A new 34-membered N6O4-donor macrocycle : synthetic, X-ray and solvent extraction studies
Marco
Wenzel
ab,
Kerstin
Gloe
a,
Karsten
Gloe
*a,
Gert
Bernhard
b,
Jack K.
Clegg
c,
Xue-Kui
Ji
c and
Leonard F.
Lindoy
*c
aDepartment of Chemistry and Food Chemistry, TU Dresden, 01062, Dresden, Germany. E-mail: Karsten.Gloe@chemie.tu-dresden.de
bInstitute of Radiochemistry, Research Centre Dresden-Rossendorf, 01314, Dresden, Germany
cCentre for Heavy Metals Research, School of Chemistry, University of Sydney, NSW, 2006, Australia
First published on 11th September 2007
Abstract
The synthesis and crystal structure of a new 34-membered N6O4-donor macrocycle 2 is reported. Solvent extraction experiments (water/chloroform) indicated that 2 acts as an efficient extractant towards silver(I) and zinc(II) at pH values beyond 6 while the extraction of iodide and chromate occurs below this pH. A competitive metal extraction experiment at pH 7.2 in which the perchlorate salts of cobalt(II), nickel(II), copper(II), zinc(II) and cadmium(II) were present together in the aqueous phase led to the following order of increasing extraction efficiency: cobalt(II) < nickel(II) < zinc(II) < copper(II) ∼ cadmium(II). A substantial synergistic enhancement of zinc(II) salt extraction was observed for a dual-host extraction system using the macrocycle 2 as cation binder and the tripodal thiourea ligand 3 as anion receptor; in particular, a notable extraction of zinc(II) sulfate was obtained.
Introduction
Mixed donor macrocyclic ligands such as 11,2 have long been used as receptors for a wide range of metal ions, often yielding metal complexes that exhibit enhanced kinetic and thermodynamic stabilities (the macrocyclic effect) as well as enhanced metal ion specificity towards individual metal ions.3 Large N,O-donor cyclic ligands have also been employed for binding metal ions, neutral molecules and anionic species. For these, the type of complex formed has been demonstrated to be strongly dependent on the number and arrangement of the respective donor atoms as well as on the nature of the spacer groups separating the donors.4 Herein we report the synthesis of the new 34-membered N6O4-donor macrocycle 2, a [2 + 2] analogue of 1. We also report the results of liquid–liquid extraction experiments used to explore the ability of 2 to act as both a cation receptor and an anion receptor under different pH conditions and compare these results with those for such of the known 17-membered N3O2-donor macrocycle 1.1
The potential of polyaza receptors to bind both cations5 and anions6–8 continues to receive considerable attention, with the relative binding behaviour being quite dependent on the protonation state of the amine sites present and thus on the pH employed for the studies. In contrast to the normally strong coordinate bonds used for binding of cations to such ligands, anion binding tends to be weaker and typically involves multiple hydrogen bond formation acting in concert with electrostatic interactions. The achievement of an appropriate balance of electrostatic and hydrogen bonding interactions between the anion and the aza ligand is normally a requirement for the occurrence of significant anion complexation. It needs to be noted that in particular instances π-interactions of various types may also contribute to the overall stability of such host–guest species.
Macrocycle 2 appeared suitable for use in solvent extraction studies for binding both cations and anions since it contains six nitrogen donors spanning a range of basicities that are available as both metal binding sites (in their non-protonated form) or as positively charged hydrogen-bond donors (in their protonated form) for a suitable anionic guest. Four ether functions are also present as potential metal binding sites; alternatively they may act as hydrogen bond acceptors for a protonated anionic guest. Further, the cyclic nature of this receptor coupled with the lipophilicity arising from the presence of six aromatic groups appear to be additional features that make 2 attractive as a potential solvent extractant for cation/anion binding studies. An interesting possibility to enhance the aqueous–organic phase transfer of cations in presence of hydrophilic anions with neutral macrocyclic ligands as 2 is based on their combination with a neutral anion receptor in a dual-host system.8,9 For this purpose we tested the previously reported tripodal thiourea ligand 3.10

Experimental
All reagents and solvents were purchased from commercial sources and were used without further purification. NMR spectra were recorded on Bruker Avance DPX200, DPX300 or DPX400 spectrometers. Low resolution electrospray ionisation mass spectra (ESI MS) were obtained on a Finnigan LCQ-8 spectrometer.Synthesis of 2
Macrocycle 2 was synthesised by an adaptation of the procedure previously reported for obtaining the corresponding [1 + 1] macrocycle 1.1 In contrast to the previous procedure where 1 was found to be the major product formed in the presence of manganese(II) as a templating ion, the reaction was performed in the present study in the absence of a metal template and under higher dilution conditions. The major product was found to be the corresponding [2 + 2] macrocyclic condensation product 2.2,6-Bis(2′-formylphenoxymethyl)pyridine (1.042 g, 3.0 mmol) and molecular sieves (20 g) were stirred in warm methanol (400 ml) for ∼0.5 h. Ethane-1,2-diamine (0.180 g, 3.0 mmol) in methanol (200 ml) was added dropwise at 50 °C over 1.5 h. The reaction mixture was refluxed for a further 30 min and allowed to cool. Sodium borohydride solid (1.1 g, 30 mmol) was added incrementally with stirring over several minutes. The reaction solution was refluxed again for 4 h, then filtered through Celite 521 which was washed thoroughly with absolute ethanol (3 × 20 ml). The filtrate was taken to dryness on a rotary evaporator. Water (100 ml) was added, the pH adjusted to ∼12, and the macrocyclic product was extracted into dichloromethane (3 × 50 ml). The organic phases were combined and dried over anhydrous sodium sulfate. The solution was filtered and the filtrate taken to dryness on a rotary evaporator to yield the product which was recrystallised from ethyl acetate to yield 2 as a white solid.
Yield 0.48 g (45%); mp 225–228 °C. MS (EI) m/z 751.4; C46H51N6O4 requires 751.4. Found: C, 72.8; H, 6.5; N, 10.7%. Calc. for C46H50N6O4·1/2H2O: C, 72.70; H, 6.76; N, 11.06%. 1H NMR (CDCl3) δ 2.77 (s, CH2CH2, 8 H), 3.87 (s, CH2N, 8 H), 5.16 (s, CH2O, 8 H), 6.88–6.96 (m, phenyl, 8 H), 7.18–7.26 (m, phenyl, 8 H), 7.40–7.45 (m, pyridine, 6 H), 13C NMR (CDCl3) δ 48.6, 49.7, 70.3, 111.5, 119.9, 121.0, 128.4, 128.7, 130.4, 137.6, 156.5, 156.8.
Synthesis of 3
The tripodal ligand 3 has been described previously10 but no full synthetic (nor characterisation) details have been reported, only reference to the synthesis of the analogous tris-urea ligand derivative being given.11 The following procedure was used for its preparation in the present study. Butyl isothiocyanate (3.53 g, 30.0 mmol) in dichloromethane (100 ml) was added dropwise to tris(2-aminoethyl)amine (1.54 g, 10.0 mmol) in dichloromethane (100 ml) and the reaction mixture was stirred for 3 h at room temperature. The solvent and excess reagent were removed under reduced pressure to yield 3 as a viscous yellow liquid.Yield, 4.34 g (86%). MS (EI) m/z 492.2; C21H45N7S3 requires 491.8. Found: C, 49.84; H, 9.04; N, 18.95%. Calc. for C21H45N7S3·H2O: C, 49.47; H, 9.29; N, 19.23%. 1H NMR (DMSO-d6) δ 0.85 (t, CH3, 9H), 1.28 (m, CH2, 6H), 1.44 (q, CH2, 6H), 3.08 (s, NCH2CH2, 6 H), 3.45 (s, CH2NH, 6H), 7.47 (s, NH, 3H), 7.77 (s, NH, 3H), 13C NMR (DMSO-d6) δ 182.6, 52.5, 43.4, 30.9, 19.6, 13.7.
X-Ray crystal structural data for 2
These were collected on a Bruker-Nonius APEX2-X8-FR591 diffractometer employing graphite-monochromated Mo-Kα radiation generated from a rotating anode (0.71073 Å) with ω and ψ scans.12 Data were collected at 150 K to approximately 56° 2θ. Data integration and reduction were undertaken with SAINT and XPREP13 and subsequent computations were carried out using the WinGX-32 graphical user interface.14 The structure was solved by direct methods using SIR97.15 Multi-scan empirical absorption corrections were applied to the data set using SADABS.16 Data were refined and extended with SHELXL-97.17 Non-hydrogen atoms were refined anisotropically. Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. Nitrogen bound hydrogen atoms were first located in the difference Fourier map before refinement with bond length restraints. Despite numerous recrystallisation attempts, the crystals employed in the study were extremely small, weakly diffracting and decayed rapidly out of solvent. As a result useful data was only recorded to a maximum of 50° 2θ despite long exposure times and equipment equipped with a rotating anode generating 5 kW. This is reflected in higher than ideal Rint values and a higher than ideal observed data/unique data ratio. Rint values at a resolution above 1.09 Å are well below 0.06. This, with good residuals, reflects the correct assignment of Laue class and space group.R1 = ∑||Fo| – |Fc||/∑|Fo| for Fo > 2σ(Fo); wR2 = (∑w(Fo2 – Fc2)2/∑(wFo2)2)1/2, all reflections w = 1/[σ2(Fo2) + (0.0618P)2 + 0.0000P] where P = (Fo2 + 2Fc2)/3.
CCDC reference number 658209.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b710641f
Liquid–liquid extraction
Water–chloroform extraction experiments were performed at 25 ± 1 °C in microcentrifuge tubes (2 ml) accompanied by mechanical shaking. The phase ratio V(org) : V(w) (500 µl each) was 1 : 1. The initial concentration of the cation or anion in the aqueous phase was 1 × 10–4 M. In the case of cation extraction, NaClO4 was also present at 5 × 10–3 M. The ligand concentration in the organic phase was 1 × 10–3 M. The shaking time was 30 min except for the competitive extraction experiment where 24 h was employed. Citric acid/NaOH (pH = 2.1–4.8), MES/NaOH (pH = 5.2–6.8) and HEPES/NaOH (pH = 7.1–8.0) buffer systems were used to adjust the pH of the aqueous phase in the extraction experiments. The equilibrium pH was confirmed in each case with the aid of an InLab micro pH electrode.After extraction, all two-phase samples were centrifuged and the phases separated. The distribution ratio D of the particular cation or anion (D = cion(org)/cion(w)) of interest was obtained from the concentrations in both phases determined radiometrically by γ-radiation measurements using 110mAg, 65Zn, 125I or 51Cr radioisotopes in a NaI(TI) scintillation counter (Cobra II/Canberra-Packard).18 For the competitive cobalt(II), nickel(II), copper(II), zinc(II) and cadmium(II) extraction experiment, the concentrations of each metal ion in the aqueous phase before and after extraction were determined using an ICP-MS (ELAN 9000/Perkin Elmer) spectrometer.
Results and discussion
Synthesis
The reported synthesis of the 17-membered N3O2-donor macrocycle 1 involves the use of a manganese(II) templated reaction between 2,6-bis(2′-formylphenoxymethyl)pyridine and ethane-1,2-diamine to yield the corresponding [1 + 1] cyclic diimine which was then reduced using sodium borohydride to produce 1 in 50% yield.1 In the present study the corresponding reaction between 2,6-bis(2′-formylphenoxymethyl)pyridine and ethane-1,2-diamine in the absence of the manganese(II) bromide template was investigated. In order to favour the possible production of larger oligomers over the [1 + 1] product, approximately seven-fold higher dilution conditions were employed over those used in the template procedure to produce 1. This modified procedure led to isolation of the corresponding [2 + 2] macrocyclic condensation product 2 which was recrystallised from ethyl acetate and isolated in 45% yield. The ESI mass spectrum indicated that a minor amount of the [1 + 1] adduct, 1, along with other by-product(s) were present as contaminants in the crude reaction product but these were successfully removed in the recrystallisation step. Single crystals of 2, suitable for X-ray analysis, were grown by slow diffusion of diethyl ether vapour into a chloroform solution of the above product. As expected, the 1H NMR spectrum of 2 is very similar to that of 1.Structure
The X-ray structure of 2 is presented in Fig. 1 and confirms its [2 + 2] stoichiometry. The molecule is located around an inversion centre and adopts an open structure with the pyridyl nitrogen atoms pointing away from the centre of the ring. The arrangement is stabilised by intramolecular hydrogen bonding involving the ethane-1,2-diamine fragments (N(3)–H(2A)⋯N(2)) as well as by the presence of short C–H(phenylene)⋯O(ether) interactions, (C(3)–H(3A)⋯O(1) and C(5)–H(5A)⋯O(2)). | ||
| Fig. 1 ORTEP plot of 2 with 50% probability ellipsoids. Dashed lines indicate intramolecular hydrogen bonds. Symmetry operator used to generate equivalent atoms: 1 – x, 1 – y, 1 – z. | ||
In the structure no direct π–π stacking interactions are evident. However short C–H(methylene)⋯π contacts present (C(6)–H(6A)⋯Cg(1), C(23)–H(23A)⋯Cg(2)) result in the formation of a ribbon-like arrangement throughout the structure (Fig. 2 and Table 1). These ribbons are arranged in a two-dimensional fishbone pattern. Each fishbone layer lies in the bc-plane and stack along the a-axis. No significant interactions between adjacent ribbons or layers is observed.
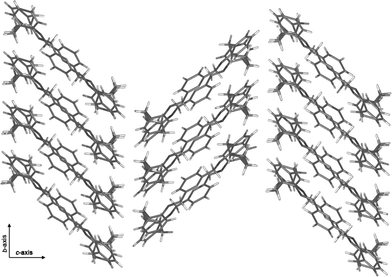 | ||
| Fig. 2 C–H(methylene)⋯π contacts lead to ribbons, which are arranged in layers of a fishbone motif throughout the crystal. Schematic representation of part of one layer, viewed along the a-axis. | ||
| Intramolecular D–H⋯A interactions | |||||
|---|---|---|---|---|---|
| D–H | A | D–H | H⋯A | D⋯A | D–H⋯A |
| N(3)–H(2N) | N(2) | 0.87 | 2.56 | 2.983 | 111 |
| C(3)–H(3A) | O(1) | 0.95 | 2.45 | 2.762 | 152 |
| C(5)–H(5A) | O(2) | 0.95 | 2.41 | 2.732 | 154 |
Liquid–liquid extraction
In order to investigate the solvent extraction properties of 2 towards selected cations and anions, single-ion extraction experiments for silver(I) (pH 6.1) and zinc(II) (pH 7.2) as well as iodide (pH 5.5) and chromate (pH 5.5) were preformed using the previously described radiotracer technique.18 The aqueous phase pH values were chosen for the silver and zinc extractions to avoid complications arising from the onset of metal ion hydrolysis.The anion extractions were performed at the lower pH of 5.5 since the amine functions of 2 will be significantly protonated at this pH and hence available as hydrogen-bond donors for anion binding. It needs to be noted that the use of 'chromate' in the following discussion refers to a pH-dependent equilibrium mixture of CrO42–, HCrO4– and Cr2O72– in the respective aqueous phases.19 Considering the low Cr(VI) concentration and the weak acidic conditions employed, as well as the single negative charge on the chromate anion HCrO4–, it appears that this ion may be the one extracted; this observation accords with the extraction results discussed below.
The extraction results are summarised in Fig. 3. Under the pH conditions mentioned above, the use of 2 results in the quantitative extraction of silver(I), while zinc(II) is extracted at 82%. The observed percentage extractions of iodide and chromate under these conditions were 40 and 16%, respectively; the relative extraction efficiencies thus fall in the order predicted by the Hofmeister series reflecting the lipophilicity differences of both anions.20
![Percentage extraction of chromate, iodide, zinc(ii) and silver(i) by 1 and 2. Cation extraction: [Zn(ClO4)2, AgClO4] = 1 × 10–4 M, [NaClO4] = 5 × 10–3 M, pH = 6.1 (MES/NaOH buffer) for silver(i) and pH = 7.2 (HEPES/NaOH) for zinc(ii); Anion extraction: [NaI, Na2CrO4] = 1 × 10–4 M, pH = 5.5 (MES/NaOH buffer); [1, 2] = 1 × 10–3 M in CHCl3; T = 25 ± 1 °C.](/image/article/2008/NJ/b710641f/b710641f-f3.gif) | ||
| Fig. 3 Percentage extraction of chromate, iodide, zinc(II) and silver(I) by 1 and 2. Cation extraction: [Zn(ClO4)2, AgClO4] = 1 × 10–4 M, [NaClO4] = 5 × 10–3 M, pH = 6.1 (MES/NaOH buffer) for silver(I) and pH = 7.2 (HEPES/NaOH) for zinc(II); Anion extraction: [NaI, Na2CrO4] = 1 × 10–4 M, pH = 5.5 (MES/NaOH buffer); [1, 2] = 1 × 10–3 M in CHCl3; T = 25 ± 1 °C. | ||
It is interesting to note that comparative extraction experiments under similar conditions using the smaller [1 + 1] macrocycle 1 (see Fig. 3) show similar high extraction for silver(I) (99%) whereas for the other ions significantly lower values were obtained: zinc(II) 4%, iodide 23% and chromate 3%. Clearly a number of factors, such as different lipophilicities, different number of donors in their donor sets and perhaps also different stoichiometries of extracted species, will influence the extraction behaviour of these isomeric ligand systems and hence any attempt at direct (quantitative) comparison of the respective systems is inappropriate in the absence of more detailed studies.
Competitive cation extraction
In order to probe the actual metal ion selectivity of 2, a competitive extraction experiment (water/chloroform) was carried out at pH 7.2 (HEPES/NaOH) in which equal concentrations of the perchlorate salts of cobalt(II), nickel(II), copper(II), zinc(II) and cadmium(II) (each at 1 × 10–4 M) were present in the aqueous phase. The concentration of 2 in the chloroform phase was 1 × 10–3 M. The sequence of the percent extraction observed follows the order cobalt(II) (11%) < nickel(II) (50%) < zinc(II) (78%) < copper(II) (100%) ∼ cadmium(II) (99%). This order agrees with the Irving–Williams order of stability constants for the first row transition elements (as also obtained experimentally for ligand 1).1 Also the reversion of the normal stability sequence of Zn(II) over Cd(II) has been reported already for the macrocycle 1.1 Generally, from these results it can be postulated that the extraction order for 2 is controlled mainly by differences in the stability constants, with other factors playing only a minor role.pH Dependence of anion extraction
In order to determine the optimum pH conditions for anion extraction by 2, extraction experiments were carried at intervals over the pH range 2.0–8.0 in the case of iodide and 3.3–7.6 for chromate. The buffer systems citric acid/NaOH, MES/NaOH and HEPES/NaOH were employed to adjust the respective pH values. Plots for percentage iodide and chromate extraction against the equilibrium pH of the aqueous phase are shown in Fig. 4. The maximum extraction for iodide was ∼60% and this value was maintained over the approximate pH range 2–5. The near constant extraction value for this range suggests that two factors influencing the efficiency of anion extraction – ionic attraction caused by higher protonation of the N-donor functions and higher water solubility arising from the generation of charged species – may to some extent cancel each other over this pH range. For pH values higher than 5.0, iodide extraction progressively decreases until extraction is <5% at a pH >7.6. As a consequence, the stripping of iodide from the organic phase will thus be possible under moderately basic conditions.![pH dependence of the extraction of iodide (■) and chromate (◆). [NaI, Na2CrO4] = 1 × 10–4 M; [2] = 1 × 10–3 M in CHCl3; T = 25 ± 1 °C; pH = 2.0–4.8 (citric acid/NaOH buffer), pH = 5.0–6.5 (MES/NaOH buffer), pH = 7.1–8.0 (HEPES/NaOH buffer).](/image/article/2008/NJ/b710641f/b710641f-f4.gif) | ||
| Fig. 4 pH dependence of the extraction of iodide (■) and chromate (◆). [NaI, Na2CrO4] = 1 × 10–4 M; [2] = 1 × 10–3 M in CHCl3; T = 25 ± 1 °C; pH = 2.0–4.8 (citric acid/NaOH buffer), pH = 5.0–6.5 (MES/NaOH buffer), pH = 7.1–8.0 (HEPES/NaOH buffer). | ||
In the case of chromate the total extraction achieved is smaller. Maximum extraction of 30% was observed at pH 3.3 and by a pH of 7.3 this had fallen to less than 1%, with a plateau of about 18% occurring between pH 4.2–5.5. The presence of the pH-dependent chromate equilibrium mentioned above needs to be considered in relation to this result. At higher pH values the CrO42– species will dominate and the double charge is expected to inhibit extraction efficiency, whereas at lower pH values interference from the oxidising character of the Cr2O72– ion becomes important: precipitate formation at the interface and a significant colour change was observed below pH ∼3. It is perhaps significant that the equilibrium concentration of the mono-protonated species, HCrO4–, is substantial19 over the pH range for which chromate is observed to be most efficiently extracted.
Stoichiometry of the extracted anion species
Extraction experiments over a range of ligand concentrations (5 × 10–4–2.5 × 10–3 M) were carried out in order to probe the anion : ligand ratio present in the extracted anion complexes. Provided a ‘simple’ equilibrium is involved, the slope (s) of the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Danionvs. logL(org) plot yields this ratio directly. Plots for iodide and chromate are given in Fig. 5, where Danion is the distribution ratio of the anion between the organic and aqueous phases.
Danionvs. logL(org) plot yields this ratio directly. Plots for iodide and chromate are given in Fig. 5, where Danion is the distribution ratio of the anion between the organic and aqueous phases.
![Plots for the variable ligand concentration experiments for 2 with iodide (■) and chromate (◆). [NaI, Na2CrO4] = 1 × 10–4 M; pH = 5.5 (MES/NaOH buffer); [2] = 5 × 10–4–2.5 × 10–3 M in CHCl3; shaking time 30 min; T = 25 ± 1 °C, slopes: s (iodide) = 0.98, s (chromate) = 1.15.](/image/article/2008/NJ/b710641f/b710641f-f5.gif) | ||
| Fig. 5 Plots for the variable ligand concentration experiments for 2 with iodide (■) and chromate (◆). [NaI, Na2CrO4] = 1 × 10–4 M; pH = 5.5 (MES/NaOH buffer); [2] = 5 × 10–4–2.5 × 10–3 M in CHCl3; shaking time 30 min; T = 25 ± 1 °C, slopes: s (iodide) = 0.98, s (chromate) = 1.15. | ||
The observed slopes for iodide (∼1.0) and chromate (∼1.2) indicate the formation of 1 : 1 (anion : ligand) complexes in the respective organic phases. The similarity of the slope for chromate to that for (singly charged) iodide suggests that in the former case the mono-protonated species, HCrO4–, is likely the favoured extracted species under the conditions employed.
Dual-host extraction
The use of a dual-host extraction system, namely one extractant for the cation and one for the anion, as a strategy for achieving metal salt extraction in two-phase systems has been of increasing interest over recent years.8,9 In the present study we have applied such a strategy to the extraction of zinc(II) sulfate using both 2 and the tripodal tris-thiourea derivative 3; the latter has been well documented previously to act as an anion-binder in non-aqueous media.10 Subsequent comparative experiments were also performed in which the more lipophilic anions NO3–, ClO4– and Pic– were present in the aqueous phase. In these experiments NaClO4, NaNO3 or picric acid (HPic) was added to the aqueous phase containing zinc sulfate (at 1 × 10–4 M) to produce 2 × 10–4 M concentrations of each new anion. Na2SO4 was also similarly added to the initial zinc sulfate solution to produce a solution that was overall 3 × 10–4 M with respect to sulfate. In all cases a pH of 7.7 (HEPES/NaOH buffer) was used to ensure that no significant anion extraction was exhibited by 2 (see earlier discussion).A series of comparative extraction experiments involving the use of 2 and 3 alone as well as in combination under otherwise comparable conditions was undertaken. Clearly, the results show a significant synergistic enhancement of zinc(II) extraction (see Table 2) when both receptors 2 and 3 (64–76%) were present in the chloroform phase in comparison to the analogous experiments in the presence of either 2 or 3 alone (with the respective individual percentage extractions summed). This fact illustrates convincingly the efficiency of the studied dual-host system. It is especially noticeable that the use of the dual-host system compensates for the pronounced hydrophilicity of sulfate and achieves comparable percentage extraction for all anions investigated.
Under the conditions employed, the zinc(II) extraction efficiencies by 2 alone follow the Hofmeister series20 reflecting the hydrophilicity sequence of the counter anions employed: sulfate (7%) < nitrate (8%) < perchlorate (25%) < picrate (29%), whereas in the case of 3 alone, as expected, the metal extraction percentages are only small.
Concluding remarks
The present study clearly confirms that under appropriate pH conditions the new macrocyclic reagent 2 acts as both a metal ion and anion extractant for the cation and anion types investigated. The extraction efficiency of 2 is higher for all systems studied than for the smaller macrocycle 1. Further, at pH 7.7, when 2 is combined with the known anion-binding tripodal thiourea ligand 3, the resulting dual-host system exhibits substantial synergism towards metal salt extraction in the presence of a range of anions of varying size, charge and lipophilicity.Acknowledgements
We thank the Australian Research Council and the Deutsche Forschungsgemeinschaft for support.References
- (a) D. E. Fenton, B. P. Murphy, A. J. Leong, L. F. Lindoy, A. Bashall and M. McPartlin, J. Chem. Soc., Dalton Trans., 1987, 2543 RSC; (b) N. A. Bailey, D. E. Fenton, S. J. Kitchen, T. H. Lilley, M. G. Williams, P. A. Tasker, A. J. Leong and L. F. Lindoy, J. Chem. Soc., Dalton Trans., 1991, 627 RSC; (c) D. E. Fenton, Pure Appl. Chem., 1993, 65, 1493 CAS.
- E. Bértolo, R. Bastida, A. de Blas, D. E. Fenton, C. Lodeiro, A. Macías, A. Rodríguez and T. Rodríguez-Blas, J. Inclusion Phenom. Macrocycl. Chem., 1999, 35, 191 Search PubMed.
- (a) L. F. Lindoy, The Chemistry of Macrocyclic Ligand Complexes, Cambridge University Press, Cambridge, UK, 1989 Search PubMed; (b) J. D. Chartres, L. F. Lindoy and G. V. Meehan, Coord. Chem. Rev., 2001, 249, 216.
- (a) K. R. Adam, G. Anderegg, K. Henrick, A. J. Leong, L. F. Lindoy, H. C. Lip, M. McPartlin, R. J. Smith and P. A. Tasker, Inorg. Chem., 1981, 20, 4048 CrossRef CAS; (b) M. W. Hosseini, J. Comarmond and J.-M. Lehn, Helv. Chim. Acta, 1989, 72, 1066 CAS; (c) C. Bazzicalupi, A. Bencini, E. Berni, S. Ciattini, A. Bianchi, C. Giorgi, P. Paoletti and B. Valtancoli, Inorg. Chim. Acta, 2001, 317, 259 CrossRef CAS; (d) C. Lodeiro, R. Bastida, E. Bertolo, A. Macías and A. Rodríguez, Polyhedron, 2003, 22, 1701 CrossRef CAS; (e) C. Lodeiro, J. L. Capelo, E. Bértolo and R. Bastida, Z. Anorg. Allg. Chem., 2004, 630, 1110 CrossRef CAS; (f) C. Bazzicalupi, A. Bencini, A. Bianchi, C. Duce, P. Fornasari, C. Giorgi, P. Paoletti, R. Pardini, M. R. Tinè and B. Valtancoli, Dalton Trans., 2004, 463 RSC; (g) E. Bértolo, R. Bastida, D. E. Fenton, C. Lodeiro, A. Macías, A. Rodríguez, F. Li, R. Delgado, A. Coelho, M. G. B. Drew and V. Félix, Tetrahedron, 2006, 62, 8550 CrossRef CAS.
- See, for example: (a) A. Bianchi, M. Micheloni and P. Paoletti, Coord. Chem. Rev., 1991, 110, 17 CrossRef CAS; (b) L. F. Lindoy, Pure Appl. Chem., 1997, 69, 2179 CrossRef CAS; (c) J. R. Price, M. Fainerman-Melnikova, R. R. Fenton, K. Gloe, L. F. Lindoy, T. Rambusch, B. W. Skelton, P. Turner, A. H. White and K. Wichmann, Dalton Trans., 2004, 3715 RSC; (d) F. Li, R. Delgado and V. Felix, Eur. J. Inorg. Chem., 2005, 4550 CrossRef CAS; (e) B. Antonioli, D. J. Bray, J. K. Clegg, K. Gloe, O. Kataeva, L. F. Lindoy, J. C. McMurtrie, P. J. Steel, C. J. Sumby and M. Wenzel, Dalton Trans., 2006, 4783 RSC , and references therein.
- (a) Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. Garcia-Espana, Wiley-VCH, New York, 1997 Search PubMed; (b) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, RSC Publishing, Cambridge, 2006 Search PubMed.
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS; (b) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 487 CrossRef; (c) C. A. Ilioudis and J. W. Steed, J. Supramol. Chem., 2001, 1, 165 CrossRef CAS; (d) J. M. Llinares, D. Powell and K. Bowman-James, Coord. Chem. Rev., 2003, 240, 57 CAS; (e) J. M. Llinares and K. Bowman-James, in Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, Marcel Dekker, New York, 2004, p. 1170 Search PubMed; (f) S. Kubik, C. Reyheller and S. Stüwe, J. Inclusion Phenom. Macrocycl. Chem., 2005, 52, 137 Search PubMed; (g) E. García-España, P. Díaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952 CrossRef CAS.
- (a) K. Wichmann, B. Antonioli, T. Söhnel, M. Wenzel, K. Gloe, J. R. Price, L. F. Lindoy, A. J. Blake and M. Schröder, Coord. Chem. Rev., 2006, 250, 2987 CrossRef CAS; (b) K. Gloe, B. Antonioli, K. Gloe and L. F. Lindoy, in Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, Marcel Dekker, New York, 2006, online version Search PubMed; (c) K. Gloe, H. Stephan and M. Grotjahn, Chem. Eng. Technol., 2003, 26, 1107 CrossRef CAS.
- K. Kavallieratos, R. A. Sachleben, G. J. van Berkel and B. A. Moyer, Chem. Commun., 2000, 187 RSC.
- (a) K.-S. Jeong, K.-M. Hahn and Y. L. Cho, Tetrahedron Lett., 1998, 39, 3779 CrossRef CAS; (b) A. Arduini, G. Giorgi, A. Pochini, A. Secchi and F. Ugozzoli, J. Org. Chem., 2001, 66, 8302 CrossRef CAS; (c) A. Arduini, E. Brindani, G. Giorgi, A. Pochini and A. Secchi, J. Org. Chem., 2002, 67, 6188 CrossRef CAS.
- Bruker-Nonius, APEX v2.1, SAINT v.7 and XPREP v.6.14. Bruker AXS Inc., Madison, WI, USA, 2003.
- Bruker-Nonius, SAINT and XPREP. Bruker AXS Inc. Madison, WI, USA, 2003.
- WinGX-32: System of programs for solving, refining and analysing single crystal X-ray diffraction data for small molecules: L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giocavazzo, A. Guagliardi, A. G. C. Moliterni, G. Polidori and S. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, SADABS: Empirical Absorption and Correction Software, University of Göttingen, Germany, 1999–2003 Search PubMed.
- G. M. Sheldrick, SHELXL-97: Programs for Crystal Structure Analysis, University of Göttingen, Germany, 1997 Search PubMed.
- F. Werner and H.-J. Schneider, Helv. Chim. Acta, 2000, 83, 465 CrossRef CAS.
- H. Stephan, S. Juran, B. Antonioli, K. Gloe and K. Gloe, in Analytical Methods in Supramolecular Chemistry, ed. Ch. Schalley, Wiley-VCH, Weinheim, 2006, p. 79 Search PubMed.
- (a) T. Shen-Yang and L. Ke-An, Talanta, 1986, 33, 775 CrossRef; (b) M. M. Hoffmann, J. G. Darab and J. L. Fulton, J. Phys. Chem. A, 2001, 105, 1772 CrossRef CAS.
- (a) F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 247; (b) W. Kunz, P. Lo Nostro and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 1 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |