financial support from The Wellcome Trust.Notes and references
- (a) G. A. Lemieux and C. R. Bertozzi, Trends Biotechnol., 1998, 16(12), 506 CrossRef CAS; (b) K. L. S. E. Kiick, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(1), 19 CrossRef CAS; (c) L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34 CrossRef CAS; (d) J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1(1), 13 CrossRef CAS; (e) P.-C. Lin, S.-H. Ueng, M.-C. Tseng, J.-L. Ko, K.-T. Huang, S.-C. Yu, A. K. Adak, Y.-J. Chen and C. C. Lin, Angew. Chem., Int. Ed., 2006, 45(26), 4286 CrossRef CAS; (f) S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446(7139), 1105 CrossRef; (g) W. P. Heal, S. R. Wickramasinghe, R. J. Leatherbarrow and E. W. Tate, Org. Biomol. Chem., 2008, 6(13), 2308 RSC.
- (a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266(5186), 776 CrossRef CAS; (b) P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS; (c) V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2007, 46(10), 1667 CrossRef CAS.
- (a) C. J. Noren, J. M. Wang and F. B. Perler, Angew. Chem., Int. Ed., 2000, 39(3), 451; (b) D. S. Y. Yeo, R. Srinivasan, G. Y. J. Chen and S. Q. Yao, Chem.–Eur. J., 2004, 10(19), 4664 CrossRef CAS; (c) J. A. Camarero, Y. Kwon and M. A. Coleman, J. Am. Chem. Soc., 2004, 126(45), 14730 CrossRef CAS; (d) J. P. Pellois and T. W. Muir, Curr. Opin. Chem. Biol., 2006, 10(5), 487 CrossRef CAS; (e) K. A. Pickin, S. Chaudhury, B. C. R. Dancy, J. J. Gray and P. A. Cole, J. Am. Chem. Soc., 2008, 130(17), 5667 CrossRef CAS; (f) R. K. McGinty, J. Kim, C. Chatterjee, R. G. Roeder and T. W. Muir, Nature, 2008, 453(7196), 812 CrossRef CAS.
- D. Macmillan and L. Arham, J. Am. Chem. Soc., 2004, 126(31), 9530 CrossRef CAS . A similar method, operating without the aid of a polyhistidine tag, was also published by Lu and co-workers: G. Pal, F. Santamaria, A. A. Kossiakoff and W. Lu, Protein Expression Purif., 2003, 29, 185 Search PubMed.
- (a) T. J. Tolbert and C.-H. Wong, Angew. Chem., Int. Ed., 2002, 41(12), 2171 CrossRef CAS; (b) D. Macmillan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2004, 43, 1355 CrossRef CAS; (c) G. K. Busch, E. W. Tate, P. R. J. Gaffney, E. Rosivatz, R. Woscholskib and R. J. Leatherbarrow, Chem. Commun., 2008, 3369 RSC.
- (a) B. Witkop, Science, 1968, 162, 318 CrossRef CAS; (b) B. J. Smith, The Protein Protocols Handbook, ed. J. M. Walker, Humana Press, Totowa, N. J., 2002, p. 493 Search PubMed.
- Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121(50), 11684 CrossRef CAS.
- D. Macmillan and J. Blanc, Org. Biomol. Chem., 2006, 4, 2847 RSC.
- S. B. Krantz, Erythropoietin, Blood, 1991, 77(3), 419 CAS.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128(20), 6640 CrossRef CAS.
- J. P. Boissel, W. R. Lee, S. R. Presnell, F. E. Cohen and H. F. Bunn, J. Biol. Chem., 1993, 268(21), 15983 CAS.
- D. Macmillan, R. M. Bill, K. A. Sage, D. Fern and S. L. Flitsch, Chem. Biol., 2001, 8(2), 133 CrossRef CAS.
- (a) K. W. Harris, R. A. Mitchell and J. C. Winkelmann, J. Biol. Chem., 1992, 267, 15205 CAS; (b) T. Kitamura, A. Tojo, T. Kuwaki, S. Chiba, K. Miyazono, A. Urabe and F. Takaku, Blood, 1989, 73, 375 CAS.
- Erythropoietins and Erythropoiesis, ed. G. Molineux, M. A. Foote and S. G. Elliott, Berkhäuser-Verlag Press, Switzerland, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS and LC-MS characterisation for thioester 2 and the M54L EPO mutant. See DOI: 10.1039/b811501j |
| This journal is © The Royal Society of Chemistry 2008 |