Depiction of metabolome changes in histidine-starved Escherichia coli by CE-TOFMS†
Yoshiaki
Ohashi
ab,
Akiyoshi
Hirayama
b,
Takamasa
Ishikawa
a,
Seira
Nakamura
a,
Kaori
Shimizu
b,
Yuki
Ueno
a,
Masaru
Tomita
ab and
Tomoyoshi
Soga
*ab
aHuman Metabolome Technologies, Inc., Tsuruoka, Yamagata 997-0052, Japan
bInstitute for Advanced Biosciences, Keio University, 246-2 Mizukami, Kakuganji, Tsuruoka, Yamagata 997-0052, Japan. E-mail: soga@sfc.keio.ac.jp; Fax: +81-235-29-0534; Tel: +81-235-29-0528
First published on 15th November 2007
Abstract
Metabolic changes in response to histidine starvation were observed in histidine-auxotrophic Escherichia coli using a capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS)-based metabolomics technique. Prior to the analysis, we prepared an E. coli metabolome list of 727 metabolites reported in the literature. An improved method for metabolite extraction was developed, which resulted in higher extraction efficiency in phosphate-rich metabolites, e.g., ATP and GTP. Based on the results, 375 charged, hydrophilic intermediates in primary metabolisms were analysed simultaneously, providing quantitative data of 198 metabolites. We confirmed that the intracellular levels of intermediates in histidine biosynthesis are rapidly accumulated in response to a drop in histidine level under histidine-starved conditions. Simultaneously, disciplined responses were observed in the glycolysis, tricarboxylic acid cycle, and amino acid and nucleotide biosynthesis pathways as regulated by amino acid starvation.
Introduction
Metabolomics, the unbiased determination of metabolite levels, is expected to be a valuable approach for the characterisation of bioprocesses in combination with genomics, transcriptomics, and proteomics. Different from the other “omics”, metabolomics includes methodological problems derived from heterogeneity in chemical properties, that is, it involves the development of extraction methods of metabolites that can be applied to a broad range of chemical species, and analytical methods that are achieved by as few processes as possible. However, it is impossible for all metabolites to be detected by single analytical method at present.The chemical properties of metabolites are widely diverse, but the majority of them have a few common properties in terms of physico-chemical traits: molecular weight less than 500, high hydrophilicity,1 and charge in aqueous solution.2 On the basis of these properties, we have developed a metabolome analysis method using capillary electrophoresis electrospray ionization mass spectrometry (CE-MS) to determine the majority of metabolic intermediates.3–7 We found 150 metabolites in sporulating Bacillus subtilis, and analysed the changes in profiles in major energy metabolisms.6 However, it is unclear how many metabolites in bacterial cells can be determined by our analytical system. Extraction of metabolites from cells is also one of the most dominant factors in metabolomics. Extraction must be adapted to the target metabolites, organisms, and analytical methods employed. To date, many extraction methods have been comparatively studied, with cold methanol extraction,8,9 and acidic acetonitrile extraction10 as the recommended techniques. We have employed a quenching method by pure methanol followed by methanol-water-chloroform extraction to remove hydrophobic metabolites.6 While this method satisfies particular constraints in CE-MS analysis, the efficiency and data reproducibility is not sufficient especially in phosphate-rich metabolites, e.g., ATP. In this study, we modified the method to improve these problems, and applied it to determine the intermediates of histidine biosynthesis in Escherichia colihistidine-auxotroph.
The stringent response is known to be a starvation-stimulated adaptation mechanism which includes functional arrests of chromosome replication, cell division, transcription, translation, and metabolisms.11 Translation arrest induced by amino acid starvation on ribosomes activates RelA protein , resulting in temporal accumulation of a bacterial alarmone, guanosine 3′-diphosphate 5′-diphosphate (ppGpp). Recently, transcriptome and proteome analyses have been demonstrated in bacterial stringent responses, indicating the possibility that metabolisms are dramatically influenced by amino acid starvation.12–14 However, no metabolomics approach has been used, raising a question of how to change the metabolism in the starved, auxotrophic situation in bacteria.
In this study, we prepared a complete metabolite list for E. coli by searching databases and the literature. The method of metabolome extraction was also improved, realising good metabolome analyses using capillary electrophoresis electrospray ionization time-of-flight mass spectrometry (CE-TOFMS). We applied the method to the analysis of the E. colihistidine-auxotroph and demonstrated through the analysis of 375 metabolites including primary metabolism intermediates that the metabolome profile of E. coli is dramatically changed during amino acid starvation. The quantitative data obtained for 198 hydrophilic, charged metabolites provides new insights in bacterial starvation.
Results and discussion
Target metabolites of CE-MS based metabolomics
The definition of the metabolome that is widely accepted in various fields is the entire set of low-molecular weight intermediates in metabolisms including amino acids, amines , nucleotides , sugars , lipids , and other substances. Generally, DNA, RNA , and protein are excluded from the definition, but their digests are occasionally recognised to be the targets of metabolomics. These metabolites are widely diverse in their chemical properties, i.e., polarity that is a determinant of solubility, electronic charge of ions, volatility, and molecular weight. Metabolites found in E. coli cells are summarised in the EcoCyc database,15,16 and their physico-chemical properties have been previously observed.1 Prior to the metabolome analysis, we prepared a list of E. coli metabolites to understand the target metabolites in CE-TOFMS based metabolomics. Our survey of the known E. coli metabolites, carried out through a database and literature search (see Experimental), resulted in a list of metabolome including 727 metabolites (supplementary Table 1† ). Of these, 453 (62%) belong to primary metabolites and their degradation intermediates (classes I and II), which are expected to be found in cells grown in minimal medium (Fig. 1A). Since most of the primary metabolites, for which the number is calculated as 92% (hydrophilic in Fig. 1B) × 96% (anion, cation, and nucleotide in Fig. 1C) = 88% (402 metabolites), are hydrophilic and charged, it is expected that CE-TOFMS, which principally targets water-soluble, charged metabolites is an extremely promising analysis method for metabolomics in primary pathways. Since some of the metabolites (e.g., acetic acid) cannot be detected rationally, the number of the target metabolites in CE-TOFMS analysis was 375, which corresponds to 83% of the primary metabolites in E. coli.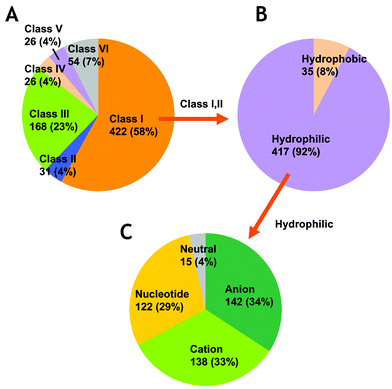 | ||
| Fig. 1 Classification of E. coli metabolites. A. Distribution of metabolites by metabolic pathways. The classes are described in the Experimental section. B. Hydrophobicity of metabolites in classes I and II. H2O was excluded from the calculation. C. Distribution of analytical modes in CE-TOFMS analysis for hydrophilic metabolites in classes I and II. The neutral metabolites cannot be analysed by CE-MS. | ||
Optimisation of metabolome extraction procedure
A problem was inherent in our previous studies regarding bacterial metabolome analyses.5,6 The extraction efficiencies of phosphate-rich metabolites such as ATP were often relatively low, affecting reproducibility of the analyses. We considered that the problem may be due to the interaction between phosphate groups of metabolites and phospholipids in the cell membrane. To improve this problem, we introduced sonication treatment during methanol extraction of metabolites from the cells (Fig. 2 and Experimental section). By this additional process, cells are peeled from the filter and completely suspended in methanol. Here, an ultrasonic syringe is suitable rather than a cell disruptor, since the methanol is transpired by excessively strong ultrasonic of cell disruptor, resulting in fluctuation and enhanced error of the metabolomics data (data not shown). To compare the extraction efficiencies, we used four methods for the methanol extraction process; (i) with sonication (improved method), (ii) without sonication (conventional method), (iii) with sonication, but cells are removed by brief centrifugation before chloroform addition (sonicated cell removal method), and (iv) with incubation at −80 °C for 12 h, but cells are removed (cold method).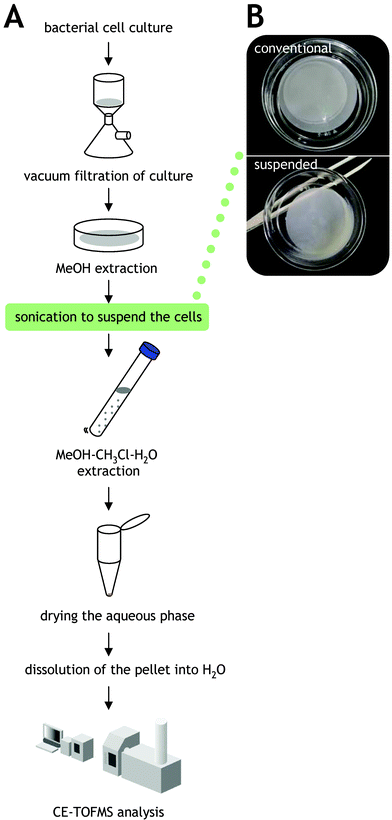 | ||
| Fig. 2 Metabolome extraction procedure modified in this study. A. Schematic of the metabolome extraction procedure. Modified point is indicated in the green box. B. Cell suspension prepared by sonication of methanol. | ||
To examine the reproducibility of the improved method, we repeatedly collected the data obtained in the improved method (the data are distinguished as “1st” and “2nd”). Fig. 3A shows the reproducibility and improvement of extraction efficiency by sonication in nucleotide metabolites containing phosphate groups. The metabolome source was E. coli W3110 grown in rich medium at 37 °C until the middle-logarithmic growth phase. Strikingly, extraction efficiency of GTP was improved 130-fold relative to the conventional method reported previously.6 For adenosine-phosphates, ATP, ADP, and AMP, the signal intensities were enhanced 39-, 17-, and 1.6-fold, respectively, by the improved method. Enhancement of detected signal intensities depended on the number of phosphate groups in the metabolites, indicating that efficient extraction of phosphate-rich metabolites is difficult by methanol extraction alone. It has been assumed that the phenomenon is due primarily to interaction of the metabolites with cell membranes by the intense negative charge of phosphate, but further investigations are needed. The improvement of extraction efficiency was not achieved by the sonicated cell removal method or cold method (Fig. 3A). This result suggests that the cells (and enzymes in the cells) fixed in methanol are damaged or inverted by contact with chloroform, and release the metabolites including phosphate groups into the solvent. The extraction efficiency of most amino acids was independent of the extraction methods except for lysine and arginine (Fig. 3B). This suggests that extraction of basic metabolites is also conditional in methanol extraction. Another basic amino acid, histidine, was efficiently extracted by all methods, possibly due to its more feeble imidazole group charge. As the isoelectric points of arginine, lysine, and histidine are 10.76, 9.74, and 7.59, respectively, the extremely intense positive charges of arginine and lysine most likely interact with the phosphate group in the phospholipids of the cell membrane. On the other hand, a problem was raised in the improved method. In some metabolites, NADH, NADPH, and coenzyme A (CoA), the extraction efficiency was worsened relative to that of the conventional method (Fig. 3A). Levels of oxidised forms of these metabolites, NAD+, NADP+, and CoA dimer (CoA–CoA) were increased in the improved method. This was not found in the other methods. Total amounts of these oxidised and reduced forms of metabolites were enhanced in the improved method (data not shown), suggesting that the extraction of them is basically improved. However, decreased levels of the oxidised forms most likely suggest that the reduced forms of the metabolites are oxidised during preparation procedures. Although it is not clear why oxidation of the metabolites is accelerated in the improved method, enhanced levels of some oxidants, e.g., flavin nucleotides may stimulate the oxidation.
![Improved extraction efficiency of nucleotide metabolites by the improved method developed in this study. A. Comparison of the detected levels of nucleotide metabolites. The number of phosphate groups that are contained in the metabolite is shown in parentheses. B. Comparison of the detected levels of amino acids. ●, area ratio of [improved method (1st)] to [improved method (2nd)], that is, the reproducibility of the improved method; △, area ratio of [improved method (1st)] to [conventional method]; □, area ratio of [improved method (1st)] to [sonicated cell removal method]; ○, area ratio of [improved method (1st)] to [cold method]. See text for details of the extraction procedures.](/image/article/2008/MB/b714176a/b714176a-f3.gif) | ||
| Fig. 3 Improved extraction efficiency of nucleotide metabolites by the improved method developed in this study. A. Comparison of the detected levels of nucleotide metabolites. The number of phosphate groups that are contained in the metabolite is shown in parentheses. B. Comparison of the detected levels of amino acids. ●, area ratio of [improved method (1st)] to [improved method (2nd)], that is, the reproducibility of the improved method; △, area ratio of [improved method (1st)] to [conventional method]; □, area ratio of [improved method (1st)] to [sonicated cell removal method]; ○, area ratio of [improved method (1st)] to [cold method]. See text for details of the extraction procedures. | ||
Determination of histidine-biosynthesis intermediates using improved methods
Histidine is synthesised via ten steps which are evolutionarily conserved in all organisms that synthesise histidine.17 However, standard chemicals of most of the intermediates are not commercially available, making pathway analysis difficult (Fig. 4A). Providing the migration times of the intermediates in CE-TOFMS analysis enables the analysis of histidine biosynthesis as a part of metabolome analysis. Since, most intermediates in histidine biosynthesis are phosphate-including complex compounds which are detected by CE-TOFMS in nucleotide mode, we performed CE-TOFMS analyses of the metabolites extracted by the improved preparation method presented here. To accumulate the histidine intermediates, we employed an E. coli mutant (JW2002) carrying a null mutation in the hisDallele that codes for histidine dehydrogenase catalysing the conversion of L-histidinol to L-histidine viaL-histidinal.18 | ||
| Fig. 4 Signal identification of histidine biosynthesis intermediates. A. Histidine biosynthesis pathway in E. coli. Metabolites that are detected in nucleotide mode by CE-TOFMS analysis are indicated in blue. Red circles show the metabolites for which standard chemicals are not commercially available. L-Histidinal coloured gray cannot be detected because it is not released during the overall reaction catalysed by histidine dehydrogenase.31 B. Accumulation of intermediates of histidine biosynthesis after histidine downshift. The data are indicated as the area values relative to that of internal standard. C. Electropherograms of the intermediates obtained by CE-TOFMS analyses. The metabolites except for L-histidinol and L-histidine were detected with nucleotide mode. The data are indicated as the signal intensities (arbitrary unit). | ||
The histidine biosynthesis pathway is repressed, as a posttranslational regulation, by histidine at the first step of the pathway,19 and is down-regulated by attenuation of the gene expression under conditions of excess histidine.20 The expression of hisoperon is also induced by amino acid starvation in a ppGpp-mediated manner.21 Hence, it is expected that amino acid starvation stimulates the accumulation of histidine biosynthesis intermediates. E. coli JW2002 was grown in a minimal medium supplemented with histidine and then resuspended in a minimal medium without histidine at the middle-to-late logarithmic growth phase. Accumulation of histidine biosynthesis intermediates are shown in Fig. 4B. The intermediates for which standard chemicals are unavailable were identified based on their calculated m/z values (Fig. 4C). A downshift of histidine in the medium activates histidine biosynthesis provoking accumulation of its intermediates. Actually, the intracellular level of histidine dropped rapidly within 30 min of the downshift, resulting in the activation of the histidine biosynthesis pathway. Though phosphoribosyl-ATP was not detected here, chronological accumulation profiles of the intermediates were observed. The phosphoribosylpyrophosphate (PRPP) level was increased at the onset of starvation (0–15 min), and then slightly decreased in concert with the drop of histidine concentrations (30–60 min). These results are consistent with the previous observations,19 and clearly indicate the accumulation of intermediates in histidine biosynthesis. Additionally, among the intermediates only PRPP and L-histidinol are commercially available at present, and thus the results yield important information of CE-TOFMS analysis in histidine biosynthesis.
Metabolome profiles under histidine starvation conditions
Amino acid starvation induces the accumulation of ppGpp, stimulating stringent response in cells.11 Actually, ppGpp levels were significantly increased after the histidine downshift (supplementary Table 2† ). This observation led us to view all the metabolite changes during histidine starvation as the stringent response. We determined the metabolome profiles including 375 metabolites belonging to classes I and II (supplementary Table 2). Of the metabolites, a total of 198 were successfully detected in cells during histidine starvation, and are depicted in the E. coli metabolic pathway map shown in Fig. 5 (high resolution, printable version is available as supplementary Fig. 1† ). Of the metabolites detected in cells, 45 were determined based on their m/z values because the standard chemicals are not available. At a glance, dramatic changes were found in regional primary metabolisms, but most of the metabolites were not changed. Since the metabolic network in E. coli is generally robust against environmental perturbations,22 the metabolic changes found here seem to involve homeostasis of metabolic processes.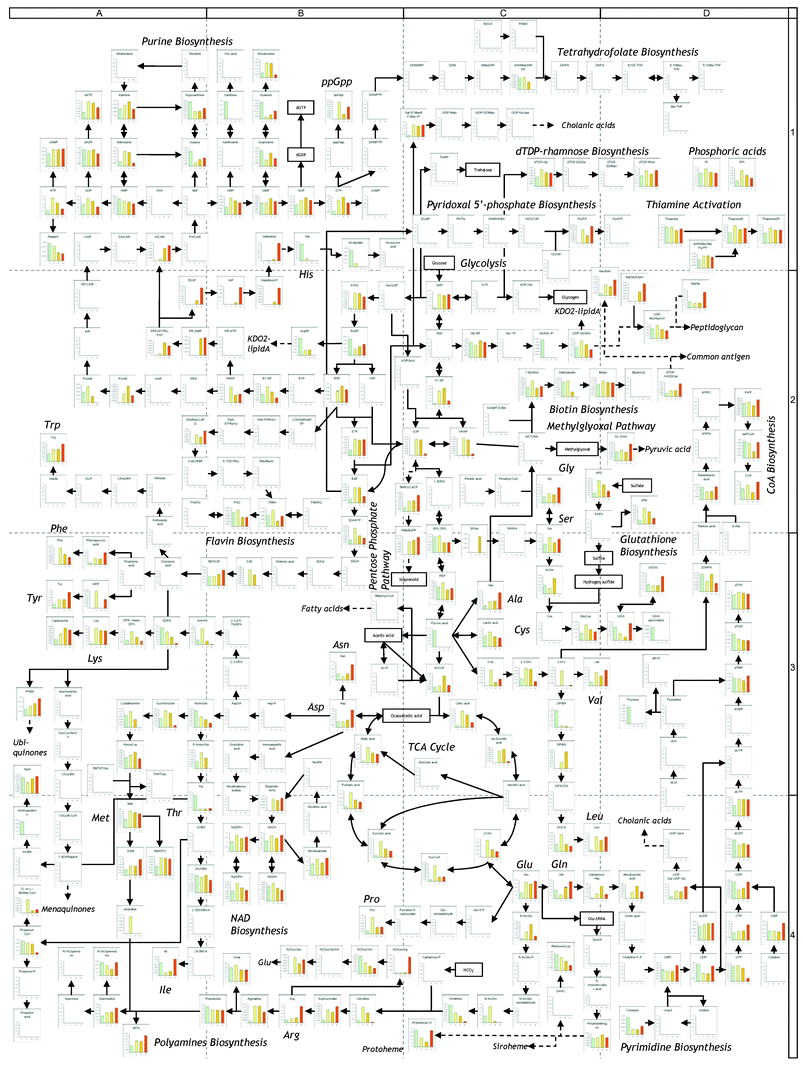 | ||
| Fig. 5 Metabolomic profile of E. coli after histidine downshift. The data of each metabolite level are summarized in supplementary Table 2. Enlarged, printable version is available as supplementary Figure 1.† | ||
To identify the metabolic changes, we performed clustering analysis of the metabolome data using hierarchical clustering analysis (HCA), generating five significant clusters. Clustering analysis is applicable and valuable in transcriptomic analysis, since the accumulation of a specific mRNA directly shows the activation of the gene expression.23 However, it should be noted in metabolomics that the accumulation of a metabolite does not indicate activation of the metabolic pathway, it should be estimated in combination with the change of its down- and upstream metabolites. The clusters generated here are shown in Fig. 6 and listed in Table 1. The metabolites for which levels were unchanged were excluded from HCA. Clusters 1, 2, and 3 resembled each other in the profiles and tend to be accumulated after histidine downshift (Fig. 6B). Cluster 1 metabolites were temporary accumulated during 15 to 30 min after the histidine downshift, and then decreased (Fig. 6B). Cluster 2 metabolites were rapidly accumulated at 15 min and maintained thereafter (Fig. 6B). Accumulations of cluster 3 metabolites were delayed to 60 min (Fig. 6B). Clusters 4 and 5 tended to be decreased gradually and rapidly, respectively (Fig. 6B).
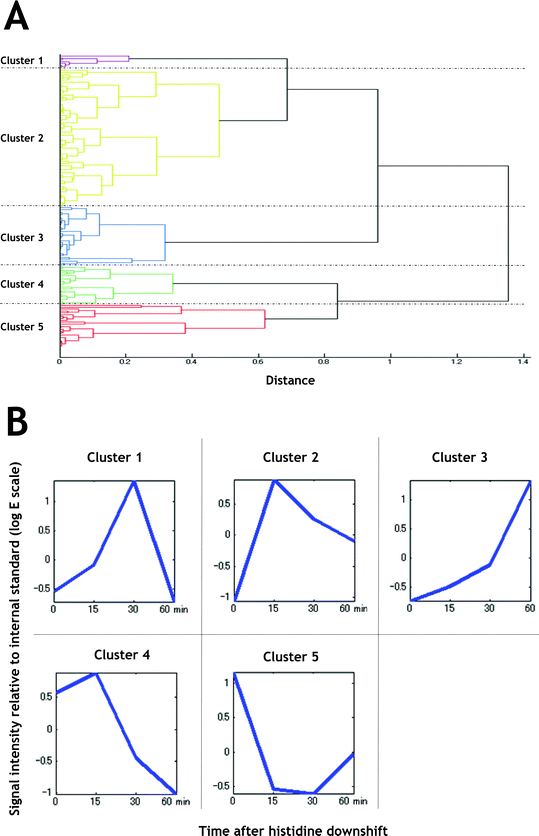 | ||
| Fig. 6 Clustering analysis of metabolites for which intracellular levels were changed during histidine starvation. A. Tree view of distances between the clusters generated by HCA. B. Average view of the clusters. Signal intensities relative to that of internal standard were standardised. | ||
During stringent responses, DNA replication, transcription, translation, and cell division are arrested in a ppGpp-dependent manner.11 Actually, growth was seen to be arrested by monitoring optical density at 600 nm at the onset of histidine starvation (data not shown). High-energy carrier molecules, ATP and GTP, were accumulated slightly (cluster 2), suggesting that DNA replication, and transcription of ribosomal RNAs are stopped, as described elsewhere.11 This is considered to be a reservation for further growth when de novohistidine biosynthesis is activated (growth cannot occur because of hisD disruption in the cells). The building blocks of DNA, dNTP, were maintained after histidine downshift. Amino acid levels were significantly accumulated after downshift: most are distributed to clusters 2 and 3. In particular, accumulation of lysine, leucine, arginine, and phenylalanine was prominent. Of amino acids, threonine, except histidine, was assigned to cluster 5, indicating threonine was consumed after histidine-downshift. These results suggest that threonine biosynthesis from aspartic acid was shut down during starvation, resulting in the drop of isoleucine and following stimulation of valine and leucine biosynthesis.24
Next, we focused on the behavior of cluster 2 metabolites which were clustered with ppGpp in order to discuss the candidate pathways under stringent control (Table 1). The first half of glycolysis, from glucose-6-phosphate (G6P) to glyceraldehyde-3-phosphate (G3P) and dihydroacetonephosphate (DHAP), was accumulated in concert with the intermediates in the pentose phosphate pathway (Fig. 5, positions B-2 and C-2). This effect seems to involve increased histidine biosynthesisviaPRPP. However, the middle of glycolysis, from 3- or 2-phosphoglyceric acid (3- or 2-PG) to phosphoenolpyruvic acid (PEP), was unchanged (Fig. 5, position C-3), while late step of glycolysis, pyruvic acid, dropped remarkably (Fig. 5, position C-3). These results suggest that the behavior of glycolysis is complicated during histidine starvation: that is, the first half is activated with the pentose phosphate pathway, but the flux is not extended to pyruvic acid synthesis. Moreover, the entire tricarboxylic acid (TCA) cycle intermediates were distributed into cluster 2, indicating that the TCA cycle was activated to yield precursors of downstream pathways and consumes intracellularpyruvic acid. Although we cannot exclude the possibility that the aforementioned pathways are regulated in a ppGpp-independent manner, the results provide new insights for understanding the stringent response in bacteria. Metabolomics observations in the relA mutant are needed to unveil the mechanism of stringent response in metabolism.
Experimental
Preparation of E. colimetabolite list
Metabolites in E. coli were selected from the EcoCyc15,16,25 and KEGG26,27 databases. The metabolites listed in the databases were identified one by one in the literature. Finally, 727 metabolites were selected as E. coli metabolites (supplementary Table 1† ) and then classified based on the metabolism pathways as follows. Class I metabolites belong to primary metabolisms including biosynthesis of building block metabolites of cells, energy metabolisms such as the glycolysis and TCA cycle, and other biosynthesis pathways. Class II includes the intermediates of degradation pathways of primary metabolites, e.g., amino acid degradation. The metabolites in classes I and II are expected to be found in cells grown in general minimal medium. Class III involves the degradation pathways of environmental compounds which are not included in general minimal medium. Class IV includes secondary metabolism or unconventional metabolisms. Class V includes the metabolites found in E. coli cells previously, but the pathways are unknown. Class VI metabolites are intermediates and products of in vitro enzymatic reactions using putative gene products.Bacterial strains and growth conditions
E. coli W3110 (laboratory stock) grown in Luria-Bertani (LB) liquid medium28 was used for optimisation of metabolome extraction procedure. The hisD deletion mutant, JW2002, was distributed from the Keio collection.29 The cells were pre-grown on LB plates at 37 °C for 12 h. The fresh colonies that appeared were inoculated into M9 liquid medium28 containing 50 µg ml−1 of L-histidine, and this was followed by incubation at 37 °C with shaking. When the cells reached the middle-to-late logarithmic phase (optical density at 600 nm = 0.8), they were harvested by brief centrifugation, washed with M9 medium without histidine, and then suspended in the M9 medium. The culture was continuously shaken at 37 °C until the sampling times.Metabolite extraction procedure
Intracellular metabolites were extracted as described previously with some modifications.6 Schematic of the metabolome extraction procedure is shown in Fig. 2. Cells grown as described above were collected for metabolome analysis at indicated times. Culture including approximately 109 cells (calculated as optical density at 600 nm × sampling volume of culture (ml) = 20) was filtered by a vacuum filtration system using a 0.4 µm pore size filter. The residual cells on the filter were washed twice with 5 ml of Milli-Q water. The filter was immersed in 2 ml of methanol including 5 µM each of internal standards, methioninesulfone and D-camphor-10-sulfonic acid (CSA). The dish was sonicated for 30 s using an Elma Transsonic T460/H ultrasonic syringe (not an ultrasonic cell disrupter) (Elma Hans Schmidbauer GmbH & Co., Singen, Germany) to suspend the cells completely. Effects of this process on metabolite extraction efficiency are discussed in the results and discussion section. A 1.6 ml portion of the methanol cell suspension was transferred to a Falcon Blue Max Jr., 352097 centrifugal tube (15 ml) (Becton Dickinson & Co., New Jersey, USA), and mixed with 1.6 ml of chloroform and 640 µl of Milli-Q water. After vortexing well, the mixture was centrifuged at 4600 × g and 4 °C for 5 min. The aqueous layer (750 µl) was distributed to three Amicon Ultrafree-MC ultrafilter tips (Millipore Co., Massachusetts, USA) and centrifuged at 9100 × g and 4 °C for approximately 2 h. The filtrate was dried and preserved at −80 °C until CE-MS analysis. Prior to analysis, the sample was dissolved in 25 µl of Milli-Q water.Instrumentation
CE-TOFMS was carried out using an Agilent CE Capillary Electrophoresis System equipped with an Agilent 6210 Time-of-Flight mass spectrometer, Agilent 1100 isocratic HPLC pump, Agilent G1603A CE-MS adapter kit, and Agilent G1607A CE-ESI-MS sprayer kit (Agilent Technologies, Waldbronn, Germany). The system was controlled by Agilent G2201AA ChemStation software version B.03.01 for CE (Agilent Technologies, Waldbronn, Germany). Data acquisition was performed by Analyst QS Build: 7222 software for Agilent TOF (Applied Biosystems, California, USA/MDS Sciex, Ontario, Canada).CE-TOFMS conditions
Separations and detections of metabolites were basically performed as described previously for cationic metabolites,3,6 anionic metabolites,4,6 and nucleotides .30 For cationic metabolites, capillary electrophoreses were performed using a fused silica capillary. The electrolyte was 1 M formic acid. Methanol-water (50% v/v) containing 0.5 µM reserpine (the lock mass for exact mass measurements) was delivered as the sheath liquid at 10 µl min−1. For anionic metabolites, a polymer coated SMILE(+) capillary (Nacalai tesque, Kyoto, Japan) was used. The electrolyte was 50 mM ammonium acetate (pH 8.5). Ammonium acetate (5 mM) in 50% (v/v) methanol-water containing 1 µM reserpine was delivered as the sheath liquid at 10 µl min−1. For nucleotides , separations were performed using a fused silica capillary. The electrolyte was 50 mM ammonium acetate (pH 7.5). The sheath liquid for anionic metabolites was used as the electrolyte. The capillary was pretreated with preconditioning buffer including 25 mM ammonium acetate and 75 mM sodium phosphate at pH 7.5. Pressure of 50 mbar was applied to inlet capillary during run to reduce the analysis time. For all analytical modes, inner diameter and total length of capillary are 50 µm and 100 cm, respectively. The applied voltage was set at +30 kV and −30 kV for cation and anion modes and nucleotide mode, respectively.Electrosplay ionisation-TOFMS was operated in the positive ion mode (4 kV), the negative ion mode (3.5 kV), and the negative ion mode (3.5 kV) for cationic metabolites, anionic metabolites, and nucleotides , respectively. A flow rate of heated dry nitrogen gas (heater temperature 300 °C) was maintained at 10 psig. Exact mass data were acquired over a 50–1000 m/z range.
Cluster analysis
The metabolome data set was prepared by selection of metabolites based on a coefficient of variation of more than 0.2 to exclude the metabolites for which levels were unchanged. HCA was performed on the data set using MATLAB 2007a (The Math Works, Massachusetts, USA). Distances between the metabolites were calculated by the equation, correlation coefficient + 1.Conclusion
In this study, we produced a list of previously reported E. coli metabolites together with analytical information for CE-TOFMS-based metabolome analysis. This list is also useful for other analytical methods using MS as a detector. The metabolome extraction method was also improved, realising quantitative analyses of phosphate-rich metabolites and other hydrophilic, charged metabolites. However, this method cannot be applied to the analysis of metabolites oxidised in the metabolome pool. Extraction methods inhibiting oxidation of the metabolites are needed to determine them. We successfully determined 198 metabolites in primary metabolisms of E. colihistidine-auxotroph. The metabolome profiles were analysed by mapping data to metabolic pathways and HCA, suggesting separated activation of amino acid biosynthesis pathways, glycolysis, and the TCA cycle. Although more detailed studies using the relA mutant is required to unveil the entire view of stringent responses mediated by ppGpp, we believe that metabolome profiling should provide new insights into bacterial adaptation to environmental changes.Acknowledgements
We are grateful to Hirotada Mori, Tomoya Baba, and Kenji Nakahigashi for providing the E. coli mutant strain and to Masatomo Hirabayashi and Yuji Sakakibara for providing excellent technical expertise in CE-TOFMS analysis. We also thank Mineo Morohashi for helpful discussions.References
- I. Nobeli, H. Ponstingl, E. B. Krissinel and J. M. Thornton, J. Mol. Biol., 2003, 334, 697–719 CrossRef CAS.
- M. R. Monton and T. Soga, J. Chromatogr., A, 2007, 1168, 237–246 CrossRef CAS.
- T. Soga and D. N. Heiger, Anal. Chem., 2000, 72, 1236–1241 CrossRef CAS.
- T. Soga, Y. Ueno, H. Naraoka, Y. Ohashi, M. Tomita and T. Nishioka, Anal. Chem., 2002, 74, 2233–2239 CrossRef CAS.
- T. Soga, Y. Ueno, H. Naraoka, K. Matsuda, M. Tomita and T. Nishioka, Anal. Chem., 2002, 74, 6224–6229 CrossRef CAS.
- T. Soga, Y. Ohashi, Y. Ueno, H. Naraoka, M. Tomita and T. Nishioka, J. Proteome Res., 2003, 2, 488–494 CrossRef CAS.
- T. Soga, R. Baran, M. Suematsu, Y. Ueno, S. Ikeda, T. Sakurakawa, Y. Kakazu, T. Ishikawa, M. Robert, T. Nishioka and M. Tomita, J. Biol. Chem., 2006, 281, 16768–16776 CrossRef CAS.
- S. G. Villas-Bôas, J. Højer-Pedersen, M. Åkesson, J. Smedsgaard and J. Nielsen, Yeast, 2005, 22, 1155–1169 CrossRef CAS.
- R. P. Maharjan and T. Ferenci, Anal. Biochem., 2003, 313, 145–154 CrossRef.
- J. D. Rabinowitz and E. Kimball, Anal. Chem., 2007, 79, 6167–6173 CrossRef CAS.
- M. Cashel, D. R. Gentry, V. J. Hernandez and D. Vinella, in Escherichia coli and Salmonella: cellular and molecular biology, American Society for Microbiology Press, Washington DC, USA, 1992, 2nd edn Search PubMed.
- C. Eymann, G. Homuth, C. Scharf and M. Hecker, J. Bacteriol., 2002, 184, 2500–2520 CrossRef CAS.
- O. Brockmann-Gretza and J. Kalinowski, BMC Genomics, 2006, 7, 230 CrossRef.
- A. Hesketh, J. Chen, J. Ryding, S. Chang and M. Bibb, Genome Biol., 2007, 8, R161 Search PubMed.
- I. M. Keseler, J. Collado-Vides, S. Gama-Castro, J. Ingraham, S. Paley, I. T. Paulsen, M. Peralta-Gil and P. D. Karp, Nucleic Acids Res., 2005, 33, D334–337 CAS.
- http://ecocyc.org/ .
- A. Nagai, E. Ward, J. Beck, S. Tada, J. Y. Chang, A. Scheidegger and J. Ryals, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4133–4137 CrossRef CAS.
- J. C. Loper and E. Adams, J. Biol. Chem., 1965, 240, 257–268.
- H. S. Moyed and M. Friedman, Science, 1959, 129, 968–969 CrossRef CAS.
- M. E. Winkler, D. J. Roth and P. E. Hartman, J. Bacteriol., 1978, 133, 830–843 CAS.
- J. C. Stephens, S. W. Artz and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4389–4393 CrossRef CAS.
- N. Ishii, K. Nakahigashi, T. Baba, M. Robert, T. Soga, A. Kanai, T. Hirasawa, M. Naba, K. Hirai, A. Hoque, P. Y. Ho, Y. Kakazu, K. Sugawara, S. Igarashi, S. Harada, T. Masuda, N. Sugiyama, T. Togashi, M. Hasegawa, Y. Takai, K. Yugi, K. Arakawa, N. Iwata, Y. Toya, Y. Nakayama, T. Nishioka, K. Shimizu, H. Mori and M. Tomita, Science, 2007, 316, 593–597 CrossRef CAS.
- J. Quackenbush, Nat. Rev. Genet., 2001, 2, 418–427 CrossRef CAS.
- H. E. Umbarger, in Escherichia coli and Salmonella: cellular and molecular biology, American Society for Microbiology Press, Washington DC, USA, 1992, 2nd edn Search PubMed.
- C. A. Ouzounis and P. D. Karp, Genome Res., 2000, 10, 568–576 CrossRef CAS.
- H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono and M. Kanehisa, Nucleic Acids Res., 1999, 27, 29–31 CrossRef CAS.
- http://www.genome.jp/kegg/ .
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. USA, 1989, 2nd edn Search PubMed.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2 Search PubMed 2006.0008.
- T. Soga, T. Ishikawa, S. Igarashi, K. Sugawara, Y. Kakazu and M. Tomita, J. Chromatogr., A, 2007, 1159, 125–133 CrossRef CAS.
- A. Kheirolomoom, J. Mano, A. Nagai, A. Ogawa, G. Iwasaki and D. Ohta, Arch. Biochem. Biophys., 1994, 312, 493–500 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary tables and high quality figures. See DOI: 10.1039/b714176a |
| This journal is © The Royal Society of Chemistry 2008 |