Molecular design of inhibitors against the Mproprotein of the severe acute respiratory syndrome (SARS) virus
Jesus
Olivero-Verbel
a,
Isaías
Lans
b,
Emiliano
Martinez
b,
Isaura
Ospino
b,
Angelica
Padilla
b and
Ricardo
Vivas-Reyes
*b
aEnvironmental and Computational Chemistry Group, Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Cartagena, Campus de Zaragocilla, Cartagena, Colombia Web: www.reactivos.com
bTheoretical and Quantum Chemistry Group, Facultad de Ciencias Naturales y Exactas, Universidad de Cartagena, Campus de Zaragocilla, Cartagena, Colombia. E-mail: rvivasr@unicartagena.edu.co
First published on 8th November 2007
Abstract
Several outbreaks of Severe Acute Respiratory Syndrome (SARS) have alerted the health systems of the world this century. The treatment of SARS patients with inhibitors, targeting proteins similar to those observed in this coronavirus, has not achieved satisfactory results, and currently there is no effective treatment for its control. The main proteinase of SARS coronavirus (3CLpro or Mpro) is crucial for its replication process, and therefore it is a potential target for the development of anti-SARS drugs. This protease is constituted of two protomers (A and B), displaying an active site formed by the catalytic dyad His-41 Cys-145. In this work, the structure of Mpro (PDB 1UJ1) has been optimized using an MMFF94s force field and docked with AG7088, an inhibitor of the human rhinovirus 3C protease. This protein has structural and functional similarities with SARS Mpro. Docking between AG7088 and the protomers A and B of Mpro, using the FlexX/Cscore program of SYBYL 7.0, showed that the calculated protein–ligand binding affinity was more favorable for the complex with Protomer A than for Protomer B. In order to find a good inhibitor with a high protein binding affinity (BA), 1250 molecules derived from AG7088 were docked with protomer A of Mpro. Molecules (83 inhibitors) with BAs lower than −31 kcal mol−1 (BA of AG7088), and also capable of interacting with the protein within the active site pocket, were considered possible inhibitors of Mpro. The average BA of ligands in all complexes was −36 kcal mol−1, and the best presented a BA of −49 kcal mol−1, suggesting that this example could act as a good Mpro competitive inhibitor. The ligand, named GQAC-02441, was also docked with the non-optimized structure of 1UJ1, generating satisfactory BA values. Taken together, our results propose a new high affinity ligand for SARS Mpro, with inhibitory properties independent of slight structural changes in the protease.
Introduction
The last reported SARS outbreak occurred in 2003,1 and the risk of infection has still not been eliminated. This fact has driven considerable efforts to study the SARS virus, to be sufficiently prepared for if it appears again. This type of study can be performed in different ways; one of them is to carry out computational studies that may help to understand some of the fundamental aspects of virus control by using inhibitors of viral proteins.The main proteinase of the SARS coronavirus is called 3CLpro or Mpro.2 This proteinase plays an importance roll in the replication process of the virus, and is formed by two subunits or protomers (PA and PB) displaying an active site containing a catalytic dyad formed by His-41 and Cys-145.3–6 Because Mpro is fundamental for viral maturation, it is considered an important target for the design of drugs against SARS and other infections caused by coronaviruses.3,7 A molecule called AG7088 (Fig. 1) is an anti-rhinovirus compound that inhibits the replication of several viruses, such as the transmissible gastroenteritis virus (TGEV), the murine hepatitis virus (MHV), the bovine coronavirus (BCoV) and human rhinoviruses (HRV), amongst others.8 Therefore, it has been proposed as a starting point for the design of anti-SARS molecules.3 The selection of this compound was based on the functional and structural similarities that exist between the active site of the protease 3CLpro in rhinoviruses and the Mpro of SARS-CoV.3,9–12
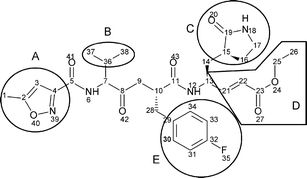 | ||
| Fig. 1 Structure of AG7088. Lateral groups susceptible to structural modifications are enclosed and named as A–E. | ||
Having a template molecule and a recognized active site for a target protein, the design of possible inhibitors for the Mpro of SARS-CoV could be performed using computational and bioinformatic tools. One of the most widely used approaches for molecular design includes the use of protein–ligand docking.13 This technique allows the generation of multiple docking conformations for each ligand molecule on the protein.14 Depending on the software, conformational searching can be achieved using different methods, such as genetic algorithms,15,16 Monte Carlo searches,17,18 simulated annealing,17 distance geometry,19 or incremental construction,20,21 amongst others. However, independent of the applied method, the process of finding optimal conformers must be guided by a scoring function (energy function), which is used to evaluate the physicochemical interactions between the protein and the ligand. The most frequently used scoring functions used are: Chemscore,22 D-score,23 PMF-score24 and Gscore.25 Each has its own criteria for model evaluation, and includes energies of hydrophilic or hydrophobic interactions, van der Waals energies and de-solvation or solvation energies, amongst others.26 Moreover, the use of a consensus score for evaluating ligand–receptor interactions seems to be more robust and accurate than any particular function.13
Methods
Optimization of the crystalline structure of Mpro
The 3D structure of SARS Mpro was obtained from the Protein Data Bank (PDB entry code 1UJ1). Modelling, visualization and optimization were performed with the SYBYL suite of programs (Tripos, Inc., St. Louis, MO). Hydrogen atoms and lone pairs were added to the X-ray crystal structure using the SYBYL Biopolymer and Build/Edit menu tools. The initial orientation of the hydrogen atoms were set to be the same as those used by dictionary definitions.27 Each one of the protomers of SARS Mpro were individually optimized with the molecular force field MMFF94s28 to an energy gradient of 0.05 kcal mol−1 Å−1 using the conjugate gradient method,29 and a maximum displacement gradient cut-off of 0.01 Å.Docking Mpro–AG7088
The structure of the ligand ethyl 4-[2-[(4-fluorophenyl)methyl]-6-methyl-5-(5-methyloxazol-3-yl) carbonylamino-4-oxo-heptanoyl] amino-5-(2-oxopyrrolidin-3-yl)-pentanoate (AG7088) was optimized using the MMFF94s force field.28 Employing the FlexX program,20 this geometry was submitted to exhaustive conformational sampling on optimized structures of SARS Mpro protomers A and B (PA and PB). The search generated 100 plausible conformations for AG7088 at the active site, which was defined as all those residues found within 6.5 Å of Cys-145 and His-41 on both protomers. FlexX automatically placed ligands inside this pre-defined active site by considering both geometrical and electronic constraints. The applied scoring functions were the total score (from FlexX), the empirical scoring function Chemscore,22 D-score, Gscore and the knowledge-based potential PMF-score,24 all of them implemented within the Cscore (Consensus Score) module in SYBYL, allowing the most robust and accurate possible evaluation of the conformations for ligand–receptor interactions.30 However, when different conformations had the same Cscore, the consensus suggested by Wang and co-workers (2003)26 was applied. Sometimes, even the use of two consensus scores assigned equal values for different conformations. In this case, in order to select the best conformation, the Chemscore function was used as the definitive criterion.Design of possible inhibitors
Based on the conformations with the best scoring values, once AG7088 had been docked with the Mpro protomers, some structural modifications of AG7088 (Fig. 1) were considered in order to design molecules with possible anti-SARS activity. These changes were made based on the detected interactions between AG7088 and the residues at the active site of PA of Mpro (PA-Mpro), and were intended to increase both the electrostatic and hydrophobic interactions. To facilitate the design, some specific substituents were used as lateral groups (A–E) along the linear skeleton of the inhibitor. In total, 1250 structures were tested through docking procedures with the PA-Mpro. Two basic criteria were used to include changes to the ligand structure: the achievement of better interactions with PA-Mpro (Table 1, blue structures) and the use of lateral groups present in other high-binding inhibitors (red structures).

|
Docking of Mproinhibitors to Mpro protomers
The FlexX algorithm was used to dock the structures of the theoretical ligands into the ligand-binding pocket of PA and PB of Mpro. For each conformer, the binding affinity was evaluated by following the same criteria as those applied for the docking of AG7088.Statistical analysis
Box-Plot analysis was used to display the distribution of binding affinity data for the designed theoretical ligands. The resulting graph plots a five number summary: minimum, first quartile, median, third quartile and maximum; extremely high maximums or low minimums being called outliers.Results and discussion
Optimization of Mpro and docking to AG7088
Once the optimization process (after the adding of hydrogen atoms on both protomers of Mpro) had been completed, a quality assessment of the models was performed in order to find indicators of poorly calculated/folded regions. This was achieved by comparing Ramachandran plots, calculated using Rampage (Table 2), and computing root mean square deviation (RMSD) values by superposing optimized and X-ray structures (Table 3).| Residues by regions | PA | PB | ||
|---|---|---|---|---|
| Non-optimized | Optimized | Non-optimized | Optimized | |
| Favoured region | 290 (97.0%) | 273 (91.3%) | 283 (94.3%) | 278 (92.7%) |
| Allowed region | 8 (2.7%) | 25 (8.4%) | 13 (4.3%) | 19 (6.3%) |
| Outlier region | 1 (0.3%) | 1 (0.3%) | 4 (1.3%) | 3 (1.0%) |
| Protomers | RMSD/Å | |||
|---|---|---|---|---|
| Cα | Backbone | Side chains | All | |
| PA | 1.0737 | 1.1077 | 1.5957 | 1.3663 |
| PB | 1.1524 | 1.1626 | 1.7348 | 1.4688 |
Optimized structures of protomers were also subjected to an additional check with Prosa 200331 software, which uses knowledge-based potentials to generate scores reflecting the quality of protein structures in terms of a Z-score. It indicates the overall model quality and measures the deviation of the total energy of the structure with respect to an energy distribution derived from random conformations.32 The results showed that for PA, the overall model qualities of the non-optimized and optimized geometries presented Z-scores of −7.29 and −7.08, respectively, while for PB, the values were 7.66 and −7.01, respectively. This indicates that the optimization, after adding hydrogens, did not significantly modify the 3D-structure of the protomers, as seem in the quality graph generated with a window of 50 residues (Fig. 2) and the superposition of protein structures for both protomers (Fig. 3).
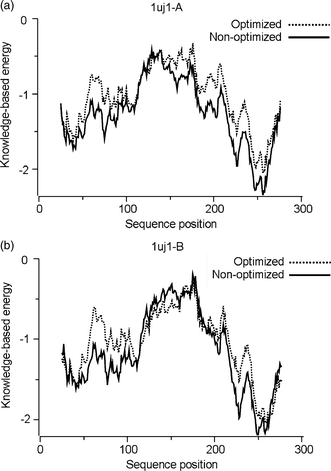 | ||
| Fig. 2 Local model quality graph generated for PA and PB of SARS Mpro. | ||
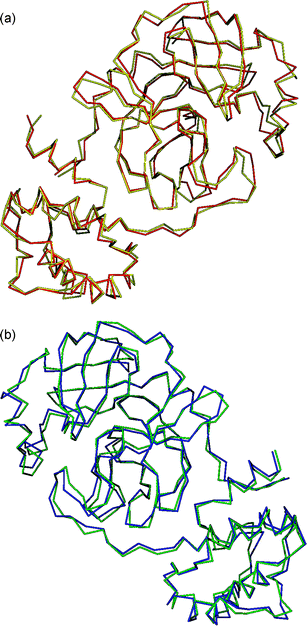 | ||
| Fig. 3 Superpositioning of optimized (yellow and blue) and non-optimized (red and green) structures of PA (top) and PB (bottom). | ||
Once structural models for both protomers of Mpro were obtained, scoring functions for the docking of AG7088 revealed that the ligand has the best binding affinity when bound to PA (Table 4). PB had two favorable function scores, whereas PA had three; additionally, only PA reached a Cscore value of 5.
| Conformation | Total score | Gscore | PMF-score | D-score | Chemscore | Cscore |
|---|---|---|---|---|---|---|
| A_001 | −4.9 | −206.7 | −25.6 | −154.3 | −31.0 | 5 |
| A_002 | −4.9 | −198.9 | −26.5 | −151.8 | −30.4 | 5 |
| B_001 | −5.9 | −233.6 | 25.4 | −139.5 | −24.1 | 4 |
| B_003 | −5.5 | −214.7 | 21.7 | −139.6 | −24.8 | 4 |
The binding site of PA-Mpro, and the complex formed with AG7088 after optimization and docking using FlexX, are shown in Fig. 4a and 4b, respectively. PA displays a cavity with differentiated sites for ligand binding. Once the complex is formed, this cavity is filled by the ligand, but some possible sites still seem to be partially empty. However, this coupling interaction is enough to inhibit the enzymatic activity of this molecule.
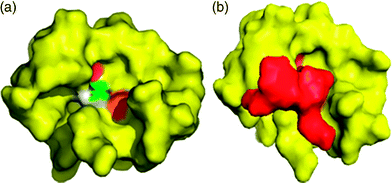 | ||
| Fig. 4 Docking between the optimized PA-Mpro and AG7088. (a) Model of PA showing the amino acids present in the active site in different colors. (b) Complex formed by PA-Mpro (yellow) and AG7088 (red). | ||
The contact area for the complex PA-Mpro:AG7088 is shown in Fig. 5. An important feature of the activity of AG7088 is its capacity to form a covalent bond through C21 to a cysteine group of the protein.33,34 This interaction does not occur in this theoretical docking complex. The distance between C21 and Cys-145 is 4.66 Å, suggesting that the binding probability to PA-Mpro is low. However, other reported interactions between AG7088 and proteins similar to Mpro are indeed present in the complex.33,34 These are hydrogen bonds with His-163 and Glu-166, as well as for His-41. Hydrophobic interactions include Met-165 and Phe-181 with C2 and C1, respectively, and Met-49 with the fluorophenyl ring.
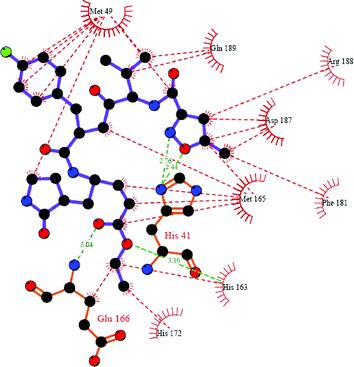 | ||
| Fig. 5 Protein–ligand interactions present in the complex PA-Mpro:AG7088, as displayed by Ligplot35 and SYBYL 7.0. Red lines represent hydrophobic interactions, whereas hydrogen bonds are showed in green. | ||
The docking process performed using PB showed a different pattern to that seen with PA. AG7088 does not fit completely into the active site of PB, and some empty spaces can be visualized (Fig. 6). Moreover, the calculated FlexX/Chemscore binding affinity is less favorable for the complex with the PB (−25 kcal mol−1) than that measured for PA (−31 kcal mol−1). These observations may suggest that the protomers have different conformations, and therefore unequal proteolytic activities, in agreement with what has been reported previously by Yang and co-workers.7 Based on the differences in binding affinities and conformational gaps, it is evident from both a geometric and energetic point of view, that PA generates the best complex with the ligand. Accordingly, all successive calculations were carried out using PA-Mpro.
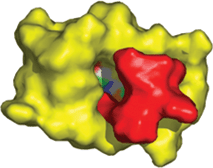 | ||
| Fig. 6 Docking between the PB of Mpro (yellow) and AG7088 (red). It is clear that the inhibitor does not fit completely into the active site (shown with different colors on the protein). | ||
Design of theoretical inhibitors
In total, 1250 molecules were generated and optimized using the MMFF94s force field. The optimal conformations during the docking were searched, applying the consensuses previously described. To determine the ligand that may be an effective inhibitor, the following factors were taking in to account: 1. Molecules whose docking to PA-Mpro could provide Cscore values equal to five; 2. Ligands that interact with the protein within the active site pocket in such way that they may generate competitive inhibition; 3. The newly designed inhibitors must have better Chemscore binding affinities than AG7088 (−31 kcal mol−1). Protein ligands that fulfilled these criteria are presented in Table 5. These 83 molecules could be considered as theoretical inhibitors of PA-Mpro.| Ligand | BA/kcal mol−1 | Ligand | BA/kcal mol−1 | Ligand | BA/kcal mol−1 | Ligand | BA/kcal mol−1 |
|---|---|---|---|---|---|---|---|
| 122 | −33 | 1423 | −38 | 3401 | −41 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 041 041 |
−41 |
| 144 | −33 | 1430 | −35 | 3411 | −34 | 11043 |
−34 |
| 222 | −32 | 2420 | −39 | 3441 | −39 | 11344 |
−32 |
| 322 | −32 | 2422 | −31 | 4000 | −34 | 11422 |
−40 |
| 341 | −33 | 2441 | −48 | 4012 | −32 | 11443 |
−32 |
| 402 | −38 | 3003 | −35 | 4021 | −36 | 12002 |
−35 |
| 423 | −38 | 3021 | −32 | 4023 | −32 | 12221 |
−33 |
| 444 | −44 | 3023 | −34 | 4213 | −32 | 12402 |
−36 |
| 1000 | 34 | 3041 | −33 | 4233 | −32 | 12412 |
−37 |
| 1020 | −34 | 3112 | −34 | 4343 | −33 | 13001 |
−37 |
| 1023 | −41 | 3121 | −31 | 4403 | −33 | 13002 |
−36 |
| 1040 | −49 | 3140 | −39 | 10002 |
−33 | 13011 |
−34 |
| 1041 | −39 | 3141 | −39 | 10023 |
−32 | 13013 |
−35 |
| 1043 | −46 | 3220 | −36 | 10044 |
−34 | 13021 |
−39 |
| 1044 | −46 | 3221 | −35 | 10240 |
−34 | 13042 |
−41 |
| 1121 | −31 | 3222 | −34 | 10421 |
−40 | 13043 |
−35 |
| 1123 | −31 | 3233 | −33 | 10424 |
−39 | 13044 |
−35 |
| 1144 | −35 | 3244 | −33 | 11012 |
−32 | 13143 |
−34 |
| 1341 | −33 | 3340 | −43 | 11022 |
−37 | 13144 |
−41 |
| 1343 | −39 | 3341 | −46 | 11023 |
−33 | 13202 |
−36 |
| 1401 | −31 | 3343 | −34 | 11040 |
−38 |
A Box-Plot analysis (Fig. 7) was performed on the binding affinity data of the new inhibitors (Table 5). The graph reveals that compounds named GQAC-0140 (BA −49 kcal mol−1) and GQAC-02441 (BA −48 kcal mol−1) are outliers. Theoretically, this implies that these two molecules have excellent inhibitory potencies against PA-Mpro, and when compared to the average (−36 kcal mol−1), the binding affinity is almost 36% greater.
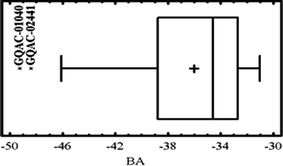 | ||
| Fig. 7 Box-Plot of binding affinities (BA) for the ligands designed for PA-Mpro. | ||
Despite the favourable energetic considerations observed for these two molecules in terms of binding to PA-Mpro, only GQAC-02441 fits into the active site of PB with a better binding affinity (−41 kcal mol−1) than AG7088 (−25 kcal mol−1).
In order to gather additional theoretical evidence on the inhibitory activities of the predicted compounds, GQAC-01040 and GQAC-02441 were docked onto the non-optimized form (crystal structure) of PA-Mpro. The rationale was that if both compounds could dock onto the surface features previously specified for the receptor binding pocket, it is possible to infer that their activities would not vary with slight structural modifications of the protein. The results of the docking procedure revealed that only GQAC-02441 entered the binding site for both the crystal and the optimized structure (Fig. 8), and at the same time, it also had a better binding affinity (−37 kcal mol−1) than AG7088 (−34 kcal mol−1) for the protein in the crystal conformation.
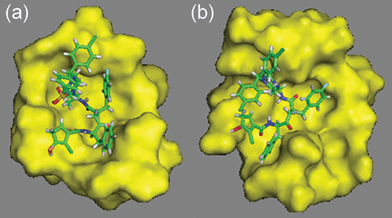 | ||
| Fig. 8 Docking of GQAC-02441 to PA-Mpro using the optimized PA-Mpro (a) and the crystal structure (b). | ||
It was also established that GQAC-02441 did not present equal conformations to the two 3D-structures of PA-Mpro (optimized and crystal 1UJ1). This is not surprising, as ligands can change their conformation when binding to a protein.36
When comparing GQAC-02441 with the Mpro rhinovirus inhibitor (AG7088), several structural similarities could be identified, in particular, the motif –CO–X–C(Bn)CO–, where X can be either –CH2– or –NH–. It is possible to hypothesize that there are geometrical requirements for inhibitors to enter the active site of proteases with similarities to Mpro of SARS (HIV and Rhinovirus), since inhibitors to these proteins, such as Saquinavir12 and compound I,34 also have this common structural feature (Fig. 9). This has also been shown by Shie and co-workers,37 who proposed a potent anilide inhibitor (inhibition constant Ki = 0.03 µM) against the SARS 3CL protease, which contains this motif. In addition, docking analysis for GQAC-02441 and Mpro showed that this substructure could fit in close proximity to His-41 on the active site (Fig. 10), in the same way as some inhibitors do to PA-Mpro proteases, establishing an interaction between the phenyl and the His-41 ring.34 Interestingly, docking calculations performed on GQAC-02441 and AG7088 with Mpro showed that the two rings, His-41 and fluorophenyl, are closer in PA-Mpro:GQAC-02441 than in PA-Mpro:AG7088.
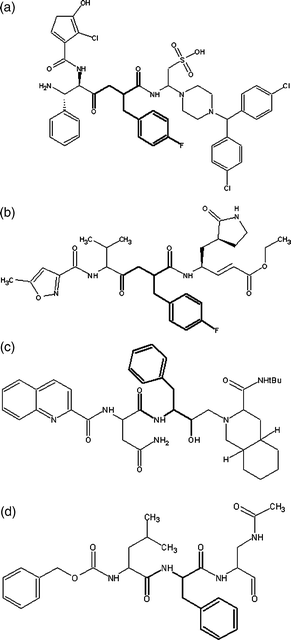 | ||
| Fig. 9 Structural similarities between ligands that bind Mpro and structural-related proteins: (a) GQAC-02441, (b) AG7088, (c) Saquinavir, (d) 1. | ||
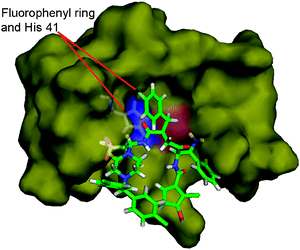 | ||
| Fig. 10 Fluorophenyl ring of GQAC-02441 is located near His-41 on the active site of PA-Mpro. | ||
In terms of Mpro inhibition, a possible advantage of GQAC-02441 over AG7088 might be linked to its sulfonate and amine groups, which could form multiple hydrogen bonds with residues His-41, Tyr-54, Asp-187, Cys-145, Gly-143 and Ser-144 (Fig. 11). Hydrogen bonds are more common in complex Mpro:GQAC-02441 than in Mpro:AG7088, increasing the strength of the interaction.38,39 In addition, a large number of hydrophobic interactions in the Mpro:GQAC-02441 complex could also provide additional complex stability 40–42 (Fig. 12).
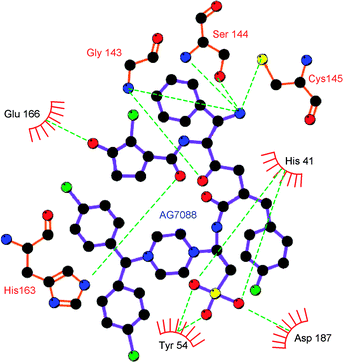 | ||
| Fig. 11 Hydrogen bonds (green dotted lines) in the complex formed between the PA of SARS Mpro and GQC-02441. | ||
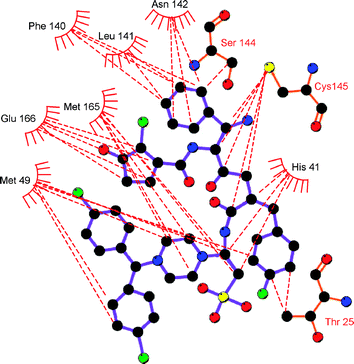 | ||
| Fig. 12 Hydrophobic contacts (red dotted lines) in the complex formed between the PA of SARS Mpro and GQC-02441. | ||
Conclusions
In summary, based on our results, we conclude that:• AG7088 would exert its inhibitory activity mainly on PA, since this ligand shows a greater binding affinity than that elicited by PB.
• Inhibitors of proteases structurally similar to Mpro have the motif –CO–X–C(Bn)CO–. This suggests that this structure might constitute a structural requirement for high affinity binding to the active site of Mpro.
• The molecule called GQAC-02441 has a great probability of competitively inhibiting SARS Mpro.
Acknowledgements
The authors are grateful to COLCIENCIAS–Universidad de Cartagena (grants 1107-05-14663 and 1107-05-13692), Bogotá-Cartagena, Colombia, as well as the Program of Young Investigators, sponsored by both institutions.References
- S. Sirois, D. Q. Wei, Q. Du and K. C. Chou, J. Chem. Inf. Comput. Sci., 2004, 44, 1111–1122 CrossRef CAS.
- K. Anand, J. Gottfried, R. Jeroen, G. Stuart, J. Ziebuhr and R. Hilgenfeld, EMBO J., 2002, 21, 3213–3224 CrossRef CAS.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS.
- D. Matthews, W. Smith, R. Ferre, B. Condon, D. Budahazi, W. Sisson, J. Villafranca, C. Janson, H. McElroy and C. Gribskov, Cell, 1994, 77(5), 761–771 CrossRef CAS.
- C.-Y. Chou, H.-C. Chang, W.-C. Hsu, T.-Z. Lin, C.-H. Lin and G.-C. Chang, Biochemistry, 2004, 43(47), 14958–14970 CrossRef CAS.
- K. Fan, L. Ma, X. Han, H. Liang, P. Wei, Y. Liu and L. Lai, Biochem. Biophys. Res. Commun., 2005, 329, 934–940 CrossRef CAS.
- H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld and Z. Rao, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190–13195 CrossRef CAS.
- R. Leung, W. Li, T. F. Tam and K. Karimian, Curr. Med. Chem., 2002, 9(9), 979–1002 CrossRef.
- L. Zalman, M. Brothers, P. Dragovich, T. Zhou, T. Prins, S. Worland and A. Patick, Chemotherapy, 2000, 44(5), 1236–1241 Search PubMed.
- A. K. Patick, S. L. Binford, M. A. Brothers, R. L. Jackson, C. E. Ford, M. D. Diem, F. Maldonado, P. S. Dragovich, R. Zhou, T. J. Prins, S. A. Fuhrman, J. W. Meador, L. S. Zalman, D. A. Matthews and S. T. Worland, Antimicrob. Agents Chemother., 1999, 43(10), 2444–2450 CAS.
- E. L. C. Tan, E. E. Ooi, C. Y. Lin, H. C. Tan, A. E. Ling, B. Lim and L. W. Stanton, Emerging Infect. Dis., 2004, 10(4), 581–586 Search PubMed.
- A. K. Patick and K. E. Potts, Clin. Microbiol. Rev., 1998, 11(4), 614–627 CAS.
- M. Stahl and M. Rarey, J. Med. Chem., 2001, 44, 1035–1042 CrossRef CAS.
- L. A. Ian, T. P. Nikolay and M. D. Philip, J. Med. Chem., 2005, 48, 6585–6596 CrossRef.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- D. S. Goodsell and A. J. Olson, Proteins: Struct., Funct., Genet., 1990, 8, 195–202 CAS.
- J. Y. Trosset and H. A. Scheraga, J. Comput. Chem., 1999, 20, 412–427 CrossRef CAS.
- A. R. Leach and A. S. Smellie, J. Chem. Inf. Comput. Sci., 1992, 32, 379–385 CrossRef CAS.
- M. Rarey, B. Kramer, T. Lengauer and G. Klebe, J. Mol. Biol., 1996, 261, 470–489 CrossRef CAS.
- G. Klebe and T. Mietzner, J. Comput. Aided Mol. Des., 1994, 8, 583–606 CrossRef CAS.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, J. Comput. Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS.
- J. Kuntz, J. M. Blaney, S. J. Oatley, R. Langridge and T. E. A. Ferrin, J. Mol. Biol., 1982, 161, 269–288 CAS.
- I. Muegge and C. A. Ivonne, J. Med. Chem., 1999, 42, 791–804 CrossRef CAS.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- R. Wang, Y. Lu and S. Wang, J. Med. Chem., 2003, 46, 2287–2303 CrossRef CAS.
- SYBYL 7.0, Tripos Inc., St. Louis, MO, USA, 2004 Search PubMed.
- T. A. Halgren, J. Comput. Chem., 1999, 20(7), 720–729 CrossRef CAS.
- W. H. Press, B. P. Flannery, S. A. Teukolsky and W. T. Vetterling, Numerical Recipes in C, The Art of Scientific Computing, Cambridge University Press, 1988 Search PubMed.
- R. D. Clark, A. Strizhev, J. M. Leonard, J. F. Blake and J. B. Matthew, J. Mol. Graphics Modell., 2002, 20(4), 281–295 CrossRef CAS.
- M. J. Sippl, Proteins: Struct., Funct., Genet., 1993, 17, 355–362 CrossRef CAS.
- M. Wiederstein and M. J. Sippl, Nucleic Acids Res., 2007, 35, W407–W410 CrossRef.
- S. C. Lovell, I. W. Davis, W. B. Arendall III, P. I. W. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Proteins: Struct., Funct., Genet., 2003, 50(3), 437–450 CrossRef CAS.
- E. G. Hutchinson and J. M. Thornton, Protein Sci., 1996, 5, 212–220 CAS.
- D. Matthews, P. Dragovich, S. E. Webber, S. A. Fuhrman, A. K. Patick, L. S. Zalman, T. F. Hendrickson, R. A. Love, T. J. Prins, J. T. Marakovits, R. Zhou, J. Tikhe, C. E. Ford, J. W. Meador, R. A. Ferre, E. L. Brown, S. L. Binford, M. A. Brothers, D. M. Delisle and S. T. Worland, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11000–11007 CrossRef CAS.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., 1995, 8, 127–134 CrossRef CAS.
- J. Shie, J. Fang, C. Kuo, T. Kuo, P. Liang, H. Huang, W. Yang, C. Lin, J. Chen and Y. W. C. Wong, J. Med. Chem., 2005, 48, 4469–4473 CrossRef CAS.
- G. M. Crippen, J. Med. Chem., 1979, 22(8), 988–997 CrossRef CAS.
- A. Senes, J. Ubarretxena-Belandia and D. M. Engeman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9056–9061 CrossRef CAS.
- C. Pace, B. Shirley, M. McNutt and K. Gajiwala, FASEB J., 1996, 10, 75–83 CAS.
- J. Kellis, K. Nyberg, D. Sali and A. R. Fersht, Nature, 1998, 333, 784–786.
- P. D. Ross and M. V. Rekharsky, Biophys. J., 1996, 71, 2144–2154 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |