Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity
Mark R.
Wilson
*,
Justin J.
Yerbury
and
Stephen
Poon
School of Biological Sciences, University of Wollongong, Northfields Ave, Wollongong 2522, New South Wales, Australia. E-mail: mrw@uow.edu.au; Fax: +61 2 42214135; Tel: +61 2 42214534
First published on 19th November 2007
Abstract
The in vivo formation of fibrillar proteinaceous deposits called amyloid is associated with more than 40 serious human diseases, collectively referred to as protein deposition diseases. In many cases the amyloid deposits are extracellular and are found associated with newly identified abundant extracellular chaperones (ECs). Evidence is presented suggesting an important regulatory role for ECs in amyloid formation and disposal in the body. A model is presented which proposes that, under normal conditions, ECs stabilize extracellular misfolded proteins by binding to them, and then guide them to specific cell receptors for uptake and subsequent degradation. Thus ECs and their receptors may be critical parts of a quality control system to protect the body against dangerously hydrophobic proteins /peptides. However, it also appears possible that in the presence of a high molar excess of misfolded protein , such as might occur during disease, the limited amounts of ECs available may actually exacebate pathology. Further advances in understanding of the mechanisms that control extracellularprotein folding are likely to identify new strategies for effective disease therapies.
 Mark R. Wilson | Mark Wilson is a Professor of Biological Sciences at the University of Wollongong, Australia. He obtained his PhD in Biology from the University of Sydney in 1985. His research is focussed on identifying and characterising quality control mechanisms for extracellular protein folding. |
 Justin J. Yerbury | Justin Yerbury is a graduate student in the School of Biological Sciences at the University of Wollongong. He received his Honours Degree in Biology from the University of Wollongong in 2004. His research focuses on the role of extracellular chaperones in amyloid formation and toxicity. |
 Stephen Poon | Stephen Poon received his PhD in Biological Sciences in 2003 from the University of Wollongong, where he characterized the chaperone action of the extracellular chaperone, clusterin. Since then he has undertaken a postdoctoral role at the University of Cambridge, under the supervision of Prof. Christopher Dobson and Dr Jesús Zurdo, investigating the amyloid formation propensity of several therapeutically important peptides (e.g. calcitonin, GLP-1 and glucagon). Current studies include assessing the possible roles of clusterin in amyloidosis. |
1.0 Introduction
The term “molecular chaperone” was first developed in the late 1970's when referring to the ability of nucleoplasmin to inhibit inappropriate interactions between histones and DNA.1 The meaning of this term is continuing to evolve but two key properties of molecular chaperones are (i) selective binding to non-native protein conformations to form stable complexes, and (ii) inhibition of the irreversible aggregation of non-native protein conformations.2 Many molecular chaperones maintain other proteins in “folding-competent” conformations, which are returned to the native conformation by the involvement of other refolding chaperones. Previous studies have overwhelmingly focussed on intracellular molecular chaperones and their roles in protein folding within the cell. The discoveries that a range of serious human diseases are related to protein aggregation phenomena, and that molecular chaperones affect these processes, has led to a recent explosion of research in this area. The pathology of more than 40 human degenerative diseases is associated with the deposition of fibrillar proteinaceous aggregates called amyloid. Collectively referred to as protein deposition diseases, these include various sporadic (e.g. Alzheimer's (AD) and Parkinson's (PD) diseases), familial, and transmissible degenerative disorders (e.g. spongiform encephalopathies such as Creutzfeldt-Jakob disease (CJD)), many of which affect, amongst other tissues and organs, the brain and the central nervous system (CNS). In many cases, molecular chaperones have been found physically associated with amyloid deposits, although the reason for this association remains to be established.It has only recently become apparent that extracellular counterparts to the intracellular molecular chaperones exist. The role of these abundant extracellular chaperones (ECs) in the formation of amyloid deposits in vivo is emerging as an exciting field. In this review, we briefly introduce the molecular basis of amyloid formation, examine the available evidence that ECs influence this process, and discuss the potential roles of ECs in amyloid formation and disposal in vivo.
2.0 Amyloids
2.1 Amyloid formation
The hallmark of a wide range of debilitating and incurable human pathologies is the abnormal presence of extracellular deposits in a variety of organs and tissues, including the brain and CNS (e.g. AD, CJD), peripheral organs such as the heart, liver, and spleen (e.g. systemic amyloidoses), and skeletal tissues and joints (e.g. haemodialysis-related amyloidosis). There are many excellent reviews of the molecular events underlying amyloid formation (e.g.);3 only a very brief overview is provided here.Amyloid fibrils arise when a specific protein or protein fragment converts from an otherwise soluble form into insoluble filamentous aggregates. It appears that the aggregation behaviours of various amyloid-forming peptides and proteins are strikingly similar. The kinetics of amyloid formation (as measured by Thioflavin T fluorescence or light scattering) is generally characterized by an initial ‘lag’ or nucleation phase, followed by a rapid exponential ‘growth’ or polymerization phase,4 and lastly, by a plateau phase in which no further polymerization occurs (Fig. 1). The lag (or nucleation) phase is defined as the time required for the formation of the soluble (prefibrillar) oligomers or nuclei. This phase begins with the destabilization and partial unfolding of the native protein , leading to the formation of an ensemble of intermediately folded species. Structural perturbation leading to protein unfolding can be achieved by exposing the native protein to one or more chaotropic conditions, including elevated temperature, low pH, oxidative stress, molecular crowding, and protease-mediated degradation.5 A subset of proteins within this ensemble will undergo non-ordered aggregation to form nuclei. After nucleation and immediately preceding the polymerization phase, the nuclei transform into an ensemble of various assemblies called protofibrils that exhibit increased levels of complexity. During the polymerization phase, these protofibrils rapidly grow to form well-ordered protofilaments which laterally associate to give rise to mature amyloid fibrils.6
 | ||
| Fig. 1 A schematic representation of the amyloid forming pathway. The formation of amyloid fibrils occurs via a nucleation dependent mechanism in which soluble prefibrillar oligomers or nuclei are formed during the lag phase. These oligomeric species then act as templates to sequester other aggregation-prone intermediates, leading to rapid fibril growth (represented by the exponential elongation phase) and subsequently, to the formation of insoluble mature amyloid fibrils. The plateau phase represents the steady state when maximum fibril growth has been reached. Adapted from ref. 3. | ||
Typically, amyloid fibrils exist as long, unbranched, but often twisted structures that are 6–12 nanometres in diameter, bearing a characteristic ‘cross-β’ X-ray fibre diffraction pattern.7 Each fibril is usually comprised, within its core structure, of two to six ‘protofilaments’ that wind around each other to form supercoiled rope-like structures. Protofilaments are believed to be principally stabilized by intra- and intermolecular interactions (e.g. hydrogen bonds) and are composed of β-sheets whose strands lie perpendicular to the long axis of the fibril.8,9
2.2 Causes of amyloid pathology
Numerous theories have been advanced to account for the pathology of amyloid diseases, many focusing on the toxic nature of amyloids and how this relates to neuronal loss in the brain (reviewed in Stefani and Dobson).10 In some cases, disease pathology might simply be attributed to the shear bulk of deposited material present in the affected organs and tissues, which can cause physical disruption to the cellular architecture, subsequently leading to organ dysfunction (e.g. some of the systemic amyloidoses). However, an alternative widely accepted theory suggests that soluble, toxic protein aggregates are the primary cause of pathogenesis in disorders such as AD. An increasing quantity of data suggests that pre-fibrillar structures (e.g. oligomers and protofibrils) are toxic, rather than the mature fibrils into which they develop.11 The demonstration that these species are, in addition to being highly cytotoxic, readily diffusible throughout the brain may explain why neuronal loss is commonly observed at sites distant from those of the amyloid deposits. By proposing that the primary toxic species are the early aggregates, this theory also provides a plausible explanation for the lack of correlation between the extent of deposition of mature fibrils in the form of amyloid plaques in the diseased brain and the severity of clinical symptoms.123.0 Extracellular chaperones
There is evidence to suggest that there are control mechanisms for protein folding in extracellular spaces. For example, although cerebrospinal fluid (CSF) is regularly released into the venous system through the arachnoid villi, the half lives of proteins in the CSF differ from protein to protein , and even differ between various proteolytic fragments of the same original protein ,13 suggesting a selective mechanism of removal. In the event of a large-scale presentation of extracellular non-native protein(s), such as might occur during amyloid disease, only physically abundant extracellular chaperone(s) could reasonably be expected to provide an effective line of defense. “Normally intracellular” chaperones (e.g. Hsp70) are present extracellularly at very low (ng ml−1) levels, thus, their capacity would be quickly exceeded. It is therefore likely that bulk processing of non-native proteins is dealt with by much more abundant proteins with chaperone properties, hereafter referred to as extracellular chaperones (ECs), which have only recently been identified.Four secreted glycoproteins , clusterin,14 haptoglobin,15 α2-macroglobulin (French et al., unpublished work) and serum amyloid P component (SAP),16 have been shown to exhibit chaperone properties in vitro. The first three of these proteins share functional characteristics with the small heat shock proteins in that they are able to efficiently stabilize misfolded proteins to prevent them aggregating but are not capable of independently refolding proteins .
3.1 Clusterin
Clusterin is secreted from many different cell types and is found in human plasma, CSF and seminal fluid at concentrations of approximately 100, 2, and 1000 µg ml−1,17–19 respectively. The expression of clusterin is increased in a wide variety of models of stress and disease, including withdrawal of growth factors and exposure to noxious agents.20 The transcriptional regulator heat shock factor 1 (HSF1) binds to a highly conserved 14 bp element in the clusterin promoter21 and activates expression of both clusterin and heat shock proteins (which protect cells from stresses).21 Clusterin is encoded by a single gene and the translated product is internally cleaved to produce its α and β subunits, and glycosylated, prior to secretion from the cell. The exclusively N-linked glycosylation is variable in nature and extent, ranging from 17–27% (by weight).22 In aqueous solution at physiological pH, clusterin exists in a range of oligomeric forms; mildly acidic pH favours partial dissociation of oligomers into individual α–β heterodimers.23 Current insights into clusterin structure are largely reliant upon predictions based on sequence analyses, which suggest that the protein has significant contiguous regions of disordered conformation that separate other regions of well-defined secondary structure, such as amphipathic α-helical regions and coiled-coil α-helices.24,25 The high level of disorder, variable glycosylation and tendency to form oligomers have so far limited attempts to structurally characterise clusterin.Recent studies have demonstrated that clusterin has chaperone activity with a potent ability to influence the amorphous and fibrillar aggregation of many different proteins . Clusterin inhibits stress-induced protein aggregation by ATP-independent binding to exposed regions of hydrophobicity on non-native proteins to form soluble, high molecular mass complexes.26–28 Immunoaffinity depletion of clusterin from human plasma renders proteins in this fluid more susceptible to aggregation and precipitation.29 Clusterin lacks the ability to independently refold heat-stressed, non-native enzymes but, like the small heat shock proteins , is able to preserve heat-inactivated enzymes in a state competent for subsequent ATP-dependent refolding by Hsc70.27 However, because there is no currently known abundant refolding-competent EC, the physiological significance of this remains uncertain. During amorphous aggregation of proteins , clusterin appears to interact with slowly aggregating species on the off-folding pathway. Interestingly, where tested, clusterin has been found associated with all amyloid deposits.28
By complexing with misfolded extracellularproteins , ECs like clusterin may mediate their cellular uptake and degradation.30 Clusterin has long been known to interact with the cell surface receptor megalin (LRP2) and to complex with Aβ to mediate its uptake by megalin and subsequent degradation.31 It also interacts with other members of the low density lipoprotein (LDL) receptor family — it binds to chicken LR8 and an LDLR-related protein ,32 and uptake of clusterin-leptin complexes by apoER2 and VLDLR has been proposed to facilitate leptin clearance.33 Furthermore, clusterin and LRP1/megalin have been implicated in the clearance of cellular debris by fibroblasts and epithelial cells.34
3.2 Haptoglobin
Haptoglobin (Hp) is a secreted acidic glycoprotein produced mainly in the liver and found in most body fluids of humans and other mammals. Normally, it is present in human plasma at 300–2000 µg ml−135 and CSF at 0.5–2 µg ml−1.36 However, the levels of Hp in human plasma are increased up to 8-fold during various physiological stresses (e.g. inflammation), leading to it being designated as an “acute phase protein ”.35,37 Hp is encoded by a single gene; uniquely, in humans, there are two principal alleles (Hp1 and Hp2), which results in individual humans expressing one of three major Hp phenotypes (Hp 1-1, Hp 2-1, Hp 2-2). In all cases, Hp can be represented as a multimer of an αβ subunit. In its simplest form (Hp 1-1), Hp consists of a disulfide-linked (α1)2β2 structure (∼100 kDa). However, in Hp 2-1 and Hp 2-2, an additional cysteine residue in the α2 chain allows the formation of a complex series of various sized disulfide-linked αβ polymers (∼100 to ∼500 kDa).Hp binds with extremely high affinity to hemoglobin (Hb) (KD ∼ 10−15 M).35 Formation of the Hp–Hb complex inhibits Hb-mediated generation of lipid peroxides and hydroxyl radical, which is thought to occur in areas of inflammation.37 Hp has also been implicated in immune regulation38 and shown to inhibit cathepsin B activity.39 Binding of Hp to human neutrophils has been reported to inhibit respiratory burst activity.40 In addition, neutrophils have been shown to take up exogenous Hp and store it within cytoplasmic granules — they subsequently secrete it into the local extracellular environment in response to a variety of pro-inflammatory stimuli (e.g. yeast, TNFα, or the chemotactic peptide fMLP).41,42 Thus, the available evidence indicates that Hp is likely to play an important role in suppressing inflammatory responses.
Human Hp specifically inhibits the precipitation of a wide variety of proteins induced by a range of stresses.15,43 All three human Hp phenotypes exert this chaperone action, although at equivalent mass concentrations, at least for one substrate protein tested, Hp1-1 was the most efficient. Like clusterin, Hp forms stable, soluble high molecular mass complexes with misfolded proteins . Also like clusterin, Hp lacks ATPase activity and has no independent ability to refold misfolded proteins . The possibility that Hp holds misfolded proteins in a state competent for refolding by other chaperones is currently untested. Immunoaffinity depletion of Hp from human serum significantly increased the amount of protein that precipitated in response to stresses.15 Thus, Hp has the ability to protect many different proteins from stress-induced amorphous precipitation and its effects in whole human serum suggest that this activity is likely to be relevant in vivo. Currently, there are no published studies of the effects of Hp on amyloid formation, although Hp is found associated with Aβ amyloid deposits in vivo.44
When complexed to Hb, Hp is known to bind to the CD163 cell surface receptor.45 Other receptors to which Hp binds are the CD11b/CD18 integrin (Mac-1/CR3), which also binds denatured proteins and the iC3b fragment of complement,46 and the CD22 B lymphocyte receptor. Unidentified specific Hp binding sites have also been reported to occur on neutrophils40 and mast cells.47 Thus, it appears feasible that Hp might interact with one or more of these receptors to mediate the clearance and degradation of misfolded extracellularproteins .
3.3 α2-Macroglobulin (α2M)
α2-Macroglobulin (α2M) is a major human blood glycoprotein, comprised of ∼10% carbohydrate by mass. It is assembled from four identical 180 kDa subunits into a 720 kDa tetramer; the 180 kDa subunits are disulfide bonded to form dimers, which non-covalently interact to yield the final tetrameric quaternary structure.48 α2M is present in human plasma and CSF at 1500–200049 and 1–3.6 µg ml−1,50 respectively. It has a well-known ability to inhibit a broad spectrum of proteases, which it accomplishes using a unique trapping method. When exposed to a protease, α2M undergoes limited proteolysis at its bait region leading to a large conformational change, physically trapping the protease within a steric “cage”.49 The trapped protease forms a covalent linkage with α2M by reacting with an intramolecular thiol ester bond to yield a conformationally altered form known as “activated” or “fast” α2M (α2M*), which exposes a receptor recognition site for low density lipoprotein receptor related protein (LRP).49 By directly interacting with the thiol ester bond, small nucleophiles such as methylamine can also activate α2M.51Aside from its interactions with proteases, α2M binds to Aβ peptide, prion protein and β2-microglobulin, which are associated with Alzheimer’s disease,52 spongiform encephalopathies53 and dialysis related amyloidosis,54 respectively, to cytokines and growth factors,55 and to a range of hydrophobic molecules including endotoxin, phenyl-Sepharose, and liposomes.56 The binding to hydrophobic molecules does not inhibit the trapping of proteases and is not known to be associated with any conformational changes.56 In addition, α2M is found associated with amyloid deposits in AD and spongiform encephalopathies.53,57 Previous work has indicated that α2M-polypeptide complexes are immunogenic.58,59 α2M bound peptides are internalised by LRP and fragments of the peptide are subsequently re-presented on the cell surface. This response is identical to the one elicited by peptides chaperoned by intracellular heat shock proteins .60 A further hint that α2M might have chaperone properties came from the observation that it inhibits the aggregation of Aβ and protects cells from Aβ toxicity.61 It was recently shown that α2M has a promiscuous ATP-independent chaperone action similar to that of both clusterin and haptoglobin. It forms stable complexes with misfolded proteins to inhibit their stress-induced aggregation and precipitation but is unable to independently effect their refolding (French et al., unpublished work). α2M is the first known mammalian protein with both protease inhibitor and chaperone-like activities.
3.4 Serum amyloid P component
SAP is a member of the pentraxin family of proteins which are characterised by five identical subunits noncovalently associated to form a disc-like structure. The SAP pentamer consists of five 25 kDa subunits rich in anti-parallel β-strands, each containing 204 amino acids, an invariant N-linked biantennary oligosaccharide (constituting more than 8% of the mass of the molecule),62 and a single intra-chain disulfide bond.63 It has been proposed that SAP circulates as a decamer with two pentameric discs noncovalently bound face to face.63,64 Other reports claim that the decameric form of SAP is obtained only upon purification.65,66 SAP is synthesised and catabolized in the liver, and is present in human plasma and CSF at concentrations of ∼40 µg ml−167 and 8.5 µg ml−1,68 respectively. Whereas SAP is an acute-phase protein in mice,69 in humans its plasma concentration is not significantly elevated during acute inflammation.70 Each subunit monomer has two Ca2+-binding sites and shows Ca2+-dependent binding to many different ligands, including certain oligosaccharides , glycosaminoglycans,71 fibronectin,72 C-reactive protein ,73 aggregated IgG,74 C1q,75 complement C4-binding protein ,75 DNA,76chromatin,77 histones,78 and phosphoethanolamine-containing compounds such as phosphatidylethanolamine.63 Interaction with these ligands localises SAP to elastic microfibrils,79 glomerular and alveolar basement membrane, arterioles, bronchioles, sarcolemma of cardiac and smooth muscle,80 and all forms of amyloid.81Previous studies of the chaperone properties of SAP are insufficient in number and depth to allow unequivocal classification of SAP, together with clusterin, haptoglobin and α2M as a genuine EC. However, one study showed that when added to a refolding buffer containing denatured lactate dehydrogenase (LDH), SAP markedly enhanced the yield of active LDH; the reaction was Ca2+-independent and supra-stoichometric, a ratio of SAP pentamer to LDH substrate of 10 : 1 was needed to recover 25% of enzyme reactivity.16 This suggests that SAP is an inefficient refolding-competent chaperone. It is important to note that this in vitro activity was demonstrated in the absence of ATP and any “helper” chaperones. Clearly, further studies of the chaperone properties of SAP and how they might relate to known ECs will be valuable. Of particular interest in the current context, SAP has been found present in all amyloid deposits examined.82–85 The interaction between SAP and amyloid fibrils is highly specific, and the abundance of SAP in amyloid fibrils relative to its trace concentration in plasma is extraordinary.86 SAP has a protease-resistant β-pleated sheet structure that in the presence of Ca2+ is resistant to proteolysis.87 Furthermore, it has been shown that SAP inhibits the degradation of several types of amyloid fibrils by proteases. Tennent et al. 88 suggested that SAP protects amyloid from proteolytic degradation in vivo by binding to fibrils and masking fibrillar conformation.
4.0 The effects of extracellular chaperones on amyloid formation and toxicity
4.1 Effects of ECs on amyloid formation in vitro
In vivo amyloid formation and deposition is an extraordinarily complex process that can occur over much of the lifetime of an individual, thus making it difficult to monitor and study over a reasonable timescale. Although this complexity prevents the amyloid-forming process from being fully reproduced in a test tube, by examining individual steps in the process in vitro, it is still possible to gain insight into many facets of in vivo amyloid formation and deposition. In vitro, conditions can be manipulated such that fibril formation can be assessed in a manageable timeframe. Much of the current knowledge regarding the effects of ECs on amyloid formation has been obtained from in vitro studies that utilise such strategies. Whilst many studies have highlighted the ability of ECs to suppress amorphous aggregation, their effects on amyloid formation are, apart from clusterin, less well-documented. In 1994,89 it was shown that the formation of Aβ fibrils could be inhibited by sub-stoichiometric levels of clusterin, a finding which has since been confirmed by other independent studies.90,91 Since then, clusterin has been shown to potently inhibit the formation of fibrils derived from a broad range of other unrelated ‘substrates’, including prion protein Pr106–126,92 Apo C-II,93 chicken egg white lysozyme, calcitonin, κ-casein, α-synuclein, β2-microglobulin, PI3-SH3, Aβ, a model peptide CCβw,28 and the naturally occurring amyloidogenic variant of lysozyme, I56T.94 Similar inhibitory capabilities have also been documented for α2M. Like clusterin, sub-stoichiometric levels of α2M (a 1 : 8 molar ratio of α2M : substrate) were enough to completely inhibit the formation of fibrillar Aβ aggregates.90 Finally, it was shown that in vitro a 1 : 5 molar ratio of SAP to target protein completely attenuated the fibrillation of Aβ1–42 and α1-antitrypsin-derived C-terminal peptides.95 There are no published data to show whether haptoglobin has similar anti-amyloidogenic properties in vitro.Recent studies exploring the interactions between clusterin and amyloid forming proteins revealed that clusterin did not bind to the native form of the substrates tested, nor did it interact with mature fibrils.28,95 Clusterin most potently inhibited amyloid formation when it was present during the early stages of fibrillogenesis. Yet, based on results obtained by mass spectrometry, the presence of clusterin did not eliminate the appearance of a partially unfolded monomeric intermediate identified as the initial step in the pathway of lysozyme amyloidosis.94 The addition of clusterin during the elongation phase did not greatly alter the kinetics of aggregation of I56T lysozyme.94 Similarly, clusterin was more effective at suppressing fibril formation in solutions of Aβ and SH3 which had been seeded with samples taken from corresponding aggregation mixtures during the lag phase compared with those taken from the late (plateau) phase.28 This is consistent with data showing that at the same molar clusterin : substrate ratio, as the concentration of amyloid forming protein was increased, clusterin was less able to inhibit the formation of amyloid by Apo C-II, PI3-SH3 and Aβ. Increasing concentrations of amyloid forming protein increase the abundance of destabilised monomer, favouring self-association into oligomers able to nucleate the aggregation process and thereby shortening the lag phase.28,93 Taken together, the above results suggest that clusterin primarily exerts its effects on amyloid formation by binding to destabilized pre-fibrillar oligomeric species present at or near the nucleation phase. When in equilibrium with the native fold, destabilised and non-native structures can initiate aggregation when present at an abundance as low as 1%.96,97 Therefore, such species are present at concentrations low enough to account for the potent sub-stoichiometric effects of the ECs. The binding of ECs such as clusterin probably reduces the availability of these species to participate in the nucleation events that normally precede fibril formation (Fig. 2).
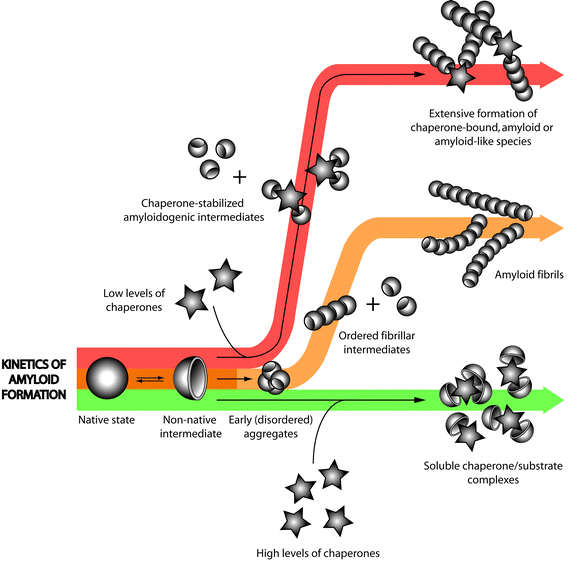 | ||
| Fig. 2 The biphasic effects of extracellular chaperones on amyloid fibril formation. The kinetics of the amyloid assembly process is often depicted as a sigmoidal curve to reflect the three distinct phases characterizing the fibril formation pathway (i.e. lag, elongation and plateau; see also Fig. 1). Complete inhibition of fibril formation can be achieved when clusterin and α2M are present at high (but usually still sub-stoichiometric) levels relative to the substrate protein . Yet, at low EC : substrate ratios, extensive amyloid or amyloid-like fibril formation is observed for some substrate proteins , suggesting that in these cases, ECs may be stabilizing an otherwise unstable conformation and promoting amyloid fibril formation. Figure reproduced with kind permission from Springer Publications. | ||
Despite many reports of the ability of ECs to inhibit fibril formation in vitro, there are indications to suggest that they may, at least under certain conditions, actually promote the formation or maintain the persistence of amyloid fibrils. α2M binds to and protects prion protein from degradation by proteinase K; this protection was not exerted by α2M binding to and inhibiting proteinase K.53 In addition, another study showed that α2M prevents the trypsin-mediated degradation of fibrils composed of either immunoglobulin lambda (λ) light chains or β2M.98 Similarly, the calcium-dependent binding of SAP to amyloid fibrils formed from Aβ, serum amyloid A protein or immunoglobulin light chain, protected the fibrils from subsequent protease-mediated degradation.88 This in turn led to suggestions that SAP may promote the persistence of amyloid deposits in vivo. For some protein substrates, at low clusterin : substrate ratios (1 : 50 to 1 : 500), clusterin promoted the formation of aggregates with an increased level of thioflavin T fluorescence, suggesting that (at these ratios) it promoted the formation of fibrils from calcitonin, α-synuclein and Aβ. At these low levels, clusterin was found incorporated into insoluble Aβ and SH3 fibrils.28 Similarly, at low SAP : Aβ ratios, SAP promoted the formation of Aβ aggregates;99 at a SAP : Aβ of 1 : 1000, short fibrillar-like structures lacking typical amyloid features were formed which often contained associated SAP molecules.95 In addition, α2M has been shown to stabilize a conformation of the prion protein resistant to proteinase K (PrPRes) that is thought to initiate aggregation and is associated with prion disease pathology.53 Taken together, these results suggest that when ECs are present at low levels relative to the substrate they may act to stabilize an otherwise unstable conformation and facilitate amyloid fibril formation (Fig. 2).
4.2 The in vitro effects of ECs on amyloid toxicity
Recent studies have indicated that the location of amyloid deposits in vivo do not correlate well with sites of neurotoxicity (e.g. in AD,100 amyotrophic lateral sclerosis (ALS),101 PD102 and familial amyloidotic polyneuropathy).103 Neuronal losses associated with these disorders may be brought about by toxicity exerted by smaller soluble aggregates (sometimes referred to as oligomers or protofibrils).3 These protofibrils have been shown to be more toxic than both the protein /peptide from which they are made and the mature fibrils constructed from them.11 If this scenario is correct, then it follows that at least under some conditions EC-mediated inhibition of protein aggregation could actually promote cytotoxicity. When tested against neuronal cell lines, under certain specific conditions, clusterin and α2M were shown to promote the neurotoxicity of Aβ (clusterin — PC12 cells;104 α2M — LAN5 cells).57 In stark contrast, using primary rat mixed neuronal cultures, others have demonstrated that clusterin and α2M can protect cells from Aβ toxicity.61,105 Even within individual studies, depending on the conditions used, clusterin was shown both to protect cells and to promote cytotoxicity. Complexes of clusterin and Aβ formed at high ratios of clusterin : Aβ (e.g. 1 : 10) were less toxic to SH-SY5Y cells than Aβ alone; in contrast, complexes formed at low ratios of clusterin : Aβ (e.g. 1 : 500) were more toxic than Aβ alone.28 Thus, the effects of ECs, and in particular clusterin, on the toxicity of aggregates are complex and have been shown to depend upon the clusterin : substrate ratio, the stage of amyloid formation at which the aggregates are formed, and the cell type.It has been postulated that the level of hydrophobicity exposed by aggregates may determine their toxicity.3 How this directly relates to the mechanism of their toxicity is uncertain and is currently a hotly researched topic, but it is probably due to interactions of hydrophobic residues with membranes or other molecules essential for normal cellular function.3 This, in part, may explain the differential effects of clusterin on toxicity. When the substrate protein is present at a high molar excess, clusterin may be unable to mask all regions of exposed hydrophobicity but instead may stabilize aggregates which retain sufficient exposed hydrophobicity to exert toxicity. In contrast, at higher but still sub-stoichiometric ratios of clusterin : Aβ, toxicity is reduced.28
Another potentially important factor to consider when trying to understand the in vivo effects of ECs on amyloid toxicity, is that in addition to their effects on the nature and size of protein aggregates, ECs may also be involved in the physical clearance of protein aggregates. This is illustrated by the demonstration that in the presence of α2M (but not otherwise), SH-SY5Y cells expressing the α2M receptor (LRP) are more resistant to Aβ toxicity than cells that do not.57 The protective effect of α2M could be inhibited by RAP (a pan-specific inhibitor of LRP ligand binding). Furthermore, α2M promoted Aβ toxicity against LRP-negative LAN5 cells but had the opposite effect with LRP-expressing LAN5 transfectants.57 Therefore, although ECs may provide cells with some protection by binding to exposed hydrophobic regions on protein aggregates, a further protective mechanism may only come into play when appropriate cell surface receptors are available to mediate uptake and degradation of EC-substrate protein complexes.
4.3 ECs and disposal of extracellular misfolded proteins
It has been proposed that ECs are scavengers for misfolded proteins in the extracellular space, inhibiting protein aggregation and deposition and guiding the “damaged” protein substrates to specific receptors for their internalization and subsequent lysosomal degradation30 (Fig. 3). This system of EC directed receptor-mediated endocytosis may play a major role in the maintenance of protein solubility in extracellular fluids, including in the brain (interstitial fluid and CSF). Evidence supporting this model includes: | ||
| Fig. 3 Receptor-mediated endocytosis and lysosomal degradation — a putative quality control mechanism for extracellularprotein folding. An extracellular quality control process was recently proposed in which extracellular chaperones (EC) facilitate the clearance of non-native protein structures viareceptor-mediated endocytosis and intracellular, lysosomal degradation. The existence of such a quality control mechanism would ensure that proteins do not persist beyond their intended lifespan in the extracellular space and normally counteract the development of protein aggregation disease pathologies. | ||
(1) In the CNS, ECs are produced locally in astrocytes and some neuron populations106–108 and can be transported from plasma across the blood brain barrier (BBB).109 The receptors LRP and megalin are expressed on the surface of cells in contact with CSF (i.e. chloroid plexus and ependymal cells110 and at the BBB109,111 and LRP is also found on the surface of astrocytes112 and microglial cells.113
(2) Clusterin and α2M are both found complexed with soluble amyloid-forming proteins in human CSF.53,114
(3) It has previously been shown that clusterin-Aβ complexes bind to megalin on the surface of mouse teratocarcinoma F9 cells, and are subsequently internalised, transported to lysosomes and degraded.31 Similarly, α2M-Aβ complexes are internalized via LRP expressed on U87 cells and are subsequently degraded.52
(4) The normally rapid removal of radiolabelled Aβ from mouse brain is significantly inhibited by the LDL family inhibitor RAP and antibodies against LRP-1 and α2M.115 Furthermore, when complexed with clusterin, the rate of clearance of Aβ1–42 from the mouse brain across the BBB into plasma is increased by more than 80% and this transport is significantly inhibited by anti-megalin antibodies.116
4.4 When quality control is compromised
If a system of extracellularprotein quality control (QC) operates and is efficient, why do protein deposits sometimes develop? Protein aggregation is generally age-related, and some proteins such as Aβ accumulate as a part of normal aging.117 In young, healthy humans the disposal of proteins such as transthyretin or peptides like Aβ appears to be efficient, since there are no apparent protein deposits. Thus, it is likely that protein deposition may represent an age-dependent overloading of the QC system that ultimately results in toxicity and neurodegeneration. If the extracellular QC system is overloaded in protein folding-related diseases, it is critical to pinpoint the factors and pathways involved to facilitate identification of new potential therapeutic strategies.One poorly understood pathway relates to the observation that under some conditions ECs can actually promote the formation of amyloid fibrils; clusterin, α2M and SAP have been shown to promote fibril formation in vitro28,53,99 (see section 4.1). In addition, in a transgenic mouse model expressing amyloid precursor protein (APP; PDAPP mice), a comparison of clusterin−/− and clusterin+/+ mice showed no difference in the amount of Aβ deposited in the brain, however the amount of thioflavin S positive material was larger in clusterin+/+ mice.118 Although there was no electron microscopy data to confirm that this was in fact amyloid fibrils, the authors suggest that this demonstrates clusterin promoted fibril formation in this model. In contrast, PDAPP apoE−/−, clusterin−/− mice had significantly increased Aβ levels in CSF and intersititial fluid and thioflavin S deposits in the brain.119 The results of this latter study were interpreted as suggesting that clusterin works cooperatively with ApoE to inhibit the formation of Aβ amyloid in the brain. The interpretations presented from these two studies are seemingly contradictory, and a clear understanding of the in vivo role of clusterin in amyloidogenesis has yet to be achieved. An important factor influencing the nature of the effect of clusterin on amyloid formation, at least in vitro, is the clusterin : substrate protein ratio. At very low ratios of clusterin : substrate, clusterin has been shown to enhance amyloid associated thioflavin T fluorescence suggesting that it increases amyloid formation, while at higher but still sub-stoichiometric ratios have the opposite effect (see section 4.1). A similar biphasic effect on amyloid formation has also been shown for the intracellular yeast chaperone hsp104.120
It is possible that at times in local microenvironments, the concentrations of ECs are low and that under these conditions they actually promote the formation of amyloid fibrils (see section 4.1). Interestingly, under conditions of high substrate protein excess, clusterin becomes incorporated into insoluble aggregates.28 Sequestration of clusterin into deposits would effectively lower its availability, and potentially further promote fibril formation. If the other ECs behave similarly, then it is no surprise that all are found associated with deposits (see sections 3.1–3.4, and Fig. 2). In humans with AD121,122 and Creutzfeldt Jacob disease121,123 the levels of clusterin mRNA are increased, however, there is no associated increase in clusterin protein levels in AD CSF124 and the level of clusterin protein in CSF is decreased by 2.5 fold in Creutzfeldt Jacob disease.125 In addition, decreased levels of clusterin in the eye are thought to be responsible for deposition of Pseudo exfoliation (PEX) material in PEX syndrome126 (again clusterin is found associated with PEX deposits).
Another factor that could favour the development of amyloid pathology is a dysfunction in the expression or function of relevant cell surface receptors. Under some conditions, ECs may stabilize toxic conformations of substrate proteins with exposed hydrophobicity (see section 4.2). If receptor-mediated clearance is inadequate, the persistence of such toxic complexes could manifest as neuronal damage. In vitro assays demonstrated that when neuronal cells were in mixed cultures with astrocytes and microglia (which express receptors capable of specifically binding to ECs, e.g. LDLR family members), by complexing with Aβ, both clusterin and α2M were neuroprotective. However, when neuronal cells were cultured alone, complexes of clusterin or α2M with Aβ were neurotoxic, suggesting that complexes of EC and aggregating protein may be toxic in the absence of receptor mediated endocytosis pathways. Moreover, there are increased plasma concentrations of several LRP ligands in AD patients, including ApoE, α1 anti-chymotrypsin and urokinase127–129 suggesting that LRP may be either overwhelmed, downregulated or faulty in AD. More recently, this has been confirmed by data showing that LRP expression at the BBB is decreased in AD patients and mouse models.130 As discussed above (see section 4.3), this is consistent with the results from mouse models where antibody-mediated blockade of megalin and LRP significantly prolonged the time taken to clear Aβ from the brain.115,116
5.0 Conclusion & future directions
In recent years our understanding of the nature and significance of amyloid formation and deposition and the role that these play in disease has taken dramatic leaps forward. Further advances in understanding of the mechanisms which control extracellularprotein folding are likely to identify new strategies for effective disease therapies. This review focused on what little is known about quality control of the folding of amyloid forming proteins in extracellular spaces. Since the EC field is still in its infancy, this review is necessarily speculative in a number of areas. However, it is quite clear that amyloid forming proteins such as Aβ and prion proteins are ‘chaperoned’ by what have become termed extracellular chaperones. It is proposed that once bound to misfolded proteins , ECs guide them to specific receptors that function to direct proteins into lysosomes for degradation. In this scenario, specific receptors such as LRP and megalin are an integral and critical part of a quality control system which provides protection against dangerously hydrophobic proteins /peptides. Since relatively little is known about ECs and their role in protein deposition diseases many future research directions are possible. Initially, it will be important to better understand the mechanism by which, under some conditions, ECs promote amyloid formation, as this has clear relevance to the development of any EC-based therapies. Certainly, it will be critical to evaluate the roles of ECs in a variety of in vivo models of protein deposition diseases. It may be possible to accelerate the pace of advances by utilizing innovative new disease models such as those developed using the fruit fly Drosophila. The short generation times and ease of genetic manipulation in these models create opportunities not accessible with mammalian systems. Much remains to be discovered, ensuring that exciting times lie ahead.References
- R. A. Laskey, B. M. Honda, A. D. Mills and J. T. Finch, Nature, 1978, 275, 416–20 CrossRef CAS.
- A. L. Fink, Physiol. Rev., 1999, 79, 425–49 CAS.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–66 CrossRef CAS.
- J. T. Jarrett and P. T. Jr. Lansbury, Cell, 1993, 73, 1055–8 CrossRef CAS.
- J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 930–2 CrossRef CAS.
- J. van Gestel and S. W. de Leeuw, Biophys. J., 2007, 92, 1157–63.
- M. Sunde and C. Blake, Adv. Protein Chem., 1997, 50, 123–59 CAS.
- L. C. Serpell, M. Sunde, M. D. Benson, G. A. Tennent, M. B. Pepys and P. E. Fraser, J. Mol. Biol., 2000, 300, 1033–1039 CrossRef CAS.
- L. C. Serpell, Biochim. Biophys. Acta, 2000, 1502, 16–30 CrossRef CAS.
- M. Stefani and C. M. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–11 CrossRef CAS.
- R. Katzman, R. Terry, R. DeTeresa, T. Brown, P. Davies, P. Fuld, X. Renbing and A. Peck, Ann. Neurol., 1988, 23, 138–44 CrossRef CAS.
- M. J. Savage, S. P. Trusko, D. S. Howland, L. R. Pinsker, S. Mistretta, A. G. Reaume, B. D. Greenberg, R. Siman and R. W. Scott, J. Neurosci., 1998, 18, 1743–52 CAS.
- D. T. Humphreys, J. A. Carver, S. B. Easterbrook-Smith and M. R.. Wilson, J. Biol. Chem., 1999, 274, 6875–81 CrossRef CAS.
- J. J. Yerbury, M. S. Rybchyn, S. B. Easterbrook-Smith, C. Henriques and M. R. Wilson, Biochemistry, 2005, 44, 10914–25 CrossRef CAS.
- A. R. Coker, A. Purvis, D. Baker, M. B. Pepys and S. P. Wood, FEBS Lett., 2000, 473, 199–202 CrossRef CAS.
- N.-H. Choi-Miura, Y. Ihara, K. Fukuchi, M. Takeda, Y. Nakano, T. Tobe and M. Tomita, Acta Neuropath., 1992, 83, 260–264 Search PubMed.
- I. B. Fritz, K. Burdzy, B. Setchell and O. Blaschuk, Biol. Reprod., 1983, 28, 1173–88 Search PubMed.
- B. Murphy, L. Kirszbaum, I. D. Walker and J. F. d'Apice, J. Clin. Invest., 1988, 81, 1858–1864 CrossRef CAS.
- D. E. Jenne and J. Tschopp, Trends Biochem. Sci., 1992, 17, 154–159 CrossRef CAS.
- D. Michel, G. Chatelain, S. North and G. Brun, Biochem. J., 1997, 328, 45–50 CAS.
- J. T. Kapron, G. M. Hilliard, J. N. Lakins, M. P. R. Tenniswood, K. A. West, S. A. Carr and J. W. Crabb, Prot. Sci., 1997, 6, 2120–2133 Search PubMed.
- T. Hochgrebe, G. J. Pankhurst, J. Wilce and S. B. Easterbrook-Smith, Biochemistry, 2000, 39, 1411–9 CrossRef CAS.
- A. K. Dunker, C. J. Brown, J. D. Lawson, L. M. Iakoucheva and Z. Obradovic, Biochemistry, 2002, 41, 6573–82 CrossRef CAS.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS.
- D. T. Humphreys, J. A. Carver, S. B. Easterbrook-Smith and M. R. Wilson, J. Biol. Chem., 1999, 274, 6875–6881 CrossRef CAS.
- S. Poon, M. S. Rybchyn, S. B. Easterbrook-Smith, J. A. Carver and M. R. Wilson, Biochemistry, 2000, 39, 15953–15960 CrossRef CAS.
- J. J. Yerbury, S. Poon, S. Meehan, B. Thompson, J. R. Kumita, C. M. Dobson and M. R. Wilson, FASEB J., 2007, 21, 2312–22 CrossRef CAS.
- S. Poon, M. S. Rybchyn, S. B. Easterbrook-Smith, J. A. Carver, G. J. Pankhurst and M. R. Wilson, J. Biol. Chem., 2002, 277, 39532–39540 CrossRef CAS.
- J. J. Yerbury, E. M. Stewart, A. R. Wyatt and M. R. Wilson, EMBO Rep., 2005, 6, 1131–6 Search PubMed.
- S. M. Hammad, S. Ranganathan, E. Loukinova, W. O. Twal and W. S. Argraves, J. Biol. Chem., 1997, 272, 18644–18649 CrossRef CAS.
- M. G. Mahon, K. A. Lindstedt, M. Hermann, J. Nimpf and W. J. Schneider, J. Biol. Chem., 1999, 274, 4036–4044 CrossRef CAS.
- T. M. Bajari, V. Strasser, J. Nimpf and W. J. Schneider, FASEB J., 2003, 17, 1505–7 CAS.
- M. M. Bartl, T. Luckenbach, O. Bergner, O. Ullrich and C. Koch-Brandt, Exp. Cell Res., 2001, 271, 130–41 CrossRef CAS.
- B. H. Bowman and A. Kurosky, Adv. Human Genet., 1982, 12, 189–261 Search PubMed.
- O. Sobek and P. Adam, J. Neurol., 2003, 250, 371–2 CrossRef CAS.
- W. Dobryszycka, Euro. J. Clin. Chem. Clin. Biochem., 1997, 35, 647–654 Search PubMed.
- H. Louagie, J. Delanghe, I. Desombere, M. De Buyzere, P. Hauser and G. Leroux-Roels, Vaccine, 1993, 11, 1188–90 CrossRef CAS.
- O. Snellman and B. Sylven, Nature, 1967, 216, 1033 CrossRef CAS.
- S. K. Oh, N. Pavlotsky and A. I. Tauber, J. Leuko. Biol., 1990, 47, 142–8 Search PubMed.
- L. Wagner, A. Gessl, S. B. Parzer, W. Base, W. Waldhausl and M. S. Pasternack, J. Immunol., 1996, 156, 1989–96 CAS.
- N. Berkova, C. Gilbert, S. Goupil, J. Yan, V. Korobko and P. H. Naccache, J. Immunol., 1999, 162, 6226–6232 CAS.
- Z. Pavlicek and R. Ettrich, Collect. Czech. Chem. Commun., 1999, 64, 717–725 CrossRef CAS.
- J. M. Powers, W. W. Schlaepfer, M. C. Willingham and B. J. Hall, J. Neuropath. Exp. Neurol., 1981, 40, 592–612 Search PubMed.
- J. H. Graversen, M. Madsen and S. K. Moestrup, Int. J. Biochem. Cell Biol., 2002, 34, 309–314 CrossRef CAS.
- G. D. Ross, Crit. Rev. Immunol., 2000, 20, 197–222 Search PubMed.
- S. M. El-Ghmati, M. Arredouani, E. M. Van Hoeyveld, J. L. Ceuppens and E. A. Stevens, Scand. J. Immunol., 2002, 55, 352–8 Search PubMed.
- P. E. Jensen and L. Sottrup-Jensen, J. Biol. Chem., 1986, 261, 15863–9 CAS.
- L. Sottrup-Jensen, J. Biol. Chem., 1989, 264, 11539–42 CAS.
- R. G. Biringer, H. Amato, M. G. Harrington, A. N. Fonteh, J. N. Riggins and A. F. Huhmer, Brief Funct. Genomic Proteomic, 2006, 5, 144–53 Search PubMed.
- M. J. Imber and S. V. Pizzo, J. Biol. Chem., 1981, 256, 8134–8139 CAS.
- M. Narita, D. M. Holtzman, A. L. Schwartz and G. Bu, J. Neurochem., 1997, 69, 1904–11 CAS.
- V. Adler and V. Kryukov, Neurochem. J., 2007, 1, 43–52 Search PubMed.
- Y. Motomiya, Y. Ando, K. Haraoka, X. Sun, H. Iwamoto, T. Uchimura and I. Maruyama, Kidney Int., 2003, 64, 2244–52 CrossRef CAS.
- J. M. Mettenburg, D. J. Webb and S. L. Gonias, J. Biol. Chem., 2002, 277, 13338–45 CrossRef CAS.
- A. J. Barrett, Meth. Enzymol., 1981, 80, 737–54 Search PubMed.
- C. Fabrizi, R. Businaro, G. M. Lauro and L. Fumagalli, J. Neurochem., 2001, 78, 406–12 CrossRef CAS.
- C. T. Chu and S. V. Pizzo, J. Immunol., 1993, 150, 48–58 CAS.
- R. J. Binder, D. Karimeddini and P. K. Srivastava, J. Immunol., 2001, 166, 4968–72 CAS.
- P. Srivastava, Nat. Rev. Immunol., 2002, 2, 185–94 CrossRef CAS.
- Y. Du, B. Ni, M. Glinn, R. C. Dodel, K. R. Bales, Z. Zhang, P. A. Hyslop and S. M. Paul, J. Neurochem., 1997, 69, 299–305 CAS.
- M. B. Pepys, T. W. Rademacher, S. Amatayakul-Chantler, P. Williams, G. E. Noble, W. L. Hutchinson, P. N. Hawkins, S. R. Nelson, J. R. Gallimore and J. Herbert, et al., Proc. Natl. Acad. Sci. USA, 1994, 91, 5602–6 CrossRef CAS.
- J. Emsley, H. E. White, B. P. Ohara, G. Oliva, N. Srinivasan, I. J. Tickle, T. L. Blundell, M. B. Pepys and S. P. Wood, Nature, 1994, 367, 338–345 CrossRef CAS.
- S. P. Wood, G. Oliva, B. P. O'Hara, H. E. White, T. L. Blundell, S. J. Perkins, I. Sardharwalla and M. B. Pepys, J. Mol. Biol., 1988, 202, 169–73 CrossRef CAS.
- I. J. Sorensen, O. Andersen, E. H. Nielsen and S. E. Svehag, Scand. J. Immunol., 1995, 41, 263–267 Search PubMed.
- J. A. Aquilina and C. V. Robinson, Biochem. J., 2003, 375, 323–328 CrossRef CAS.
- W. L. Hutchinson, G. E. Noble, P. N. Hawkins and M. B. Pepys, J. Clin. Invest., 1994, 94, 1390–6 CrossRef CAS.
- P. N. Hawkins, M. N. Rossor, J. R. Gallimore, B. Miller, E. G. Moore and M. B. Pepys, Biochem. Biophys. Res. Commun., 1994, 201, 722–6 CrossRef CAS.
- M. B. Pepys, M. Baltz, K. Gomer, A. J. Davies and M. Doenhoff, Nature, 1979, 278, 259–61 CrossRef CAS.
- M. B. Pepys, A. C. Dash, R. E. Markham, H. C. Thomas, B. D. Williams and A. Petrie, Clin. Exp. Immunol., 1978, 32, 119–24 CAS.
- H. Hamazaki, J. Biol. Chem., 1987, 262, 1456–60 CAS.
- F. C. de Beer, M. L. Baltz, S. Holford, A. Feinstein and M. B. Pepys, J. Exp. Med., 1981, 154, 1134–9 CrossRef.
- S. J. Swanson, R. B. Christner and R. F. Mortensen, Biochim. Biophys. Acta, 1992, 1160, 309–16 CAS.
- M. R. Brown and B. E. Anderson, J. Immunol., 1993, 151, 2087–95 CAS.
- I. J. Sorensen, E. H. Nielsen, O. Andersen, B. Danielsen and S. E. Svehag, Scand. J. Immunol., 1996, 44, 401–7 Search PubMed.
- M. B. Pepys and P. J. Butler, Biochem. Biophys. Res. Commun., 1987, 148, 308–13 CrossRef CAS.
- S. M. Breathnach, H. Kofler, N. Sepp, J. Ashworth, D. Woodrow, M. B. Pepys and H. Hintner, J. Exp. Med., 1989, 170, 1433–8 CrossRef CAS.
- P. S. Hicks, L. Saunero-Nava, T. W. Du Clos and C. Mold, J. Immunol., 1992, 149, 3689–94 CAS.
- S. M. Breathnach, S. M. Melrose, B. Bhogal, F. C. de Beer, R. F. Dyck, G. Tennent, M. M. Black and M. B. Pepys, Nature, 1981, 293, 652–4 CrossRef CAS.
- R. F. Dyck, C. M. Lockwood, M. Kershaw, N. McHugh, V. C. Duance, M. L. Baltz and M. B. Pepys, J. Exp. Med., 1980, 152, 1162–74 CrossRef CAS.
- M. B. Pepys, R. F. Dyck, F. C. de Beer, M. Skinner and A. S. Cohen, Clin. Exp. Immunol., 1979, 38, 284–93 CAS.
- S. M. Breathnach, B. Bhogal, R. F. Dyck, F. C. De Beer, M. M. Black and M. B. Pepys, Br. J. Dermatol., 1981, 105, 115–24 CrossRef CAS.
- F. Coria, E. Castano, F. Prelli, M. Larrondo-Lillo, S. van Duinen, M. L. Shelanski and B. Frangione, Lab. Invest., 1988, 58, 454–8 Search PubMed.
- R. N. Kalaria, P. G. Galloway and G. Perry, Neuropathol. Appl. Neurobiol., 1991, 17, 189–201 CrossRef CAS.
- G. C. Yang, R. Nieto, I. Stachura and G. R. Gallo, Am. J. Pathol., 1992, 141, 409–19 Search PubMed.
- M. Botto, P. N. Hawkins, M. C. M. Bickerstaff, J. Herbert, A. E. Bygrave, A. McBride, W. L. Hutchinson, G. A. Tennent, M. J. Walport and M. B. Pepys, Nat. Med., 1997, 3, 855–859 CrossRef CAS.
- C. M. Kinoshita, A. T. Gewurz, J. N. Siegel, S. C. Ying, T. E. Hugli, J. E. Coe, R. K. Gupta, R. Huckman and H. Gewurz, Protein Sci., 1992, 1, 700–9 CrossRef CAS.
- G. A. Tennent, L. B. Lovat and M. B. Pepys, Proc. Natl. Acad. Sci. USA, 1995, 92, 4299–4303 CrossRef CAS.
- T. Oda, G. M. Pasinetti, H. H. Osterburg, C. Anderson, S. A. Johnson and C. E. Finch, Biochem. Biophys. Res. Commun., 1994, 204, 1131–1136 CrossRef CAS.
- S. R. Hughes, O. Khorkova, S. Goyal, J. Knaeblein, J. Heroux, N. G. Riedel and S. Sahasrabudhe, Proc. Natl. Acad. Sci. USA, 1998, 95, 3275–80 CrossRef CAS.
- E. Matsubara, C. Soto, S. Governale, B. Frangione and J. Ghiso, Biochem. J., 1996, 316, 671–9 CAS.
- S. McHattie and N. Edington, Biochem. Biophys. Res. Commun., 1999, 259, 336–40 CrossRef CAS.
- D. M. Hatters, M. R. Wilson, S. B. Easterbrook-Smith and G. J. Howlett, Euro. J., 2002, Biochem(269), 2789–94 Search PubMed.
- J. R. Kumita, S. Poon, G. L. Caddy, C. L. Hagan, M. Dumoulin, J. J. Yerbury, E. M. Stewart, C. V. Robinson, M. R. Wilson and C. M. Dobson, J. Mol. Biol., 2007, 369, 157–67 CrossRef CAS.
- S. Janciauskiene, P. G. Defrutos, E. Carlemalm, B. Dahlback and S. Eriksson, J. Biol. Chem., 1995, 270, 26041–26044 CrossRef CAS.
- D. Canet, A. M. Last, P. Tito, M. Sunde, A. Spencer, D. B. Archer, C. Redfield, C. V. Robinson and C. M. Dobson, Nat. Struct. Biol., 2002, 9, 308–15 CrossRef CAS.
- G. Marcon, G. Plakoutsi, C. Canale, A. Relini, N. Taddei, C. M. Dobson, G. Ramponi and F. Chiti, J. Mol. Biol., 2005, 347, 323–35 CrossRef CAS.
- A. Gouin-Charnet, G. Mourad and A. Argiles, Biochem. Biophys. Res. Commun., 1997, 231, 48–51 CrossRef CAS.
- H. Hamazaki, Biochem. Biophys. Res. Commun., 1995, 211, 349–53 CrossRef CAS.
- M. D. Kirkitadze, G. Bitan and D. B. Teplow, J. Neurosci. Res., 2002, 69, 567–577 CrossRef CAS.
- J. P. Lee, C. Gerin, V. P. Bindokas, R. Miller, G. Ghadge and R. P. Roos, J. Neurochem., 2002, 82, 1229–38 CrossRef CAS.
- M. J. Volles and P. T. Lansbury, Jr., Biochemistry, 2003, 42, 7871–8 CrossRef CAS.
- M. M. Sousa, I. Cardoso, R. Fernandes, A. Guimaraes and M. J. Saraiva, Am. J. Pathol., 2001, 159, 1993–2000 Search PubMed.
- T. Oda, P. Wals, H. H. Osterburg, S. A. Johnson, G. M. Pasinetti, T. E. Morgan, I. Rozovsky, W. B. Stine, S. W. Snyder and T. F. Holzman, Exp. Neurol., 1995, 136, 22–31 CrossRef CAS.
- L. N. Boggs, K. S. Fuson, M. Baez, L. Churgay, D. McClure, G. Becker and P. C. May, J. Neurochem., 1996, 67, 1324–7 CAS.
- G. M. Lauro, C. Fabrizi, R. Businaro, L. Fumagalli, S. Torelli and F. Gremo, Ital. J. Neurol. Sci., 1992, 13, 661–5 CrossRef CAS.
- I. H. Zwain, J. Grima and C. Y. Cheng, Mol. Cell. Neurosci., 1994, 5, 229–237 CrossRef CAS.
- L. Klimaschewski, N. Obermuller and R. Witzgall, Cell Tissue Res., 2001, 306, 209–16 CrossRef CAS.
- B. V. Zlokovic, Life Sci., 1996, 59, 1483–97 CrossRef CAS.
- M. Z. Kounnas, C. C. Haudenschild, D. K. Strickland and W. S. Argraves, In Vivo, 1994, 8, 343–51 Search PubMed.
- J. E. Donahue, S. L. Flaherty, C. E. Johanson and J. A. Duncan, Acta Neuropathol. (Berlin), 2006, 112, 405–15 Search PubMed.
- K. Arelin, A. Kinoshita, C. M. Whelan, M. C. Irizarry, G. W. Rebeck, D. K. Strickland and B. T. Hyman, Mol. Brain Res., 2002, 104, 38–46 CrossRef CAS.
- V. Laporte, Y. Lombard, R. Levy-Benezra, C. Tranchant, P. Poindron and J. M. Warter, J. Leukoc. Biol., 2004, 76, 451–61 Search PubMed.
- J. Ghiso, E. Matsubara, A. Koudinov, N. H. Choimiura, M. Tomita, T. Wisniewski and B. Frangione, Biochem. J., 1993, 293, 27–30 CAS.
- M. Shibata, S. Yamada, S. R. Kumar, M. Calero, J. Bading, B. Frangione, D. M. Holtzman, C. A. Miller, D. K. Strickland, J. Ghiso and B. V. Zlokovic, J. Clin. Invest., 2000, 106, 1489–99 CrossRef CAS.
- R. D. Bell, A. P. Sagare, A. E. Friedman, G. S. Bedi, D. M. Holtzman, R. Deane and B. V. Zlokovic, J. Cereb. Blood Flow Metab., 2007, 27, 909–18 Search PubMed.
- H. Fukumoto, A. Asami-Odaka, N. Suzuki, H. Shimada, Y. Ihara and T. Iwatsubo, Am. J. Pathol., 1996, 148, 259–65 Search PubMed.
- R. B. DeMattos, M. A. O'dell, M. Parsadanian, J. W. Taylor, J. A. K. Harmony, K. R. Bales, S. M. Paul, B. J. Aronow and D. M. Holtzman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10843–10848 CrossRef CAS.
- R. B. DeMattos, J. R. Cirrito, M. Parsadanian, P. C. May, J. W. Taylor, J. A. K. Harmony, B. J. Aronow, K. R. Bales, S. M. Paul and D. M. Holtzman, Neuron, 2004, 41, 193–202 CrossRef CAS.
- J. Shorter and S. Lindquist, Science, 2004, 304, 1793–7 CrossRef CAS.
- J. R. Duguid, C. W. Bohmont, N. G. Liu and W. W. Tourtellotte, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7260–4 CrossRef CAS.
- P. C. May, S. A. Johnson, J. Poirier, M. Lampert-Etchells and C. E. Finch, Can. J. Neurol. Sci., 1989, 16, 473–6 Search PubMed.
- K. Sasaki, K. Doh-ura, J. W. Ironside and T. Iwaki, Acta Neuropathol., 2002, 103, 199–208 CrossRef CAS.
- A. M. Lidstrom, C. Hesse, L. Rosengren, P. Fredman, P. Davidsson and K. Blennow, J. Alzheimers Dis., 2001, 3, 435–442 Search PubMed.
- C. Piubelli, M. Zanusso, G. Fiorini, A. Milli, E. Fasoli, S. Monaco and P. G. Righetti, Proteomics, 2006, 6, S256–61 CrossRef.
- M. Zenkel, F. E. Kruse, A. G. Junemann, G. O. Naumann and U. Schlotzer-Schrehardt, Invest. Ophthalmol. Visual Sci., 2006, 47, 1982–90 CrossRef.
- T. Aoyagi, T. Wada, F. Kojima, M. Nagai, S. Harada, T. Takeuchi, K. Isse, M. Ogura, M. Hamamoto and K. Tanaka, Experientia, 1992, 48, 656–9 CrossRef CAS.
- F. Licastro, M. C. Morini, E. Polazzi and L. J. Davis, J. Neuroimmunol., 1995, 57, 71–5 CrossRef CAS.
- K. Taddei, R. Clarnette, S. E. Gandy and R. N. Martins, Neurosci. Lett., 1997, 223, 29–32 CrossRef.
- R. Deane, Z. Wu, A. Sagare, J. Davis, S. Du Yan, K. Hamm, F. Xu, M. Parisi, B. LaRue, H. W. Hu, P. Spijkers, H. Guo, X. Song, P. J. Lenting, W. E. Van Nostrand and B. V. Zlokovic, Neuron, 2004, 43, 333–44 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |