N-Heterocyclic carbene catalysed β-lactam synthesis†
Nicolas
Duguet
,
Craig D.
Campbell
,
Alexandra M. Z.
Slawin
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, UK KY16 9ST. E-mail: ads10@st-andrews.ac.uk
First published on 12th February 2008
Abstract
N-Heterocyclic carbenes promote the formal [2+2] cycloaddition of ketenes with N-tosyl imines to give the corresponding β-lactams in good to excellent isolated yields; chiral NHCs give β-lactams in high e.e. after crystallisation.
Introduction
N-Heterocyclic carbenes (NHCs) readily promote a range of organocatalytic reactions.1 The majority of these transformations are initiated by nucleophilic attack of the NHC upon an aldehyde or acylsilane to generate the corresponding acyl anion equivalent. Alternatively, the reaction of an NHC with α-functionalised aldehydes can generate homoenolates2 for use in redox reactions,3 a reaction sequence that has been utilised to promote formal [4+2]4 and [3+3]5 cycloaddition processes. As part of a research programme directed towards developing alternative uses of NHCs as organocatalysts,6 their ability to promote the formal [2+2] cycloaddition of ketenes and imines for the synthesis of β-lactams was investigated (Fig. 1). | ||
| Fig. 1 Proposed NHC catalysed β-lactam formation. | ||
The β-lactam skeleton is a widely recognised pharmacophore that has received extensive biological and synthetic investigation. A number of methodologies exist for the preparation of β-lactams,7,8 with asymmetric routes including the ring expansion of aziridines9 and chiral auxiliary methods,10 amongst others.11 The Staudinger reaction12 is perhaps the most widely recognised procedure for the synthesis of β-lactams,13 and Lectka et al.14 and Fu et al.15 have elegantly shown that chiral amines can catalyse this reaction asymmetrically. Although the addition of nucleophiles to ketenes has been utilised for a number of synthetic applications,16 only limited studies concerning the activation of a ketene with an NHC have been reported.17 We delineate herein that NHCs catalyse the formation of β-lactams from ketenes and imines and show that enantiomerically pure NHCs provide an important platform for a catalytic enantioselective variant of this reaction.
Results and discussion
Developing an NHC mediated β-lactam synthesis
At the onset of these studies, a reliable working procedure for β-lactam formation from diphenylketene and N-tosyl imine 4 using triazolinylidene NHC 2 was sought. Deprotonation of azolium salt 1 (20 mol%) with KHMDS (19 mol%) in toluene was used to pre-generate NHC 2, with the order of addition of reactants critical to the successful generation of β-lactam 5. Addition of NHC 2 to an equimolar solution of diphenylketene and imine 4, or the inverse addition, gave 25% and 75% conversion to 5 respectively after 24 hours. Addition of imine 4 to NHC 2 followed by diphenylketene gave no conversion to 5 even after 5 days, consistent with reports of N-tosyl imine inhibition of catalyst turnover in NHC reactions through irreversible nucleophilic addition.18 However, addition of diphenylketene to NHC 2, followed by imine 4, gave >95% conversion to product, giving 5 in 59% isolated yield, implying that NHC activation of the ketene is necessary to allow catalytic turnover in this reaction (Table 1, entry 1). To further test this hypothesis, addition of diphenylketene (0.2 eq) to NHC 2, followed by imine 4 (1.0 eq) and then a further portion of diphenylketene (0.8 eq) gave 90% conversion to 5. Further reaction optimisation showed that the initial addition of 1.3 eq of ketene 3 to NHC 2 in this procedure allowed full consumption of imine 4, giving 5 in 84% isolated yield (entry 2). With a working protocol in hand, the effect of solvent upon the catalytic efficiency of this process was investigated. As a standard, using 19 mol% of NHC 2 in toluene gave 66% conversion to 5 after 1 hour, while the corresponding reactions in THF or Et2O gave 83% and >95% conversion respectively. Progressively lowering the catalyst loading in Et2O showed that 4.5 mol% of NHC 2 gave >95% conversion to 5 within 1 hour, giving 5 in 93% isolated yield (entries 6–8). Lower catalyst loadings (<1 mol%) of NHC 2 can be used to generate moderate levels of conversion to β-lactam 5 but require extended reaction times (entry 9).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 (mol%) | KHMDS (mol%) | 3 (eq) | Solvent | Time/h | Product conversiona |
| a All reaction conversions and product distributions were judged by 1H NMR spectroscopic analysis of the crude reaction product. b Isolated yield of homogeneous product after chromatographic purification. | ||||||
| 1 | 20 | 19 | 1 | Toluene | 24 | >95% (59%)b |
| 2 | 20 | 19 | 1.3 | Toluene | 24 | >95% (84%)b |
| 3 | 20 | 19 | 1.3 | Toluene | 1 | 66% |
| 4 | 20 | 19 | 1.3 | CH2Cl2 | 1 | 0 |
| 5 | 20 | 19 | 1.3 | THF | 1 | 83% |
| 6 | 20 | 19 | 1.3 | Et2O | 1 | >95% |
| 7 | 10 | 9 | 1.3 | Et2O | 1 | >95% |
| 8 | 5 | 4.5 | 1.3 | Et2O | 1 | >95% (93%)b |
| 9 | 1 | 0.9 | 1.3 | Et2O | 24 | 60% |

Wilhelm and Sereda have recently shown that metal amides such as KHMDS can catalyse directly the [2+2] cycloaddition of ketenes and N-nosyl imines.19 To confirm that NHC 2 is the catalytic species in this reaction manifold, control experiments indicated that using KHMDS (5 mol%) alone gave <10% conversion to β-lactam 5 after 24 hours in Et2O. Further experiments showed that treatment of 3 and 4 with HMDS, or simply mixing 3 and 4 in Et2O, returned only starting material. Using this information, a simplistic mechanistic rationale for this process can be proposed. Deprotonation of azolium salt 1 with KHMDS generates NHC 2, that adds to diphenylketene to generate azolium enolate 6. Enolate addition to N-tosyl imine 4 gives the β-amido carbonyl 7, which upon intramolecular cyclisation produces the β-lactam 5 and regenerates the NHC 2 (Fig. 2). While this mechanism remains our current hypothesis, Fu et al. have proposed an alternative mechanism when employing N-triflyl imines in this reaction manifold that involves initial nucleophilic activation of the imine.15b Preliminary mechanistic studies cannot, as yet, unambiguously rule out this alternative pathway when employing NHC mediated catalysis, and further investigations to delineate conclusively the mechanism of this transformation are underway.
 | ||
| Fig. 2 Proposed mechanism of β-lactam synthesis using NHC 2. | ||
Probing reaction scope
The generality of this catalytic process was next examined. Using diphenylketene, a range of N-tosyl substituted imines derived from aryl (entries 1 and 2), heteroaryl (entry 3), substituted aryl (entries 4 and 5) and α,β-unsaturated aldehydes (entry 6) are readily tolerated. High levels of conversion to the desired β-lactams were readily achieved with <10 mol% of NHC 2 within one hour, although with an electron rich 4-MeOC6H4 substituent (entry 4), 9 mol% of NHC 2 and a longer reaction time of 24 hours was required, giving 10 in 59% isolated yield (Table 2).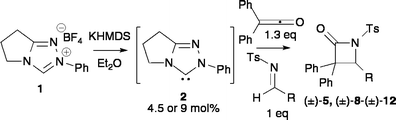
The ability of NHC 2 to catalyse the formation of β-lactams from unsymmetrical ketenes was next investigated. Addition of isobutylphenylketene13 to NHC 2 (9 mol%) followed by addition of N-tosyl imine 4 gave full conversion after one hour to the desired β-lactam 14 as a 68 : 32 mixture of syn : anti diastereoisomers in 89% isolated yield (entry 1).20 Lower catalyst loadings could be used to achieve reasonable levels of conversion in this reaction without altering the diastereoselectivity of this process (4.5 mol% of NHC 2, 16 hours, 87% conversion). This protocol proved general as a range of imines are readily tolerated, proceeding to completion using 9 mol% of NHC 2 within one hour. The desired β-lactams 14–17 are readily isolated in good yield (72–94%) as a mixture of diastereoisomers, with a preference for the syn-diastereoisomer observed in each case (Table 3).21
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time/h | Product | d.r. (syn:anti)a | Yieldb |
| a As shown by 1H NMR spectroscopic analysis of the crude reaction product. b Isolated yield of diastereoisomeric product mixture after purification. | |||||
| 1 | Ph | 1 | 14 | 68 : 32 | 89% |
| 2 | 2-Naphthyl | 1 | 15 | 63 : 37 | 89% |
| 3 | 2-Furyl | 1 | 16 | 81 : 19 | 86% |
| 4 | 4-BrC6H4 | 16 | 17 | 57 : 43 | 94% |
| 5 | (E)–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh CHPh |
1 | 18 | 76 : 24 | 72% |

Catalytic asymmetric β-lactam synthesis employing NHCs
Subsequent investigations probed the ability of enantiomerically pure NHCs to catalyse an enantioselective version of this reaction. As C2-symmetry has proven a useful tool in asymmetric catalysis,22 a C2-symmetric tetrafluoroborate NHC precatalyst 19 was prepared from (1R,2R)-cyclohexane-1,2-diammonium mono-(+)-tartrate23via bis-reductive amination and subsequent cyclisation, giving 19 in good yield over three steps (Scheme 1).
Initial results showed that NHCs 21 and 22, prepared by deprotonation of 19 and the popular triazolium salt 20 (10 mol%) with KHMDS (9 mol%) respectively, promote readily the formal cycloaddition of diphenylketene and N-tosyl imine 4, giving complete conversion to β-lactam 5 in 58% and 64% e.e. respectively and in good isolated yields (Table 4, entries 1 and 2). Preferential crystallisation of the racemate from the 64% e.e. sample, and concentration of the mother liquor, gave access to β-lactam (R)-5 in >98% e.e. The absolute configuration of 5 was proven unambiguously by X-ray crystallographic analysis (Fig. 3).‡ This procedure was subsequently applied to a number of N-tosyl imines , giving access to enantiomerically enriched β-lactams (R)-8, (S)-9 and (R)-11 in 56–75% e.e. and in good isolated yields (79–96%).24 In each case, crystallisation and re-isolation of the β-lactam from the mother liquor was used to enhance the e.e. of the β-lactam to >92% e.e. (entries 4, 5 and 8, Table 4).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Salt | R | Time/h | Product | Yielda | e.e.b | e.e.c |
| a Isolated yield of homogeneous product after chromatographic purification. b e.e. of isolated β-lactam after chromatography as shown by HPLC analysis. c e.e. of β-lactam after crystallisation and re-isolation of the mother liquor as shown by HPLC analysis. | |||||||
| 1 | 19 | Ph | 1 | (R)-5 | 90% | 58% | |
| 2 | 20 | Ph | 3 | (R)-5 | 96% | 64% | >98% |
| 3 | 19 | 2-Naphthyl | 1 | (R)-8 | 95% | 73% | |
| 4 | 20 | 2-Naphthyl | 3 | (R)-8 | 92% | 75% | >99% |
| 5 | 19 | 2-Furyl | 1 | (S)-9 | 85% | 61% | 92% |
| 6 | 20 | 2-Furyl | 3 | (S)-9 | 93% | 55% | |
| 7 | 19 | 4-BrC6H4 | 2 | (R)-11 | 79% | 56% | |
| 8 | 20 | 4-BrC6H4 | 3 | (R)-11 | 96% | 57% | >99% |

 | ||
| Fig. 3 Molecular representation of the X-ray crystal structure of (R)-5. | ||
In conclusion, we have shown that NHCs can catalyse effectively the formal [2+2] cycloaddition of ketenes and imines to generate β-lactams, and that chiral NHCs show promising levels of enantioselectivity in this reaction manifold. Current investigations are focused upon expanding the reactions of chiral NHCs to a variety of catalytic enantioselective processes.
Experimental
General experimental
All reactions involving moisture sensitive reagents were performed under an atmosphere of nitrogen via standard vacuum line techniques and with freshly dried solvents. All glassware was flame dried and allowed to cool under vacuum. Toluene, tetrahydrofuran (THF), diethyl ether (Et2O) and dichloromethane were obtained dry from a solvent purification system (MBraun, SPS-800). Potassium hexamethyldisilazide (KHMDS) was supplied as a 0.5 M solution in toluene (Aldrich), titrated before use,25 and used as a 0.45 M solution. Petrol is defined as petroleum ether 40–60 °C. All solvents and commercial reagents were used as supplied without further purification unless stated otherwise. Room temperature refers to 20–25 °C. Reflux conditions were obtained using an oil bath equipped with a contact thermometer. In vacuo refers to the use of a Büchi Rotavapor R-2000 rotary evaporator with a Vacubrand CVC2 vacuum controller or a Heidolph Laborota 4001 rotary evaporator with a vacuum controller. Analytical thin layer chromatography was performed on aluminium sheets coated with 60 F254 silica. TLC visualisation was carried out with ultraviolet light (254 nm). Column chromatography was performed on Kieselgel 60 silica in the solvent system stated. 1H and 13C nuclear magnetic resonance (NMR) spectra were acquired on a Bruker Avance II 400 (400 MHz 1H, 100 MHz 13C) spectrometer and in CDCl3 unless stated otherwise. Coupling constants (J) are reported in Hz. Infrared spectra (νmax) were recorded on a Perkin-Elmer Spectrum GX FT-IR Spectrometer using KBr discs as stated. Only the characteristic peaks are quoted. Microanalyses were carried out on a Carlo Erba CHNS analyser. Melting points were recorded on an Electrothermal apparatus and are uncorrected. Mass spectrometric (m/z) data were acquired by electrospray ionisation (ESI), electron impact (EI) or chemical ionisation (CI), either at the University of St Andrews Mass Spectrometry facility or from the EPSRC National Mass Spectrometry Service Centre, Swansea. At the University of St Andrews, low and high resolution ESI MS was carried out on a Micromass LCT spectrometer and low and high resolution CI MS was carried out on a Micromass GCT. At the EPSRC National Mass Spectrometry Service Centre, low and high resolution EI MS was carried out on a Micromass Quattro II spectrometer.General procedure 1 for the synthesis of β-lactams (±)-5 and (±)-8 to (±)-12
A 0.45 M solution of KHMDS in toluene (0.04 mL, 0.0174 mmol, 4.5 mol% or 0.08 mL, 0.0347 mmol, 9 mol%) was added to a suspension of triazolium salt 1 (5.3 mg, 0.0193 mmol, 5 mol% or 10.5 mg, 0.0386 mmol, 10 mol%) in Et2O (1.5 mL) under a nitrogen atmosphere and the mixture was stirred for 30 minutes at room temperature. A solution of diphenylketene3 (97.4 mg, 0.501 mmol, 1.3 equiv) in Et2O (1.5 mL) was added, followed immediately by the imine (0.386 mmol, 1.0 equiv) as a solid, then the reaction mixture was stirred at room temperature before concentration in vacuo.Preparation of (±)-3,3,4-triphenyl-1-tosylazetidin-2-one (±)-5
Following general procedure 1, β-lactam (±)-5 was obtained using diphenylketene3 (97.4 mg, 0.501 mmol, 1.3 equiv) and N-benzylidene-4-methylbenzenesulfonamide 4 (100.0 mg, 0.386 mmol, 1.0 equiv). The reaction mixture was concentrated after stirring for 1 hour at room temperature and the residue was purified by column chromatography (EtOAc–petroleum ether 20 : 80 then CH2Cl2–petroleum ether –EtOAc 50 : 45 : 5) to give β-lactam (±)-5 (162.6 mg, 93% yield) as a white solid (mp = 184–185 °C). IR (KBr disk) νmax 3063, 3037, 2928, 1776 (CO), 1597, 1495, 1452, 1447, 1383, 1370, 1262, 1210, 1188, 1172, 1141, 1090, 1070, 1028; 1H NMR δ 2.32 (3H, s), 5.70 (1H, s), 6.77–6.94 (7H, m), 6.98 (2H, t, J = 7.6), 7.05 (1H, t, J = 7.2), 7.11–7.27 (5H, m), 7.31 (2H, d, J = 7.2), 7.65 (2H, d, J = 8.0); 13C NMR δ 21.8 (CH3), 69.3 (CH), 72.9 (C), 127.0 (2 CH), 127.3 (CH), 127.7 (2 CH), 127.9 (3 CH), 128.0 (2 CH), 128.1 (2 CH), 128.4 (2 CH), 128.5 (CH), 129.0 (2 CH), 129.9 (2 CH), 134.0 (C), 135.4 (C), 135.9 (C), 139.0 (C), 145.4 (C), 166.9 (C); CIMS (NH3) m/z 471 (M + NH4+, 4), 257 (67), 256 (Ph2CCH+–Ph, 29), 212 (Ph2CCO + NH4+, 27), 106 (OC–N–SO2, 100), 91 (C6H5+–CH3, 77); HRMS (CI, NH3) [M + NH4+] C28H27N2O3S requires 471.1737, found, 471.1738; Anal. Calcd. for C28H23NO3S: C 74.15, H 5.11, N 3.09%, Found: C 73.84, H 4.97, N 3.12%.
Preparation of (±)-3,3-diphenyl-4-(2-naphthyl)-1-tosylazetidin-2-one (±)-8
Following general procedure 1, β-lactam (±)-8 was obtained using diphenylketene3 (97.4 mg, 0.501 mmol, 1.3 equiv) and N-(2-naphthylidene)-4-methylbenzenesulfonamide (119.3 mg, 0.386 mmol, 1.0 equiv). The reaction mixture was concentrated after stirring for 1 hour at room temperature and the residue was purified by column chromatography (EtOAc–petroleum ether 20 : 80 then CH2Cl2–petroleum ether –EtOAc 50 : 45 : 5) to give β-lactam (±)-8 (177.9 mg, 92% yield) as a white solid (mp = 182–183 °C). IR (KBr disk) νmax 3054, 1792 (CO), 1595, 1496, 1449, 1374, 1361, 1167, 1128, 1121, 1088; 1H NMR δ 2.39 (3H, s), 5.94 (1H, s), 6.81 (1H, dd, J = 8.4, 1.2), 6.83–7.03 (5H, m), 7.18 (2H, d, J = 8.0), 7.22–7.39 (4H, m), 7.39–7.51 (5H, m), 7.56 (1H, s), 7.63 (1H, dd, J = 5.6, 3.2), 7.69 (3H, d, J = 8.0); 13C NMR δ 27.7 (CH3), 69.7 (CH), 72.8 (C), 124.8 (CH), 126.3 (CH), 126.5 (CH), 127.0 (2 CH), 127.3 (CH), 127.7 (4 CH), 128.0 (4 CH), 128.1 (CH), 128.2 (2 CH), 129.0 (2 CH), 129.8 (2 CH), 131.5 (C), 132.7 (C), 133.1 (C), 135.5 (C), 135.7 (C), 139.2 (C), 145.4 (C), 166.8 (C); CIMS m/z 504 (M + H+, 18), 308 (M − Ph2CCO, 13), 307 (100), 306 (40), 198 (OC–NSO2–tol, 38), 194 (Ph2CCO, 18), 155 (SO2–tol, 16), 141 (C11H9, 5); HRMS (CI) [M + H+] C32H26NO3S requires 504.1633, found 504.1631.
General procedure 2 for the synthesis of β-lactams (±)-14 to (±)-18
A 0.45 M solution of KHMDS in toluene (0.08 mL, 0.0347 mmol, 9 mol%) was added to a suspension of triazolium salt 1 (10.5 mg, 0.0386 mmol, 10 mol%) in Et2O (1.5 mL) under a nitrogen atmosphere and the mixture was stirred for 30 minutes at room temperature. A solution of isobutylphenylketene 13 (87.3 mg, 0.501 mmol, 1.3 equiv) in Et2O (1.5 mL) was added, followed by the imine (0.386 mmol, 1.0 equiv) as a solid, then the reaction mixture was stirred at room temperature before concentration in vacuo. The initial diastereoselectivity was measured by 1H NMR on the crude product and can be improved (after purification by column chromatography ) by trituration of the product in Et2O and filtration. This procedure allowed the characterisation of the major syn diastereoisomer.Preparation of syn-3,4-diphenyl-3-isobutyl-1-tosylazetidin-2-one (±)-1415
Following general procedure 2, β-lactam (±)-14 was obtained using isobutylphenylketene 13 (87.3 mg, 0.501 mmol, 1.3 equiv) and N-benzylidene-4-methylbenzenesulfonamide 4 (100.0 mg, 0.386 mmol, 1.0 equiv). The reaction mixture was concentrated after stirring for 1 hour at room temperature (d.r. = 68 : 32 syn : anti) and the residue was purified by column chromatography (EtOAc–petroleum ether 10 : 90 then CH2Cl2–petroleum ether –EtOAc 50 : 45 : 5) to give β-lactam (±)-14 (148.7 mg, 89% yield). Trituration in Et2O gave a white solid (mp = 156–158 °C, d.r. = 89 : 11 syn : anti). IR (KBr disk) νmax 3064, 3034, 2958, 2927, 2868, 2360, 1770 (CO), 1597, 1496, 1447, 1364, 1241, 1188, 1170, 1145, 1090; 1H NMR syn: δ 0.69 (3H, d, J = 6.8), 0.87 (3H, d, J = 6.4), 1.48–1.61 (1H, m), 1.92 (1H, dd, J = 14.6, 6.0), 2.04 (1H, dd, J = 14.0, 6.4), 2.43 (3H, s), 5.00 (1H, s), 6.77 (2H, d, J = 7.6), 6.85–6.92 (2H, m), 6.95–7.05 (5H, m), 7.10 (1H, t, J = 7.4), 7.15–7.30 (2H, m), 7.71 (2H, d, J = 8.4); anti (visible peaks): δ 0.37 (3H, d, J = 6.4), 2.43 (3H, s), 5.05 (1H, s), 7.80 (2H, d, J = 8.4); 13C NMR syn: δ 21.7 (CH3), 23.7 (CH3), 23.8 (CH3), 24.9 (CH), 47.5 (CH2), 69.3 (C), 69.8 (CH), 127.0 (CH), 127.4 (2 CH), 127.6 (2 CH), 127.9 (2 CH), 127.95 (2 CH), 128.0 (2 CH), 128.4 (CH), 129.7 (2 CH), 134.2 (C), 134.9 (C), 135.7 (C), 145.2 (C), 168.1 (C); anti (visible peaks): δ 23.0, 24.0, 24.5, 42.1, 69.9, 126.1, 127.4, 127.8, 128.6, 128.8, 128.9, 129.8, 168.5.
Preparation of syn-3-isobutyl-4-(2-naphthyl)-3-phenyl-1-tosylazetidin-2-one (±)-15
Following general procedure 2, β-lactam (±)-15 was obtained using isobutylphenylketene 13 (87.3 mg, 0.501 mmol, 1.3 equiv) and N-(2-naphthylidene)-4-methylbenzenesulfonamide (119.3 mg, 0.386 mmol, 1.0 equiv). The reaction mixture was concentrated after stirring for 1 hour at room temperature (d.r. = 63 : 37 syn : anti) and the residue was purified by column chromatography (EtOAc–petroleum ether 10 : 90 then CH2Cl2–petroleum ether –EtOAc 50 : 45 : 5) to give β-lactam (±)-15 (165.8 mg, 89% yield). Trituration in Et2O gave a white solid (mp = 146–148 °C, d.r. = 90 : 10 syn : anti). IR (KBr disk) νmax 3060, 2956, 2927, 2870, 2358, 1784 (CO), 1597, 1496, 1467, 1448, 1368, 1187, 1172, 1088; 1H NMR syn: δ 0.72 (3H, d, J = 6.8), 0.91 (3H, d, J = 6.8), 1.51–1.65 (1H, m), 2.00 (1H, dd, J = 14.4, 6.0), 2.11 (1H, dd, J = 14.0, 6.4), 2.38 (3H, s), 5.18 (1H, s), 6.62 (1H, dd, J = 8.4, 1.2), 6.88–7.00 (5H, m), 7.16 (2H, d, J = 8.0), 7.32 (1H, d, J = 8.8), 7.38–7.48 (3H, m), 7.60 (1H, dd, J = 6.0, 3.2), 7.60–7.69 (3H, m); anti (visible peaks): δ 0.33 (3H, d, J = 6.4), 2.45 (3H, s), 5.21 (1H, s); 13C NMR syn: δ 21.7 (CH3), 23.7 (CH3), 23.8 (CH3), 24.9 (CH), 47.8 (CH2), 69.2 (C), 70.0 (CH), 124.9 (CH), 126.2 (CH), 126.4 (CH), 127.0 (CH), 127.4 (2 CH), 127.5 (CH), 127.6 (2 CH), 127.9 (CH), 128.0 (2 CH), 128.2 (CH), 129.7 (2 CH), 131.7 (C), 132.6 (C), 133.1 (C), 134.8 (C), 135.6 (C), 145.1 (C), 168.0 (C); CIMS m/z 484 (M + H+, 100), 287 (MH+–OCN–SO2tol, 37), 198 (OCN–SO2–tol, 40), 174 (Ph(i-Bu)CCO, 75), 154 (SO2tol, 29); HRMS (ESI+) [M + Na+] C30H29NNaO3S requires 506.1766, found, 506.1768.
General procedure 3 for the enantioselective synthesis of β-lactams (R)-5, (R)-8, (S)-9 and (R)-11
A 0.45 M solution of KHMDS in toluene (0.08 mL, 0.0347 mmol, 9 mol%) was added to a suspension of chiral imidazolinium salt 19 (15.2 mg, 0.0386 mmol, 10 mol%) or chiral triazolium salt 20 (14.5 mg, 0.0386 mmol, 10 mol%) in Et2O (1.5 mL) under a nitrogen atmosphere and the mixture was stirred for 30 minutes at room temperature. A solution of diphenylketene3 (97.4 mg, 0.501 mmol, 1.3 equiv) in Et2O (1.5 mL) was added, followed immediately by the imine (0.386 mmol, 1.0 equiv) as a solid, then the reaction mixture was stirred at room temperature before concentration in vacuo.Preparation of (R)-3,3,4-triphenyl-1-tosylazetidin-2-one 5
Following general procedure 3, the β-lactam (R)-5 was obtained using diphenylketene3 (97.4 mg, 0.501 mmol, 1.3 equiv) and N-benzylidene-4-methylbenzenesulfonamide 4 (100 mg, 0.386 mmol, 1.0 equiv). The reaction mixture was concentrated after stirring for 1 hour (using the NHC from 19) or 3 hours (using the NHC from 20) at room temperature and the residue was purified by column chromatography (EtOAc–petroleum ether 10 : 90 then CH2Cl2–petroleum ether –EtOAc 50 : 45 : 5) to give β-lactam (R)-5 as a yellow solid (158.0 mg, 90% yield, from chiral imidazolinium salt 19 and 167.2 mg, 96% yield from chiral triazolium salt 20). HPLC analysis: 64% e.e. (Daicel CHIRALCEL OD-H column, eluent: hexane–i-PrOH 90 : 10, flow: 1 mL min−1, wavelengh: 254 nm, retention times: 9.4 min (major, R) and 16.5 min (minor, S)). [α]20D +17.7 (c 1.00, CHCl3, 64% e.e.). The initial e.e. can be improved by crystallisation (CH2Cl2–hexane) and re-isolation of the β-lactam from the mother liquor, giving (R)-5 in 98% e.e. (mp = 152–154 °C, 98% e.e.). [α]20D +22.0 (c 0.63, CHCl3, 98% e.e.).Preparation of (R)-3,3-diphenyl-4-(2-naphthyl)-1-tosylazetidin-2-one 8
Following general procedure 3, β-lactam (R)-8 was obtained using diphenylketene3 (97.4 mg, 0.501 mmol, 1.3 equiv) and N-(2-naphthylidene)-4-methylbenzenesulfonamide (119.3 mg, 0.386 mmol, 1.0 equiv). The reaction mixture was concentrated after stirring for 1 hour (using the NHC from 19) or 3 hours (using the NHC from 20) at room temperature and the residue was purified by column chromatography (EtOAc–petroleum ether 10 : 90 then CH2Cl2–petroleum ether –EtOAc 50 : 45 : 5) to give β-lactam (R)-8 as a pale yellow solid (184.1 mg, 95% yield, from chiral imidazolinium salt 19 and 178.5 mg, 92% yield from chiral triazolium salt 20). HPLC analysis: 75% e.e. (Daicel CHIRALCEL OD-H column, eluent: hexane–i-PrOH 90 : 10, flow: 1 mL min−1, wavelengh: 254 nm, retention times: 10.0 min (major, R) and 20.5 min (minor, S)). [α]20D −5.0 (c 1.00, CHCl3, 75% e.e.). The initial e.e. can be improved by crystallisation (CH2Cl2–hexane) and re-isolation of the β-lactam from the mother liquor, giving (R)-8 in >99% e.e. (mp = 136–138 °C, >99% e.e.). [α]20D −5.9 (c 0.69, CHCl3, >99% e.e.).Acknowledgements
The authors would like to thank the Royal Society for a University Research Fellowship (ADS), The Leverhulme Trust (ND) and the Carnegie Trust for the Universities of Scotland (CDC) for funding, and the EPSRC National Mass Spectrometry Service Centre (Swansea).References
- For recent reviews see: D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 Search PubMed; N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS.
- For select examples see: C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 Search PubMed; S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS; A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905 CrossRef CAS.
- K. Zeitler, Angew. Chem., Int. Ed., 2005, 44, 7506 CrossRef CAS; K. Y.-K. Chow and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 8126 CrossRef CAS; N. T. Reynolds, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2004, 126, 9518 CrossRef CAS; N. T. Reynolds and T. Rovis, J. Am. Chem. Soc., 2005, 127, 16406 CrossRef; S. S. Sohn and J. W. Bode, Angew. Chem., Int. Ed., 2006, 45, 6021 CrossRef CAS.
- M. He, G. J. Uc and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 15088 CrossRef CAS; M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418 CrossRef CAS.
- A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 5334 CrossRef CAS.
- J. E. Thomson, K. Rix and A. D. Smith, Org. Lett., 2006, 8, 3785 CrossRef CAS.
- H. Gilman and M. Speeter, J. Am. Chem. Soc., 1943, 65, 2255 CrossRef CAS; D. J. Hart and D.-C. Ha, Chem. Rev., 1989, 89, 1447 CrossRef CAS.
- S. G. Davies and D. R. Fenwick, J. Chem. Soc., Chem. Commun., 1995, 1109 RSC; S. Kobayashi, T. Iimori, T. Izawa and M. Ohno, J. Am. Chem. Soc., 1981, 103, 2406 CrossRef CAS.
- S. Calet, F. Urso and H. Alper, J. Am. Chem. Soc., 1989, 111, 931 CrossRef CAS.
- D. A. Evans and E. B. Sjogren, Tetrahedron Lett., 1985, 26, 3783 CrossRef CAS; A. K. Bose, M. S. Manhas, J. M. van der Veen, S. S. Bari and D. R. Wagle, Tetrahedron, 1992, 48, 4831 CrossRef CAS.
- For select examples see: H. Fujieda, M. Kanai, T. Kambara, A. Iida and K. A. Tomioka, J. Am. Chem. Soc., 1997, 119, 2060 Search PubMed; M. Miura, M. Enna, K. Okuro and M. Nomura, J. Org. Chem., 1995, 60, 4999 CrossRef CAS; M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 4572 CrossRef CAS.
- H. Staudinger, Liebigs Ann. Chem., 1907, 356, 51 CrossRef CAS.
- C. Palomo, J. M. Aizpurua, I. Ganboa and M. Oiarbide, Eur. J. Org. Chem., 1999, 12, 3223 CrossRef; I. Ojima and F. Delalogue, Chem. Rev., 1997, 26, 377 CrossRef CAS.
- A. E. Taggi, A. M. Hafez, H. Wack, B. Young, W. J. Drury III and T. Lectka, J. Am. Chem. Soc., 2000, 122, 7831 CrossRef CAS; S. France, A. Weathermax, A. E. Taggi and T. Lectka, Acc. Chem. Res., 2004, 37, 592 CrossRef CAS.
- (a) B. L. Hodous and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 1578 CrossRef CAS; (b) E. C. Lee, B. L. Hodous, E. Bergin, C. Shih and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 11586 CrossRef CAS.
- For reviews see: T. T. Tidwell, Angew. Chem., Int. Ed., 2005, 44, 6812 Search PubMed; J. Gonda, Angew. Chem., Int. Ed., 2004, 43, 3516 CrossRef CAS.
- During the preparation of this manuscript a related protocol for the asymmetric synthesis of β-lactams from unsymmetrical ketenes and N-Boc imines was reported; Y.-R. Zhang, L. He, X. Wu, P.-L. Shao and S. Ye, Org. Lett., 2008, 10, 277. Bode et al. have also reported a related β-lactam synthesis using NHCs; M. He and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 418.
- M. He and J. W. Bode, Org. Lett., 2005, 7, 3131 CrossRef CAS.
- O. Sereda and R. Wilhelm, Synlett, 2007, 3032 CAS.
- The syn-configuration within β-lactam 14 (and 16 and 18) was determined by comparison with the literature data;1515 and 17 were assigned by analogy.
- Trituration of the isolated mixture of diastereoisomers with Et2O resulted in an enhancement of the d.r. of the product, typically giving a ∼90 : 10 syn : anti ratio of diastereoisomers; see ESI for full details†.
- T. P. Dang and H. B. Kagan, J. Chem. Soc. D, 1971, 481 RSC.
- J. F. Larrow and E. N. Jacobsen, Organic Syntheses, Coll. Vol. 10, 2004, 96 Search PubMed; J. F. Larrow, E. N. Jacobsen, Y. Gao, Y. Hong, X. Nie and C. M. Zepp, J. Org. Chem., 1994, 59, 1939 CrossRef CAS.
- The configuration within (R)-8, (S)-9 and (R)-11 was assigned by analogy to that of (R)-5.
- L. Duhamel and J.-C. Plaquevent, J. Organomet. Chem., 1993, 448, 1 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental details and spectroscopic data (1H and 13C NMR) for all products. See DOI: 10.1039/b800857b |
| ‡ CCDC reference number 671783. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b800857b |
| This journal is © The Royal Society of Chemistry 2008 |