Transcriptomic analysis of Saccharomyces cerevisiae physiology in the context of galactose assimilation perturbations†
C.
Syriopoulos
ac,
A.
Panayotarou
ad,
K.
Lai
b and
Maria I.
Klapa
*ade
aMetabolic Engineering and Systems Biology Laboratory, Institute of Chemical Engineering and High-Temperature Chemical Processes (ICE-HT), Foundation for Research and Technology – Hellas (FORTH), GR-26504, Patras, Greece. E-mail: mklapa@iceht.forth.gr; Fax: +30-2610-965223; Tel: +30-2610-965249
bThe Dr John T. MacDonald Foundation Center for Medical Genetics, Department of Pediatrics, University of Miami School of Medicine, Miami, FL 33101, USA
cDepartment of Chemical Engineering, University of Patras, GR-26504, Patras, Greece
dInterdepartmental Graduate Program “Mathematical Modelling in Modern Technologies and Finance”, National Technical University of Athens, Zografou Campus, Zografou Athens, GR-15780, Greece
eDepartment of Chemical and Biomolecular Engineering, University of Maryland, College Park, MD 20742, USA
First published on 4th July 2008
Abstract
Despite being extensively studied in various organisms due to scientific, industrial and medical interest, the galactose assimilation function and regulation, and especially its interaction with other parts of cellular physiology, have not been fully elucidated yet. The post-genomic era holistic techniques (“omics”) could assist towards this goal. In this paper, we holistically analyzed full-genomeSaccharomyces cerevisiae transcriptional profiling data concerning its glucose- and galactose-grown wild-type (WT) physiology and its glucose-grown gal7-deficient (GAL7Δ) physiology, as these were obtained in the experiment described in Lai and Elsas (Biochem. Biophys. Res. Commun. 2000, 271, 392–400). The gal7gene encodes for the second enzyme of the galactose assimilation, Leloir, pathway, and its deficiency in humans causes a potentially lethal disease, named “classic galactosemia”. Analysis of the galactose-grown compared to the glucose-grown WT physiology indicated a significant increase in the transcriptional expression of the ribosomal machinery and decrease in the transcriptional activity of the fatty acids’ β-oxidation at the peroxisomes. Comparison of GAL7Δ to WT physiology in glucose indicated a significant transcriptional increase in the mitochondrial activity and the rate of catabolism of disaccharides, including sucrose, mannose and trehalose, towards amplified biosynthesis of the main cell wall components. Comparison with other physiological conditions indicated strong correlation between the glucose-grown GAL7Δ transcriptional physiology and the WT growth under glucose derepression conditions. Finally, the acquired results and the large number of still unknown genes that were related to the galactose assimilation regulation indicated the need for further, specifically designed, perturbations and integrated analyses of multiple levels of cellular function.
Introduction
In all living cells, productive utilization of α-D-galactose requires its conversion to galactose-1-phosphate (gal-1-p) by the enzyme galactokinase, which is encoded by the gal1 (or else galK) gene. Then, in the presence of the enzyme galactose-1-phosphate uridyltransferase, which is encoded by the gal7 (or else galT) gene, gal-1-p reacts with UDP-glucose to form UDP-galactose and glucose-1-phosphate (Fig. 1).1Glucose-1-phosphate could either be converted to glucose-6-phosphate and enter the central carbon metabolism, or react with UTP through UDP-glucose pyrophosphorylase (UGP) to form a new molecule of UDP-glucose (Fig. 1).1,2UDP-galactose can act as a galactosyl donor for glycoprotein/glycolipid biosynthesis or be converted to UDP-glucose by the UDP-4-galactose epimerase, which is encoded by the gal10 (or else galE) gene (Fig. 1).3 It must be emphasized that while this particular pathway, also known as Leloir, has been mainly discussed in the context of galactose assimilation, only its first reaction is under direct regulation from galactose. The rest of the pathway is potentially active even in the absence of exogenous galactose, and is directly implicated in glycosylation reactions and cell membrane synthesis. The ability of the cells to produce UDP-galactose from UDP-glucose through UDP-4-galactose epimerase renders galactose a non-essential nutrient for the cells.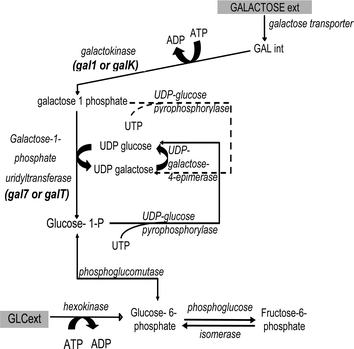 | ||
| Fig. 1 The Leloir pathway of galactose assimilation; the dashed reaction was recently discovered by Lai and Elsas.20 | ||
The Leloir pathway has been among the most studied in biology due to major scientific, industrial and medical interest. As a genetic regulatory switch,4–6 it has been investigated as a model system for the understanding of more complex genetic circuits and their regulation at all levels of cellular function.7,8 With respect to industrial bioprocessing, furthering our understanding of the Leloir pathway regulation in the context of the rest of metabolism is of high significance, because the galactose assimilation function is directly correlated with ethanol production,9 but comprehensive elucidation of this association has not been achieved yet. In humans, inherited deficiency of the galactose-1-phosphate uridyltransferase activity can lead to a potentially lethal disease called “classic galactosemia”.10 However, even a galactose-free diet, which is the current standard of care for the disease, does not prevent the development of debilitating complications in many of the patients.11–13 The cause of these complications has not been deciphered yet, thus limiting the success of any treatment.14 In summary, despite its compact appearance and the large amount of studies dealing with its regulation, current knowledge is not adequate to fully model the galactose assimilation function. Taking into consideration that most of the Leloir’s pathway activity is not related only to external galactose assimilation, but also to major cellular functions, including protein glycosylation, and glyco-/phospho-lipid formation, it is important to study galactose assimilation in the context of the entire cellular physiology and not in isolation. Well designed perturbations of the pathway’s activity and holistic analyses of their effect on the cellular function could be used to trace the role of the Leloir pathway within the cellular physiology.7 The regulation of galactose assimilation is intertwined with the complex machinery of the biological systems for nutrient sensing and regulation. The latter has been the subject of multiple studies to-date; the most recent of them have utilized holistic molecular analysis (“omic”) techniques [e.g.ref. 7, 15–19]. Indeed, systems biology approaches using Saccharomyces cerevisiae as the model eukaryotic system can assist in revealing critical regulatory mechanisms that control the assimilation and catabolism of various carbon sources, including galactose.
In the present study, we re-normalized using currently available enhanced algorithms and analyzed the full-genome transcriptional profiles of the glucose-grown and galactose-grown wild-type (WT) and the glucose-grown gal7-deficient (GAL7Δ) S. cerevisiae strains, as these were acquired in the 1999 experiment of Lai and Elsas reported in ref. 20. In that report,20 Lai and Elsas focused on the galactose assimilation genesper se in an effort to obtain indications about the potential activity of alternative to Leloir pathways and used the available transcriptional data to discover a new reaction in the Leloir pathway as shown in Fig. 1. In the present paper, the transcriptional data are analyzed for the first time systemically to reveal the major genome-wide changes in the transcriptional physiology that are triggered by (a) a change in the carbon source, i.e. growth in galactosevs.glucose, and (b) the gal7-deficiency (simulating the galactosemic genotype) in the absence of the Leloir pathway’s regulation from external galactose. The first analysis refers to the comparison of the galactose-grown (WT+GAL) to the glucose-grown WT (WT+GLC) transcriptional physiology (in the rest of the text it will be referred to as comparison 1), while the latter refers to the comparison of the glucose-grown GAL7Δ (GAL7Δ+GLC) to the WT transcriptional physiology in the same carbon source (in the rest of the text it will be referred to as comparison 2). The results are discussed in the context of the currently known physiology of galactose assimilation, in particular, and carbon source sensing and regulation, in general, at various levels of cellular function in yeast. When possible, the design of new more specific perturbations and integrative analyses of various levels of cellular physiology are proposed based on the acquired information.
Results
The first step of the holistic transcriptional profiling analysis is the identification of the genes whose expression changes significantly between the two compared physiological states. It is then through the study of the function of these genes that biologically relevant conclusions could be derived. According to the definition of the significantly over- and under-expressed genes in the present case as this is provided in the Experimental section, in comparison 1, 99 and 159, respectively, genes were identified as significantly over- and under-expressed in the [WT+GAL] with respect to the [WT+GLC] physiology among the 3741 genes that were finally considered in the transcriptomic profile (see Fig. 2). In comparison 2, 189 and 71 among the 3741 genes were, respectively, identified as significantly over- and under-expressed in the [GAL7Δ+GLC] with respect to the [WT+GLC] physiology (see Fig. 2).![The number of the significantly and moderately over- and under-expressed genes in (A) the galactose-grown [WT+GAL] with respect to the glucose-grown [WT+GLC] wild-type physiology, and (B) the glucose-grown gal7-deficient [GAL7Δ+GLC] with respect to the glucose-grown WT [WT+GLC] physiology. In all subsequent figures, the genes in the significantly overexpressed, moderately overexpressed, moderately underexpressed and significantly underexpressed categories and any molecular function associated with any of these categories will be depicted by the corresponding color (i.e. red, purple, light and dark green, respectively).](/image/article/2008/MB/b718732g/b718732g-f2.gif) | ||
| Fig. 2 The number of the significantly and moderately over- and under-expressed genes in (A) the galactose-grown [WT+GAL] with respect to the glucose-grown [WT+GLC] wild-type physiology, and (B) the glucose-grown gal7-deficient [GAL7Δ+GLC] with respect to the glucose-grown WT [WT+GLC] physiology. In all subsequent figures, the genes in the significantly overexpressed, moderately overexpressed, moderately underexpressed and significantly underexpressed categories and any molecular function associated with any of these categories will be depicted by the corresponding color (i.e. red, purple, light and dark green, respectively). | ||
The moderately over- and under-expressed genes (see Experimental section and Fig. 2) were, respectively, 179 and 281 in comparison 1, and 313 and 163 in comparison 2. Supplementary Tables 1 and 2† provide a list of the significantly and moderately, respectively, over- and under-expressed genes in both comparisons.
| Cellular component | Molecular function | Biological process | ||||||
|---|---|---|---|---|---|---|---|---|
| GO term | Over (99)↑ | Under (159)↓ | GO term | Over (99)↑ | Under (159)↓ | GO term | Over (99)↑ | Under (159)↓ |
| Cytoplasm | 56.57% (56) | 28.93% (46) | Molecular_function unknown | 30.30% (30) | 67.92% (108) | RNA metabolic process | 24.24% (24) | 4.40% (7) |
| Nucleus | 36.36% (36) | 15.72% (46) | Other | 18.18% (18) | 7.55% (12) | Ribosome biogenesis and assembly | 22.22% (22) | 0% (0) |
| Nucleolus | 15.15% (15) | 0.63% (1) | Structural molecule activity | 13.13% (13) | 0.63% (1) | Biological_process unknown | 20.20% (22) | 59.75% (95) |
| Ribosome | 15.15% (15) | 3.14% (5) | Transferase activity | 12.12% (12) | 10.06% (16) | Transport | 18.18% (18) | 4.40% (7) |
| Cellular_component unknown | 12.12% (12) | 54.72% (87) | RNA binding | 8.08% (8) | 3.14% (5) | Translation | 16.16% (16) | 1.89% (3) |
| Membrane | 12.12% (12) | 6.92% (11) | Transporter activity | 7.07% (7) | 2.52% (4) | Carbohydrate metabolic process | 9.09% (9) | 3.14% (5) |
| Mitochondrion | 7.07% (7) | 13.21% (21) | Transcription regulator activity | 5.05% (5) | 3.77% (6) | Transcription | 7.07% (7) | 4.40% (7) |
| Cell wall | 3.03% (3) | 1.26% (2) | Oxidoredutase activity | 3.03% (3) | 2.52% (4) | Response to stress | 6.06% (6) | 2.52% (4) |
| Golgi apparatus | 2.02% (2) | 0% (0) | DNA binding | 2.02% (2) | 3.77% (6) | Protein modification | 6.06% (6) | 6.29% (10) |
| Extracellular region | 1.01% (1) | 0% (0) | Nucleotidyltransferase activity | 2.02% (2) | 3.77% (6) | Other | 6.06% (6) | 8.81% (14) |
| Endoplasmic reticulum | 1.01% (1) | 1.26% (2) | Translation regulator activity | 2.02% (2) | 1.26% (2) | Cell wall organization and biogenesis | 5.05% (5) | 3.14% (5) |
| Other | 2.02% (2) | 0.63% (1) | Enzyme regulator activity | 1.01% (1) | 0.63% (1) | Amino acid and derivative metabolic process | 4.04% (4) | 2.52% (4) |
| Not_yet_annotated | 1.01% (1) | 0% (0) | Not_yet_annotated | 1.01% (1) | 0% (0) | Lipid metabolic process | 3.03% (3) | 3.14% (5) |
| Generation of precursor metabolites and energy | 2.02% (2) | 4.40% (7) | ||||||
| Cellular respiration | 0% (0) | 1.26% (2) | ||||||
| Cell cycle | 0% (0) | 5.66% (9) | ||||||
| Electron transport | 0% (0) | 0.63% (1) | ||||||
| DNA metabolic process | 0% (0) | 2.52% (4) | ||||||
| Cellular component | Molecular function | Biological process | ||||||
|---|---|---|---|---|---|---|---|---|
| GO term | Over (189)↑ | Under (71)↓ | GO term | Over (189)↑ | Under (71)↓ | GO term | Over (189)↑ | Under (71)↓ |
| Cytoplasm | 60.32% (114) | 43.66% (31) | Molecular_function unknown | 42.86% (81) | 40.85% (29) | Biological_process unknown | 32.80% (62) | 23.94% (17) |
| Mitochondrion | 37.04% (70) | 14.08% (10) | Transporter activity | 13.23% (25) | 4.23% (3) | Transport | 15.87% (30) | 7.04% (5) |
| Cellular_component unknown | 23.81% (45) | 26.76% (19) | Structural molecule activity | 11.64% (22) | 12.68% (9) | Translation | 12.17% (23) | 14.08% (10) |
| Membrane | 20.11% (38) | 12.68% (9) | Oxidoreductase activity | 6.88% (13) | 8.45% (6) | Generation of precursor metabolites and energy | 10.58% (20) | 8.45% (6) |
| Nucleus | 13.76% (26) | 19.72% (14) | Transferase activity | 3.70% (7) | 8.45% (6) | Carbohydrate metabolic process | 7.41% (14) | 7.04% (5) |
| Ribosome | 10.05% (19) | 15.49% (11) | Transcription regular activity | 3.17% (6) | 2.82% (2) | RNA metabolic process | 6.35% (12) | 8.45% (6) |
| Golgi apparatus | 2.65% (5) | 0% (0) | DNA binding | 2.65% (5) | 4.23% (3) | Response to stress | 6.35% (12) | 8.45% (6) |
| Endoplasmic reticulum | 2.12% (4) | 1.41% (1) | Other | 2.12% (4) | 2.82% (2) | Other | 5.82% (11) | 8.45% (6) |
| Cell wall | 1.59% (3) | 8.45% (6) | RNA binding | 1.59% (3) | 4.23% (3) | Cellular respiration | 5.29% (10) | 2.82% (2) |
| Other | 1.06% (2) | 2.82% (2) | Nucleotyltransferase activity | 0.53% (1) | 1.41% (1) | DNA metabolic process | 4.76% (9) | 2.82% (2) |
| Extracellular region | 0.53% (1) | 1.41% (1) | Enzyme regulator activity | 0.53% (1) | 2.82% (2) | Cell wall organization and biogenesis | 4.23% (8) | 8.45% (6) |
| Nucleolus | 0% (0) | 5.63% (4) | Translation regulator activity | 0% (0) | 1.41% (1) | Cell cycle | 3.70% (7) | 2.82% (2) |
| Transcription | 3.70% (7) | 2.82% (2) | ||||||
| Protein modification | 3.17% (6) | 2.82% (2) | ||||||
| Amino acid and derivative metabolic process | 2.65% (5) | 1.41% (1) | ||||||
| Ribosome biogenesis and assembly | 2.65% (4) | 8.45% (6) | ||||||
| Electron transport | 2.12% (4) | 0% (0) | ||||||
| Lipid metabolic process | 2.12% (4) | 1.41% (1) | ||||||
Gene Ontology analysis
Gene Ontology analysis of the significantly over- and under-expressed genes enables the identification of the biological processes or molecular functions or cellular components that are mainly represented in these gene classes. Thus, it can lead to conclusions regarding the major genome-wide transcriptional differences that are associated with the examined physiological comparisons. In this study, gene ontology analysis was carried out as described in the Experimental section. Tables 1 and 2 show the (a) cellular component, (b) molecular function, and (c) biological processgene ontology classes that were identified as associated with the significantly over- and under-expressed genes in comparisons 1 and 2, respectively. Specifically, Tables 1 and 2 indicate (i) the number of significantly over- and under-expressed genes that belonged to a certain ontology class, and (ii) the percentage of each ontology class in the total significantly over- and under-expressed gene list (called cluster frequency). While the cluster frequencies of the observed gene ontology classes can be used to extract biologically relevant conclusions, it is important to also consider the actual size of a gene ontology class, i.e. the percentage of all genes defined as belonging to this class in the entire S. cerevisiaegenome (also called background frequency). If, for example, the cluster frequency of two gene ontology classes is 10%, but the background frequency of the first is 90% and the second 2%, it is apparent that any significant conclusions should be extracted based primarily on the second ontology class. The probability for the ontology class to be by chance associated with the significantly over- or under-expressed genes is much smaller in the latter than in the former case. In this context, the Yeast Genome Database gene ontology software assigns a statistical significance p value to each ontology class taking into consideration its cluster and background frequency. Tables 3 and 4 depict the gene ontology classes with p values smaller than 0.01 that were identified as associated with the significantly over- and under-expressed genes in comparisons 1 and 2, respectively.| WT overexpressed | WT underexpressed | ||||||
|---|---|---|---|---|---|---|---|
| GO_term | Cluster frequency | Background frequency | p-value | GO_term | Cluster frequency | Background frequency | p-value |
| Component | Component | ||||||
| Cytosolic ribosome (sensu Eukaryota) | 14 out of 99 genes, 14.1% | 175 out of 7288 background genes, 2.4% | 6.82E-06 | No significant ontology term can be found for input genes | |||
| Cytosolic part | 15 out of 99 genes, 14.1% | 196 out of 7288 background genes, 2.7% | 2.81E-05 | ||||
| Cytosolic large ribosomal subunit (sensu Eukaryota) | 10 out of 99 genes, 10.1% | 96 out of 7288 background genes, 1.3% | 5.26E-05 | ||||
| Nucleolus | 15 out of 99 genes, 15.1% | 305 out of 7288 background genes, 4.2% | 0.00113 | ||||
| Large ribosomal subunit | 10 out of 99 genes, 10.1% | 141 out of 7288 background genes, 1.9% | 0.00175 | ||||
| Non-membrane-bound organelle | 29 out of 99 genes, 29.3% | 1026 out of 7288 background genes, 14.1% | 0.00444 | ||||
| Intracellular non-membrane-bound organelle | 30 out of 99 genes, 29.3% | 1026 out of 7288 background genes, 14.1% | 0.0044 | ||||
| Ribosome | 15 out of 99 genes, 15.1% | 356 out of 7288 background genes, 4.9% | 0.00699 | ||||
| Molecular function | Molecular function | ||||||
| Structural constituent of ribosome | 13 out of 99 genes, 13.1% | 230 out of 7288 background genes, 3.2% | 0.00143 | Transferase activity, transferring acyl groups, acyl groups converted into alky on transfer | 4 out 159 genes, 2.5% | 10 out of 7288 background genes, 0.1% | 0.00426 |
| Biological process | Biological process | ||||||
| Ribosome biogenesis and assembly | 22 out of 99 genes, 22.2% | 330 out of 7288 background genes, 4.5% | 8.06E-08 | No significant ontology term can be found for input genes | |||
| Ribonucleoprotein complex biogenesis | 24 out of 99 genes, 24.2% | 400 out of 7288 background genes, 5.5% | 9.24E-08 | ||||
| rRNA processing | 13 out of 99 genes, 13.1% | 173 out of 7288 background genes, 2.4% | 0.00015 | ||||
| GAL7Δ overexpressed | GAL7Δ underexpressed | ||||||
|---|---|---|---|---|---|---|---|
| GO_term | Cluster frequency | Background frequency | p-value | GO_term | Cluster frequency | Background frequency | p-value |
| Cellular component | Cellular component | ||||||
| Mitochondrial part | 45 out of 189 genes, 23.8% | 441 out of 7288 background genes, 6.1% | 9.3E-14 | Cytosolic ribosome (sensu Eukaryyota) | 9 out of 71 genes, 12.7% | 175 out of 7288 background genes, 2.4% | 0.00346 |
| Mitochondrion | 70 out of 189 genes, 37.0% | 1087 out of 7288 background genes, 14.9% | 5.43E-12 | Cytosolic part | 10 out of 71 genes, 12.7% | 196 out of 7288 background genes, 2.7% | 0.00838 |
| Mitochondrial lumen | 24 out of 189 genes, 12.7% | 166 out of 7288 background genes, 2.3% | 9.27E-10 | ||||
| Mitochondrial matrix | 24 out of 189 genes, 12.7% | 166 out of 7288 background genes, 2.3% | 9.27E-10 | ||||
| Organellar ribosome | 16 out of 189 genes, 8.5% | 83 out of 7288 background genes, 1.1% | 5.21E-08 | ||||
| Mitochondrial ribosome | 16 out of 189 genes, 8.5% | 83 out of 7288 background genes, 1.1% | 5.21E-08 | ||||
| Mitochondrial envelope | 23 out of 189 genes, 12.2% | 288 out of 7288 background genes, 4.0% | 0.00026 | ||||
| Organellar large ribosomal subunit | 9 out of 189 genes, 4.8% | 45 out of 7288 background genes, 0.6% | 0.00036 | ||||
| Mitochondrial large ribosomal subunit | 9 out of 189 genes, 4.8% | 45 out of 7288 background genes, 0.6% | 0.00036 | ||||
| Organellar small ribosomal subunit | 7 out of 189 genes, 3.7% | 34 out of 7288 background genes, 0.5% | 0.00436 | ||||
| Mitochondrial small ribosomal subunit | 7 out of 189 genes, 3.7% | 34 out of 7288 background genes, 0.5% | 0.00436 | ||||
| Cytoplasmic part | 96 out of 189 genes, 50.8% | 2681 out of 7288 background genes, 36.8% | 0.00988 | ||||
| Molecular function | Molecular function | ||||||
| No significant ontology term can be found for input genes | No significant ontology term can be found for input genes | ||||||
| Biological process | Biological process | ||||||
| Generation of precursor metabolites energy | 20 out of 189 genes, 10.6% | 237 out of 7288 background genes, 3.3% | 0.0017 | No significant ontology term can be found for input genes | |||
Analysis of the difference in the central carbon metabolism activity at the transcriptional level between the two examined physiological states
Fig. 3 and 4 show the major differences in the transcriptional activity of the central carbon metabolism between the two examined physiological states in comparisons 1 and 2, respectively. Specifically, the metabolic networks are color-coded (as described in the Experimental section and the legend of Fig. 2) to indicate the reactions whose enzymes are encoded by significantly over- or under-expressed genes. This type of graphical representation allows for (a) better visualization of the metabolic changes at the transcriptional level, and (b) potential correlation between phenomena that occur at different parts of the network and would not have been easy to relate within the context of a gene list. In addition, it enables the direct comparison with in vivo metabolic activity measurements. It needs to be underlined that the transcriptional snapshot of the metabolic physiology might be substantially different from the in vivo metabolic state of a biological system. This potential divergence needs to be taken into consideration when transcriptional profiling results are interpreted to extract biologically relevant conclusions, as it will be discussed in the context of specific examples in the following section.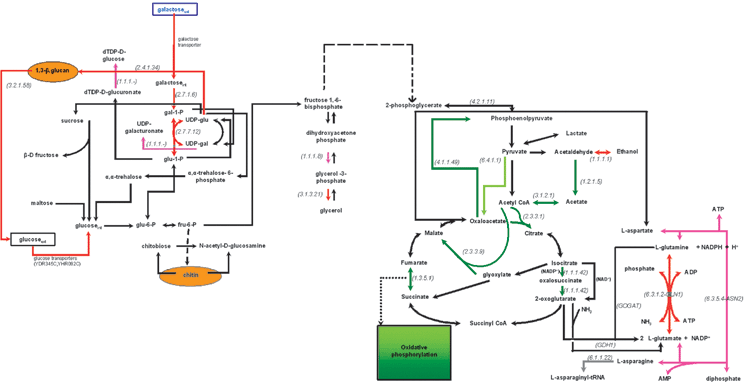 | ||
| Fig. 3 Transcriptional activity of the central carbon metabolism in the WT phenotype in 0.2% galactose with respect to 0.2% glucose based on the identified over- and under-expressed genes. Dashed arrows depict reactions that are carried out through more than one enzyme. The red and dark green arrows depict the reactions whose enzymes are encoded by over- and under-expressed genes, respectively (see legend of Fig. 2). The purple and light green arrows depict the reactions whose enzymes are encoded by the moderately over- and under-expressed genes, respectively, as explained in Fig. 2. The black arrows depict reactions whose genes were either not included in the analysis (after the normalization and filtering step) or—if included—were not identified in any of the (colored) 4 categories described above. Extracellular metabolites are in rectangular boxes, while the compounds of the cell wall are depicted in yellow ellipses. The numbers in parenthesis refer to the E.C. number of the enzyme encoded by the over- or the under-expressed gene. The reactions that are depicted with a two-color arrow (one direction in red and the other in green) are catalyzed by multiple isoenzymes in both directions. All isoenzymes catalyzing one direction of the reaction were identified as either overexpressed or underexpressed (thus each direction of the reaction is colored red or dark green, respectively), thus indicating the direction that is favored under the particular experimental conditions. | ||
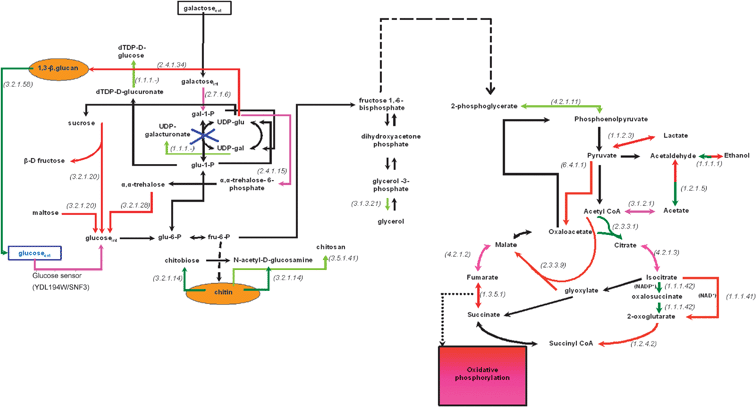 | ||
| Fig. 4 Transcriptional activity of the central carbon metabolism in the gal7-deficient with respect to the WT phenotype in 0.2% glucose based on the identified over- and under-expressed genes. Dashed arrows depict reactions that are carried out through more than one enzymes. The red and dark green arrows depict the reactions whose enzymes are encoded by over- and under-expressed genes, respectively (see legend of Fig. 2). The purple and light green arrows depict the reactions whose enzymes are encoded by the moderately over- and under-expressed, respectively, genes as explained in Fig. 2. The black arrows depict reactions whose genes were either not included in the analysis (after the normalization and filtering step) or—if included—were not identified in any of the (colored) 4 categories described above. Extracellular metabolites are in rectangular boxes, while the compounds of the cell wall are depicted in yellow ellipses. The numbers in parenthesis refer to the E.C. number of the enzyme encoded by the over- or the under-expressed gene. The reactions that are depicted with a two-color arrow (one direction in red and the other in green) are catalyzed by multiple isoenzymes in both directions. All isoenzymes catalyzing one direction of the reaction were identified as either overexpressed or underexpressed (thus each direction of the reaction is colored red or dark green, respectively), indicating thus the direction that is favored under the particular experimental conditions. | ||
Discussion
A Transcriptomic comparison of the galactose-grown vs. the glucose-grown WT phenotype
As a general comment over the particular physiological comparison, Fig. 2 indicates that the number of WT genes (among the 3741 considered in the analysis), whose expression increased significantly in the galactose with respect to the glucose media (99) is much lower than the number of genes whose expression decreased (159). This difference becomes even larger when the moderately over- (179) and under- (291) expressed genes are also considered. Indeed, the genes encoding for the first three reactions of the Leloir pathway, namely the galactose transporter (gal2), the galactokinase (gal1) and the galactose-phosphate uridyl-transferase (gal7) exhibited the highest expression ratios among all genes, i.e. 24, 40 and 40, respectively (see Supplementary Table 1† ). It needs to be underlined that all other overexpressed genes exhibited an expression ratio smaller than 8 (see Supplementary Table 1† ). The high expression ratios of gal1, gal2 and gal7 provided a strong validation checkpoint for the presented analysis. For the same physiological comparison but with other microarray platform, Lashkari et al. (1997)15 had observed expression ratios of the same order of magnitude (21.8 and 21.9, respectively) for the gal1 and gal7genes in a 2% (vs. 0.2% in the present study) carbon source concentration. The proteomic analysis of Gao et al. (2003)16 validated this direction of change at the protein level as well. In addition, the expression of the genesadh6 and hor2, encoding, respectively, an alcohol dehydrogenase (EC 1.1.1.1) and glycerol-3-phosphatase (E.C. 3.1.3.24), were observed as significantly increasing in the galactoseversus the glucose medium. Induced adh6 activity21 and ethanol and glycerol biosynthesis8,9 have been previously reported as related to galactose assimilation, providing thus an additional validation checkpoint for this data analysis.According to the gene ontology analysis, most of the overexpressed genes are related to the ribosomal function (see Table 3). More specifically, ontology analysis with respect to the cellular component indicated that these genes are related to the ribosome, or ribosomal subunits or the nucleolus. The latter is directly related to the ribosomal function as this part of the nucleus that includes genes encoding ribosomal RNA.22 The molecular function ontology classes that are primarily represented in the significantly overexpressed genes are related to structural constituents of the ribosome, and the biological functions to the ribosome or ribonucleoprotein biogenesis and assemblyand rRNA processing. In combination, these transcriptional results indicate an increased need for the WT in galactose with respect to its phenotype in glucose to synthesize ribosomal subunits and ribosomal protein complexes. Increased transcription of genes encoding ribosomal proteins had been observed for similar physiological comparison in ref. 7. However, this prominent increase in the activity of the ribosomal machinery at the transcriptional level seems in contradiction with the fact that glucose is the preferred substrate over galactose. The reason behind this phenomenon and whether (and how) this is translated to in vivocellular growth or other functions needs further investigation and integrative analyses including the protein and metabolic levels of cellular function. It is actually the opposite response to what has been observed in the transition from glucose to glycerol growth medium,17 the latter being associated with glucose derepressive conditions.
Regarding the significantly underexpressed genes, no cellular component or biological process ontology term was identified as statistically significant (see Table 4). The only molecular function class that was identified as statistically significant concerned genes encoding transferases. The transferases are enzymes that catalyze the transfer of a characteristic chemical group from one molecule to another.23 The 4 identified transferasegenes (cit2, cit3, mls1, lys) encode enzymes that during the transfer convert also the acetyl- to alkyl-group. Specifically, cit2, cit3, mls1 and lys encode, respectively, the peroxisomal and mitochondrial citrate synthase (www.yeastgenome.org), the peroxisomal malate synthase24 and the homocitrate synthase (the first reaction in the lysine biosynthesis pathway, www.yeastgenome.org), all four utilizing acetyl-CoA as one of the substrates. The decreased expression of the peroxisomal cit2 and mls1genes, which encode for the enzymes catalyzing the two reactions that “feed acetyl-CoA into the glyoxylate shunt”24 implies lower rate of β-oxidation of fatty acids inside the peroxisomes and consequently lower rate of the glyoxylate shunt at the transcriptional level in the galactoseversus the glucose growth environment. This is the first time that galactose assimilation has been directly related to lower rate of fatty acid β-oxidation within the peroxisomes, which leads to decreased production of acetyl-CoA.
The localization of the malate and citrate synthase in the peroxisome is linked to the direct utilization of the acetyl-CoA generated by the β-oxidation of fatty acids.24 Thus, through the interconnectivity of the glyoxylate and tricarboxylic acid (TCA) cycles,24 decreased production of acetyl-CoA implies decreased rate of the TCA cycle in the mitochondria as this is observed by the decreased expression of the mitochondrial citrate synthasegene in the galactoseversus the glucose medium. Moreover, 21 of the underexpressed genes were related to the mitochondria (see Table 1). This large number of underexpressed mitochondrial related genes implies impaired TCA cycle and oxidative phosphorylation activity of the WT in the galactose compared to the glucose growth environment at the transcriptional level. Specific study of the metabolically related genes showed this to be indeed the case as seen in Fig. 3, indicating consistency between the transcriptional and the metabolic activity of the TCA cycle that was reported in ref. 8. The genespck1 and pyc1 encoding the enzymes that catalyze the reactions phosphoenolpuruvate carboxykinase (first step in gluconeogenesis) and pyruvate carboxylase (anaplerotic) were also identified as underexpressed. These results agree with the proteomic analysis in ref. 16 that identified as significantly increased in galactose the concentration of the ATPase inhibitor INH1. According to ref. 25, an intrinsic ATPase inhibitor “acts to inhibit ATP hydrolysis by F1F0-ATPase upon deenergization of mitochondrial membranes”.
On the other hand, study of the metabolic map of Fig. 3 at the transcriptional level indicates an increase in the transcription of genes encoding enzymes participating in the synthesis of 1,3-β-glucan in the galactose compared to the glucose WT physiology. The 1,3-β-glucan constitutes one of the main components of the yeast cell wall, contributing significantly to its robustness.26–28 Because this observation is obtained at the transcriptional level, it is not possible to conclude whether this increase in the expression of the 1,3-β-glucan synthesisgenes is related to a stronger cell wall in the case of the galactoseversusglucose WT physiology or does actually manifest the exact opposite, i.e. that the system responds to perceived weakness in the cell wall integrity by attempting to reinforcing it. To definitely answer this question, further investigations, specifically targeting the cell wall, are needed. A relevant, interesting observation concerned the increased in the galactoseversus the glucose WT physiology expression of the gene encoding the enzyme that catabolizes extracellularly the 1,3-β-glucan to α-D-glucose. This is the first time that this is reported in the literature. It would be of significance to investigate whether under galactose substrate conditions there is indeed in vivodegradation of 1,3-β-glucan to (extracellular) glucose. Consistent with potential presence of (low levels of) extracellularglucose is the fact that two genes encoding for high affinity glucose transporters [ref. 20, 29, www.yeastgenome.org], YDR345c (hxt3) and YHR092c (hxt4), were also identified as overexpressed in the galactose compared to the glucose environment. However, the expression of hexose transporters is under the regulation of a complex carbon source sensing and catabolism machinery that has been under intense investigation [see ref. 17–19]. The overexpression of hxt3 and hxt4 could be assigned to the certain alleviation of glucose repression conditions under galactose, since the Leloir pathway bypasses the glucose transporters. The hxt3 and hxt4 were simultaneously identified as also significantly overexpressed in the transcriptional profile of the glucose-grown mig1Δ strain studied in ref. 18. Mig1 is a DNA bindingprotein that plays key role in the main glucose repression pathway.17,19 Of note, among the significantly overexpressed genes in the galactose compared to the glucose media was that encoding the dihydroorotate dehydrogenase (YKL216W/ura1), i.e. the enzyme that catalyzes the fourth reaction in the de novopyrimidine biosynthesis [www.yeastgenome.org]. The reactant of the reaction, S-dihydroorotate, is among the reporter metabolites of the mig1Δmig2Δ mutant in ref. 17. According to that study,17 a reporter metabolite is indicative of a part of the metabolism that is observed to be significantly affected by a perturbation (in this case the particular mutation). Mig2 along with Mig1P is “involved in the repression of invertase activity by high levels of glucose” [www.yeastgenome.org, ref. 17]. Despite these similarities, the bulk of the physiological changes that were observed at the transcriptional level in the galactose compared to glucose-grown WT do not correlate with the mig1Δ and mig1mig2Δ mutants in ref. 17. This indicates that further holistic analyses of the nutrient sensing and assimilation network are required.
According to current knowledge (see Fig. 3), S. cerevisiae may synthesize glutamate either through NADP-glutamate-dehydrogenase, which is encoded by the genegdh1, under conditions of high availability of NH3, or through the GS-GOGAT cycle comprising the glutamine synthetase (GS) and the glutamate synthetase (GOGAT), which are encoded by the genesgln1 and glt1, respectively, under conditions of low availability of NH3.30 In the present study, gln1 was overexpressed in the galactose compared to the glucose WT physiology, while the expression of glt1 did not change significantly between the two physiologies. Unfortunately, glt1 was filtered out of the genes considered in the present study at the normalization step. However, the asn2gene, encoding the asparaginetransaminase, was observed as overexpressed in the galactose compared to the glucose WT physiology, implying that the transaminase activity may not have been primarily carried out through the NADP-glutamate-dehydrogenase reaction. This observation along with the significantly lower rate of the TCA cycle at the transcriptional level, as discussed earlier, provides a strong indication that the galactose WT physiology uses the GS-GOGAT cycle for glutamate biosynthesis. If this is true, then the galactose grown WT physiology should be under more restricted nitrogen assimilation conditions than the glucose grown. This, however, is in need of further verification through direct metabolic activity measurements.
Finally, from Table 1 it becomes apparent that most of the genes whose expression changes significantly between the galactose and glucose WT physiology are of yet unknown function. This observation strongly supports the fact that, while among the most studied cellular processes, the regulation of galactose assimilation in the context of the rest of metabolism has not been fully elucidated yet and new discoveries are expected through further investigation.
B Transcriptomic comparison of the GAL7Δ vs. the WT physiology in glucose
Comparing the number of genes whose expression differs significantly between the GAL7Δ and the WT physiology (Fig. 2), the GAL7Δ physiology corresponds to largely more over- (189) than under- (71) expressed genes among the 3741 that were considered in the analysis. This difference increases even more when the moderately over- (313) and under- (163) expressed genes are also considered. Cumulatively, the large number of genes whose expression differs substantially between the two compared physiologies indicates that the latter are much different even in the absence of galactose. This implies that the role of the galactose-1-phosphate uridyltransferase, which is encoded by the gal7 gene, is not associated solely with galactose assimilation, but its activity is needed to accomplish essential metabolic functions even in the absence of galactose. Elucidating these functions and the role of the galactose-1-phosphate uridyltransferase is of great significance, because the gal7 deficiency is the genetic cause of the inborn metabolic disorder, galactosemia.10 Moreover, the chronic defects of galactosemic patients even under a galactose-free diet have not been fully explained yet.11–13 Further, studying the effect of the gal7 deficiency on the entire transcriptional physiology of yeast is of additional interest within the effort to decipher the complex regulatory mechanisms that govern glucose repression.17,19In the present study, ontology analysis of the significantly overexpressed genes in the GAL7Δ compared to the WT physiology indicated that the statistically significant gene categories are related to the mitochrondrial activity (see Table 4). This result implies a significantly higher respiratory activity at the transcriptional level in the GAL7Δ compared to the WT physiology. Indeed, studying more specifically the genes involved in the central carbon metabolism activity, among the overexpressed in the GAL7Δ compared to the WT physiology were identified (a) the oxidative phosphorylation genescox17, atp15 and qcr6, (b) the mitochondrial genes, idh1, kgd1, fum1, aco1, encoding for the TCA enzymes NAD(+)-dependent isocitrate dehydrogenase (EC 1.1.1.41), alpha-ketoglutarate dehydrogenase (E.C. 1.2.4.2), fumarase (E.C. 4.2.1.2) and aconitase (E.C. 4.2.1.3), respectively, and (c) the gene encoding for the anaplerotic enzyme pyruvate carboxylase (E.C. 6.4.1.1) (see Fig. 4). However, this transcriptional situation cannot directly indicate the in vivo metabolic state; it could indeed correspond to higher in vivo respiratory activity in the GAL7Δ compared to the WT physiology or could be the transcriptional response for the cellular system to overcome in vivo limitations in the respiratory activity. Further investigation of the in vivo metabolic activity is required. With respect to the underexpressed genes in the GAL7Δ compared to the WT physiology in glucose, the only statistically significant ontology category referred to genes associated with the cytosolic ribosomes (see Table 4). This result implies transcriptional decrease in the protein synthesis and cell growth in GAL7Δ compared to the WT. Table 3 shows that in this case too, there are many genes whose expression differs significantly between the GAL7Δ and WT physiology that are of yet unknown function. This provides an additional indication that the regulation of the galactose assimilation defects has not been fully elucidated yet and there is room for further investigation.
The GAL7Δ physiology at the transcriptional level was observed as related to the increased rate of biosynthesis of ethanol from acetaldehyde and acetaldehyde from acetate (Fig. 4). Interestingly, all genes encoding the isoenzymes catalyzing the production of ethanol from acetaldehyde were measured as overexpressed, while all genes encoding the isoenzymes catalyzing the opposite direction were measured as underexpressed in the GAL7Δ compared to the WT physiology. The same is true for the genes encoding the isoenzymes catalyzing the forward and reverse directions of the synthesis of acetaldehyde from acetate. This result could prove significant for the biofuel industry, if validated at the metabolic level. However, increased production of ethanol under conditions of increased activity of the TCA cycle seems contradictory. Holistic view of the transcriptional activity of the central carbon metabolism (Fig. 4) indicates that the higher transcriptional activity of the TCA cycle and oxidative phosphorylation in GAL7Δ is not actually accommodated from the available substrate and the increased activity of the glycolysis pathway; the geneeno2 (YHR174W) encoding the enzyme that catalyzes one of the glycolysis reactions (E.C. 4.2.1.11) was among those moderately underexpressed in GAL7Δ with respect to the WT physiology. Thus, other short-term sources of carbon and/or energy are to be lysed to replenish the TCA cycle activity. As discussed in the previous section, a major source of acetyl-CoA for the TCA cycle activity is the β-oxidation of fatty acids in the peroxisomes. Without this observation being conclusive for the higher transcriptional activity of the peroxisomes in the GAL7Δ with respect to the WT physiology, the expression of the yoz147Cgene encoding for the protein with the highest concentration in the peroxisomal membrane was indeed identified as significantly higher in the GAL7Δ with respect to the WT.
According to the transcriptional results shown in Fig. 4, the available extracellularglucose seems insufficient to accommodate the required metabolic functions in the GAL7Δ compared to the WT physiology even within the “upper” part of the metabolic network. This is concluded from the fact that the genes encoding the enzymes that catabolize short-term carbon storage compounds, the disaccharidessucrose, maltose and trehalose, were measured as overexpressed in the GAL7Δ compared to the WT (Fig. 4). As discussed earlier, this intracellular synthesis of additional glucose in GAL7Δ does not seem to be accommodating the higher activity of the TCA cycle. It seems to be directed towards enhancing the robustness of the cell wall; specifically, genes involved in the biosynthesis of 1,3-β-glucan were measured as overexpressed, while genes involved in the degradation of 1,3-β-glucan and chitin, the two major components of the yeast cell wall, were measured as underexpressed in the GAL7Δ compared to the WT physiology. Most of these observations, especially regarding the two separate activities taking place in parallel within the central carbon metabolism, one involving the higher rate of TCA cycle and the other the degradation of short-term storage compounds towards cell wall reinforcement, have not been reported earlier in the literature as related to the gal7 deficiency. Their significance, if validated in vivo and combined with information from other levels of cellular function, imposes further more specifically designed integrative investigations.
Comparison of the observed transcriptional profile of the glucose-grown GAL7Δ strain with other physiological conditions characterized either by growth in other carbon sources or other genetic alterations of the nutrient sensing and regulation machinery indicated strong correlation of the glucose-grown GAL7Δ transcriptional physiology with that expected under conditions of glucose derepression. This is a significant result providing insight to the role of the gal7 activity per se, but also as part of the glucose repression machinery. Specifically, it has been known18,31 that there is a decrease in the expression of the yeast’s translational machinery during and after the diauxic shift, the latter corresponding to glucose derepressive conditions, while the transcription of genes related to respiration is upregulated, these observations being in direct agreement with the glucose-grown GAL7Δ transcriptional profile. These physiological changes imply a transition from fermentative to respiratory growth. Characteristically, in the observed GAL7Δ compared to the WT transcriptional profile in glucose, the most under-expressed gene encodes for the sporulation-specific wall maturation protein SPS100, this observation indicating indeed respiratory rather than fermentative growth. Respiratory growth characterizes mainly the physiology of the WT yeast strain grown in glycerol as described in ref. 18. Similarly to the observed GAL7Δ transcriptional profile in glucose, Roberts and Hudson (2006)18 report that “in cells growing continuously in glycerol (YPG) or ethanol (YPE), the most highly upregulated genes were related to mitochondrial function or energy generation”, while “the functions of the most severely down-regulated were related to the attenuated growth rate”. Importantly, ref. 18 also reports genes encoding peroxisomal enzymes as being among the most upregulated. This result highly supports our speculation for increased transcriptional activity of the peroxisomal β-oxidation of fatty acids in the glucose-grown GAL7Δ compared to the WT. As discussed earlier, we were able to derive this speculation mainly indirectly, through the holistic study of the transcriptional physiology around the peroxisomes, since only one peroxisomalgene had been identified as overexpressed. This situation provides an additional example that indicates the significance of the systemic analysis of the physiological activity within each level and between levels of cellular function to extract important information from the available experimental data. Other common observations that support further the strong correlation between the transcriptional profiles of the glucose-grown GAL7Δ with the glycerol (or ethanol)-grown WT strain18 include the upregulation of the gene encoding the 1,3-β-D-glucan synthase and of genes related to the system’s response to oxidative and osmotic stress.
Supporting even further the conclusion that the GAL7Δ deficiency “removes” glucose repression constraints are the observed similarities between the transcriptional profiles of the glucose-grown GAL7Δ and hxk2Δ mutants as the latter is described in ref. 17. According to ref. 17, hexokinase 2 (encoded by the hxk2) is believed to be involved in the sensor of the main glucose repression pathway, key role to which is played by the DNA-bindingprotein Mig1. Thus, hxk2 deficiency resulted to the alleviation of the Mig-1 related repression of the transcription of genes involved in the metabolism of other than glucose and fructose carbon sources.17 This agrees with the observed in the glucose-grown GAL7Δ transcriptional profile overexpression of genes encoding proteins involved in the disaccharidemetabolism (fsb2, nth1). This result is very important as it directly relates for the first time the GAL7 activity with the regulation of the Mig1-related glucose repression pathway. In addition, similarly to the GAL7Δ transcriptional profile, hxk2 deficiency corresponds to “increased expression of respiratory and ATP synthesis coupled proton transport” and TCA cycle genes.17 Among the glucose sensors, the gene encoding SNF3 was identified among the moderately overexpressed (i.e. more than 2-fold increase) in the glucose-grown GAL7Δ profile. Westergaard et al. (2007)17 report “about 2-fold” upregulation of the gene encoding SNF3 in the hxk2Δ strain too. More importantly, based on the available transcriptional profiles in ref. 17, Westergaard et al. (2007) cannot determine whether the observed decreased level of respiration repression in the hxk2Δ mutant is due to a particular regulatory role of hexokinase 2 or to the decreased glycolytic flux that is direct consequence of the hxk2 deficiency. The strong similarities between the transcriptional profiles of the glucose-grown GAL7Δ strain, which was studied in the present paper, and the glucose-grown hxk2Δ strain studied in ref. 17, imply that the decreased respiration repression in both mutants is due to the decreased glycolytic flux, which was common characteristic of both profiles. It becomes apparent then how important the systemic analysis of the nutrient sensing and regulation machinery in its entirety under a variety of environmental perturbations and/or mutations is for the deconvolution of its complex mechanisms. Despite the strong similarities between the hereby studied glucose-grown GAL7Δ transcriptional physiology and the other discussed physiological conditions, the increased rate of ethanol biosynthesis at the transcriptional level was observed only in the glucose-grown GAL7Δ strain. These observed, still not directly justifiable, similarities and discrepancies between the strains along with the high number of yet unknown genes whose expression changed significantly due to the gal7 deficiency support even further the need for additional studies towards the full elucidation of the galactose assimilation regulation.
Experimental
Experimental design
The present study compares the full-genome transcriptional profiles of (a) the galactose-grown vs. the glucose-grown S. cerevisiae YPH499 wild-type (WT) strain (i.e. same strain in different substrates), and (b) the glucose-grown gal7-deficient (GAL7Δ) mutant of S. cerevisiae yJJ52 wild-type strain vs. the glucose-grown S. cerevisiae YPH499 wild-type (WT) strain (i.e. different strains in same substrate), as these were selected for study and acquired in the 1999 experiment of Lai and Elsas reported in ref. 20.DNA Microarray data acquisition
The full-genome transcriptional profile of each of the three examined conditions was acquired once using the Ye6100 full genomeS. cerevisiae microarray type of the Affymetrix platform. Array hybridization, processing (labeling and washing), quality control and scanning were carried out in 1999 at the Microarray Facility of the Emory University School of Medicine, Atlanta, GA, USA (for additional details see ref. 20). Due to the limitations of the microarray technology at the time, the yeast full genome probe sets were divided among four slides (A, B, C, D). Thus, the full genome transcriptional profile of each examined growth case had to be reconstructed from four scanned images (.DAT files) (the raw data have been uploaded to ArrayExpress under the accession number E-MEXP-1420). The .CEL data files that were provided from the facility in 1999 were generated after processing each of the scanned images using the Microarray Suite (MAS) version 4.0.Image re-processing
Due to enhancement of the image processing algorithms since 1999, the original array images were reprocessed using the relevant algorithm of the “dchip” software [www.dchip.org]. The reprocessed .CEL data files did not differ from the originally provided in 1999. This observation verified the statement in the web manual of dchip [www.dchip.org] arguing that the differences between the older and the new image processing algorithms do not affect the processing of older, low probe density microarray types as the one used in the present study. Therefore, it was decided for the original (1999) .CEL files to be used in further analysis.Gene expression and P/A call determination
In 2004, Affymetrix started using a new algorithm for the estimation of each gene’s expression and the determination of its P/A (Presence/Absence) call from the PM (perfect match) and MM (mismatch) probe intensity values. This algorithm has been incorporated in the MAS5.0 and the Affymetrix microarray analysis suite GCOS (v.1.2–3). Thus, the 1999 .CEL files were re-analyzed using the publicly available GCOS v.1.2 (beta version) software for the final .CHP files to be generated. The latter included the expression value, the standard error and the P/A call for each gene (probe set) based on the new algorithm. For the data reprocessing to take place, the .cdf file of the discontinued in 2000 Ye6100 microarray type had to be retrieved from the archives of Affymetrix; the .CDF file of a microarray type contains the topological information of each probe and probe set on the slide(s).Inter-slide normalization
Inter-slide normalization for each of the A, B, C, D slide types was carried out using the Invariant Set normalization algorithm of the dchip software in the context of the five experimental conditions discussed in the Lai and Elsas study.20 In the present work 3 out the 5 conditions are discussed; all microarrays (including those not discussed in the present study) had been processed in the same way as described in the previous sections.PM model
After the inter-slide normalization, the expression and standard error value of each gene on each microarray slide was estimated from the normalized intensities of only the PM probes of the corresponding probe set, based, on the PM-model algorithm of the dchip software. 4 PM data files (one for each of the A, B, C, D slides of one array) were generated for each of the examined experimental conditions; when unified into one file, this comprised 6594 probe sets (including repetitions of the common among the slides probe sets placed on each slide for inter-slide normalization purposes).Comparison between Ye6100 and YS98 array types
Due to developments in yeast sequencing analysis, microarray technology and yeast annotation, Affymetrix discontinued the Ye6100 array type in 2000 introducing its sequel YS98 type. For the updated annotation of yeast genes corresponding to probe sets of currently commercially available yeast array types to be used, only the Ye6100 probe sets, which compared best with their YS98 counterparts, were retained and used in further analysis. The comparison was based on the criteria of Affymetrix as stated in its “User’s Guide to Products Comparison Spreadsheets” and the “Ye6100 to YS98” file to be found on the Affymetrix “Yeast 6100 Set-Support Materials” web page [http://www.affymetrix.com/support/technical/byproduct.affx?product=ye6100]. The best comparison spreadsheet that was generated by our group comprised 3751 probe sets (genes) and is available to any user upon request. The Ye6100 PM data files that had been generated at the previous step were filtered for these 3751 genes using the “compare arrays” algorithm of the dchip software.Data filtering
Using only the PM model, potential biases in the probe set expression values due to non specificity of probes are not accounted for (there is currently debate regarding the accuracy of the available PM-MM algorithm for the non specificity estimation; thus only the PM model was used in the present analysis). Since these biases affect the low expression values more prominently, only the probe sets whose expression value was higher than 20 in all 5 experimental conditions of the Lai and Elsas study20 that were used in the normalization were considered in further analysis. In current microarray data analyses, 50 is most often used as the threshold value for filtering. However, the measured expression values are much higher than those observed in the Lai and Elsas20 study’s datasets (based on the low-density array type). After filtering, the PM datasets were comprised 3741 gene entries.Data analysis
First, the ratio of each gene’s expression in (A) the galactose-grown over the glucose-grown WT physiology ([WT+GAL]/[WT+GLC]), and (B) the glucose-grown GAL7Δ over the glucose-grown WT phsyiology ([GAL7Δ+GLC]/[WT+GLC]), was estimated. In lack of more than one transcriptional profiles for each physiological state that prevented the use of any normalization techniques, and in light of the large error of the DNA microarray analysis as bioanalytical technique, only the genes whose expression doubled or more and decreased by 50% or more in [WT+GAL] (case A) or [GAL7Δ+GLC] (case B) compared to [WT+GLC] were considered for further analysis. The observed up- or down-regulation of these genes was considered large to be due to biological reasons and not to experimental biases. As using the log2(gene expression ratio) instead of the gene expression ratio is common practice in transcriptomic analysis, the genes that were included in subsequent analysis as “overexpressed” and “underexpressed”, respectively, were those with log2(expression ratio) ≥1 and ≤−1.Any prominent biologically-relevant conclusions related to comparisons A and B were based on the genes whose expression changed significantly between the two compared physiologies. In lack of more than one transcriptional profile for each physiological state that prevented the use of any hypothesis testing methods for the identification of the significant genes, as significantly over- and under-expressed, respectively, were defined the genes with log2(expression ratio) ≥1.5 and ≤−1.5. In the figures, the significantly over- and under-expressed genes in each comparison along with any cellular function associated with them will be depicted in red and dark green, respectively. The two remaining gene classes, i.e. those with 1 ≤ log2(expression ratio) < 1.5 and −1.5 < log2(expression ratio) ≤ −1, were used to complement the physiology findings based on the significantly over- and under-expressed genes, respectively. In the rest of the text, they will be referred, respectively, as “moderately over- or under-expressed” and in the figures will be depicted in purple and light green.
The ontology analysis of the significantly over- and under-expressed genes was carried out using the online SGD Gene Ontology Slim Mapper [http://db.yeastgenome.org/cgi-bin/GO/goTermMapper] and SGD Gene ontology Term Finder [http://db.yeastgenome.org/cgi-bin/GO/goTermFinder]. According to the web page of the Yeast Genome Database, “to determine the statistical significance of the association of a particular GO term with a group of genes in the list” (see Tables 3 and 4), “GO Term Finder calculates the p-value: the probability or chance of seeing at least x number of genes out of the total n genes in the list annotated to a particular GO term, given the proportion of genes in the whole genome that are annotated to that GO Term. That is, the GO terms shared by the genes in the user’s list are compared to the background distribution of annotation. The closer the p-value is to zero, the more significant the particular GO term associated with the group of genes is (i.e. the less likely the observed annotation of the particular GO term to a group of genes occurs by chance)”. The metabolic network reconstruction shown in Figs. 3 and 4 was based on information from the KEGG (www.kegg.com) and EXPASY (www.expasy.org) databases.
Conclusions
In the present paper, we compared at the transcriptional level (A) the WT S. cerevisiae physiology in galactose with respect to glucose growth conditions, and (B) the gal7-deficient (GAL7Δ) with respect to the WT physiology in glucose growth conditions, based on the full-genome transcriptomic data that were obtained in the experiment described in ref. 15. The first comparison indicated significant increase in the transcriptional expression of the ribosomal machinery and decrease in the transcriptional activity of the fatty acids’ β-oxidation at the peroxisomes, the latter leading to decrease in the rate of the glyoxylate and tricarboxylic acid cycles at the transcriptional level. These events are accompanied by increase in the expression of genes involved in the ethanol and glycerol biosynthesis, agreeing with the current knowledge that galactose assimilation is related to ethanol production. Comparison of the GAL7Δ with respect to the WT physiology in glucose indicated significant differences, showing that the gal7 activity is essential even in the absence of galactose. Its deficiency was observed to cause significant increase in the mitochondrial activity at the transcriptional level, which was directly connected with higher transcriptional rate of the oxidative phosphorylation, TCA cycle, glyoxylate shunt and β-oxidation of fatty acids at the peroxisomes. Moreover, GAL7Δ was observed to have an increased rate of catabolism of disaccharides, including sucrose, mannose and trehalose, towards increased biosynthesis of the main components of its cell wall. Comparison of the observed glucose-grown GAL7Δ transcriptional profile with the transcriptional physiology of the WT or other mutant strains in various carbon sources indicated strong correlation between the glucose-grown GAL7Δ transcriptional profile with that expected under conditions of no glucose repression. This result can directly explain many of the observed transcriptional changes. On the other hand, there are important differences that cannot be directly justified. In addition, for both comparisons, most of the genes whose expression differed significantly between the two compared physiologies are of yet unknown function. Thus, further investigation based on specifically designed perturbations and integrated analyses of multiple levels of cellular function in the context of the entire carbon source sensing and regulation system is still needed to fully elucidate the in vivo regulation of galactose assimilation.Acknowledgements
We would like to gratefully acknowledge the financial support of FORTH/ICE-HT through junior faculty startup funding to Dr Klapa and graduate student fellowship to Mr. Syriopoulos; of NIH (Grant #:1R01 HD054744) and the American Heart Association (South-East Affiliate Scientist Development Grant # 0435267B) to Dr Lai. We are also grateful to the reviewers of this manuscript for their valuable remarks.References
- L. F. Leloir, The enzymatic transformation of uridine diphosphate glucose into a galactose derivative, Arch. Biochem., 1951, 33, 186–90 Search PubMed.
- R. G. Duggleby, Y. C. Chao, J. G. Huang, H. L. Peng and H. Y. Chang, Sequence differences between human muscle and liver cDNAs for UDPglucose pyrophosphorylase and kinetic properties of the recombinant enzymes expressed in Escherichia coli, Eur. J. Biochem., 1996, 265, 173–9 CrossRef.
- W. L. Salo, J. H. Nordin, D. R. Peterson, R. D. Bevill and S. Kirkwood, The specificity of UDP-glucose 4-epimerase from the yeast Saccharomyces fragilis, Biochim. Biophys. Acta, 1968, 151, 484–92 CAS.
- N. Yildirim and M. C. Mackey, Feedback regulation in the lactose operon: a mathematical modeling study and comparison with experimental data, Biophys. J., 2003, 84, 2841–2851 CrossRef CAS.
- G. M. Walker, Yeast Physiology and Biotechnology, John Wiley and Son Ltd., New York, 1998 Search PubMed.
- A. Platt and R. J. Reece, The yeast galactose genetic switch is mediated by the formation of a Gal4p–Gal80p–Gal3p complex, EMBO J., 1998, 17, 4086–4091 CrossRef CAS.
- T. Ideker, V. Thorsson, J. A. Ranish, R. Christmas, J. Buhler, J. K. Eng, R. Bumgarner, D. R. Goodlett, R. Aebersold and L. Hood, Integrated genomic and proteomic analyses of a systematically perturbed metabolic network, Science, 2001, 292, 929–934 CrossRef CAS.
- S. Ostergaard, L. Olsson and J. Nielsen, In vivo dynamics of galactose metabolism in Saccharomyces cerevisiae: metabolic fluxes and metabolic levels, Biotechnol. Bioeng., 2001, 73, 412–425 CrossRef CAS.
- S. Ostergaard, L. Olsson, M. Johnston and J. Nielsen, Increasing galactose consumption by Saccharomyces through metabolic engineering of the GAL gene regulatory network, Nat. Biotechnol., 2000, 18, 1283–1286 CrossRef CAS.
- K. J. Isselbacher, E. P. Anderson, K. Kurahashi and H. M. Kalckar, Congenital galactosemia, a single enzymatic block in galactose metabolism, Science, 1956, 126, 635–6 CrossRef.
- N. V. Guerrero, R. H. Singh, A. Manatunga, G. T. Berry, R. D. Steiner and L. J. Elsas, 2nd, Risk factors for premature ovarian failure in females with galactosemia, J. Pediatr., 2000, 137, 833–41 CrossRef CAS.
- A. Robertson, R. H. Singh, N. V. Guerrero, M. Hundley and L. J. Elsas, Outcomes analysis of verbal dyspraxia in classic galactosemia, Genet. Med., 2000, 2, 142–8 Search PubMed.
- A. L. Webb, R. H. Singh, M. J. Kennedy and L. J. Elsas, Verbal dyspraxia and galactosemia, Pediatr. Res., 2003, 53, 396–402 CrossRef CAS.
- K. Lai and M. I. Klapa, Alternative pathways of galactose assimilation: could inverse metabolic engineering provide an alternative to galactosemic patients?, Metab. Eng., 2004, 6, 269–244.
- D. A. Lashkari, J. L. DeRisi, J. H. McCusker, A. F. Namath, C. Gentile, S. Y. Hwang, P. O. Brown and R. W. Davis, Yeast microarrays for genome wide parallel genetic and gene expression analysis, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13057–13062 CrossRef CAS.
- J. Gao, G. Opiteck, J. M. S. Friedrichs, A. R. Dongre and S. A. Hefta, Changers in the protein expression of yeast as a function of carbon source, J. Proteome Res., 2003, 2, 643–649 CrossRef CAS.
- S. L. Westergaard, A. P. Oliveira, o. C. Br, L. Olsson and J. Nielsen, A systems biology spproach to study glucose repression in the yeast Saccharomyces cerevisiae, Biotechnol. Bioeng., 2007, 96, 134–145 CrossRef CAS.
- G. G. Roberts and A. P. Hudson, Transcriptome profiling of Saccharomyces cerevisiae during a transition from dermentative to glycerol-based respiratory growth reveals extensive metabolic and structural remodeling, Mol. Genet. Genomics, 2006, 276, 170–186 CrossRef CAS.
- I. L. Medintz, G. J. Vora, A. M. Rahbar and D. C. Thach, Transcript and proteomic analyses of wild-type and gpa2 mutant Saccharomyces cerevisiae strains suggest a role for glycolytic carbon source sensing in pseudohyphal differentiation, Mol. BioSyst., 2007, 3, 623–634 RSC.
- K. Lai and L. J. Elsas, Overexpression of human UDP-glucose pyrophosphorylase rescues galactose-1-phosphate uridyltransferase-deficient yeast, Biochem. Biophys. Res. Co., 2000, 271, 392–400 Search PubMed.
- C. Larroy, Rosario, M. Fernández, E. González, X. Parés and J. A. Biosca, Properties and functional significance of Saccharomyces cerevisiae ADHVI, Chem.-Biol. Interact., 2003, 143–144, 229–38 CrossRef CAS.
- H. Lodish, D. Baltimore, A. Berk, S. L. Zipursky, P. Matsudaira and J. Darnell, Molecular Cell Biology, Scientific American Books, Inc., US, 3rd edn, (5th printing), 1998 Search PubMed.
- L. Stryer, Biochemistry, W.H. Freeman and Company, NY, 4th edn, (2nd printing), 1995 Search PubMed.
- M. Kunze, I. Pracharoenwattana, S. M. Smith and A. Hartig, A central role for the peroxisomal membrane in glyoxylate cycle function, Biochim. Biophys. Acta, 2006, 1763, 1441–1452 CAS.
- T. Hashimoto, Y. Yoshida and K. Tagawa, Regulatory proteins of F1F0-ATPase: role of ATPase inhibitor, J. Bioenerg. Biomembr., 1990, 22, 27–38 CrossRef CAS.
- J. R. Dickinson and M. Schweizer, The metabolism and molecular physiology of Saccharomyces cerevisiae, CRC Press, Wahington, DC, 2nd edn., 2004 Search PubMed.
- E. Cabib, R. Roberts and R. Bowers, Synthesis of yeast cell wall and its regulation, Annu. Rev. Biochem., 1982, 51, 763–793 CrossRef CAS.
- H. Zlotnik, M. P. Fernandez, B. Bowers and E. Cabib, Saccharomyces cerevisiae mannoproteins form an external cell wall layer that determines wall porosity, J. Bacteriol., 1984, 3, 1018–1026.
- S. Ozkan and M. Johnston, Function and regulation of yeast hexose transporters, Microbiol. Mol. Biol. Rev., 1999, 63, 554–569 CAS.
- A. Avendano, A. Deluna, H. Olivera, L. Valenzuela and A. Gonzalez, GDH3 encodes a glutamate dehydrogenase isozyme, a previously unrecognized route for glutamate biosynthesis in Saccharomyces cerevisiae, J. Bacteriol., 1997, 179, 5594–5597 CAS.
- M. J. Brauer, A. J. Saldanha, K. Botstein and D. Dolinski, Homeostatic adjustment and metabolic remodeling in glucose-limited yeast cultures, Mol. Biol. Cell, 2005, 16, 2503–2517 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Tables 1 and 2 provide a list of the significantly and moderately, respectively, over- and under-expressed genes in both physiological comparisons studied in this paper. For ESI see DOI: 10.1039/b718732g |
| This journal is © The Royal Society of Chemistry 2008 |