Rapid microchip-based electrophoretic immunoassays for the detection of swine influenza virus†
David S.
Reichmuth
*a,
Serena K.
Wang‡
a,
Louise M.
Barrett§
a,
Daniel J.
Throckmorton
a,
Wayne
Einfeld
b and
Anup K.
Singh
a
aSandia National Laboratories, Chemical and Radiation Detection Laboratories, P.O. Box 969, Livermore, CA 94551, USA. E-mail: dreichm@sandia.gov; Fax: +1 (925) 294-3282
bSandia National Laboratories, Chemical and Biological Systems Dept., PO Box 5800, Albuquerque, NM 87185-0734, USA
First published on 19th June 2008
Abstract
Towards developing rapid and portable diagnostics for detecting zoonotic diseases, we have developed microchip-based electrophoretic immunoassays for sensitive and rapid detection of viruses. Two types of microchip-based electrophoretic immunoassays were developed. The initial assay used open channel electrophoresis and laser-induced fluorescence detection with a labeled antibody to detect influenza virus. However, this assay did not have adequate sensitivity to detect viruses at relevant concentrations for diagnostic applications. Hence, a novel assay was developed that allows simultaneous concentration and detection of viruses using a microfluidic chip with an integrated nanoporous membrane. The size-exclusion properties of the in situ polymerized polyacrylamide membrane are exploited to simultaneously concentrate viral particles and separate the virus/fluorescent antibody complex from the unbound antibody. The assay is performed in two simple steps—addition of fluorescently labeled antibodies to the sample, followed by concentration of antibody–virus complexes on a porous membrane. Excess antibodies are removed by electrophoresis through the membrane and the complex is then detected downstream of the membrane. This new assay detected inactivated swine influenza virus at a concentration four times lower than that of the open-channel electrophoresis assay. The total assay time, including device regeneration, is six minutes and requires <50 μl of sample. The filtration effect of the polymer membrane eliminates the need for washing, commonly required with surface-based immunoassays, increasing the speed of the assay. This assay is intended to form the core of a portable device for the diagnosis of high-consequence animal pathogens such as foot-and-mouth disease. The electrophoretic immunoassay format is rapid and simple while providing the necessary sensitivity for diagnosis of the illness state. This would allow the development of a portable, cost-effective, on-site diagnostic system for rapid screening of large populations of livestock, including sheep, pigs, cattle, and potentially birds.
Introduction
Intentional (agroterrorism) and unintentional spread of veterinary pathogens pose a great threat to the economy and, in the case of zoonotic diseases, to public health. Even in the case of diseases that are not a human health concern, the large economic consequences stimulate a desire for improved diagnosis and monitoring abilities. The foot-and-mouth disease (FMD) outbreak in the United Kingdom in 2001 is a prime example. During the 2001 outbreak, delays in detection and quarantine led to the slaughter of 6 million animals and over $10 billion in lost revenue and costs. It is evident from contrasting the effects of foot-and-mouth disease (FMD) outbreaks in the UK in 2001 and 2007 that early detection and intervention during an outbreak can reduce both economic impact1 and public hysteria. In the case of the 2007 outbreak, the disease appears to be confined to a much smaller number of animals. This is a result of aggressive implementation of quarantine, and most importantly by the self-reporting of diseased animals by farmers. Existing methods (PCR, ELISA, and virus isolation) to screen farm animals are slow and require extensive sample handling. Early detection and widespread screening is not possible therefore public and quarantined farms can have to wait over a week for confirmation of an outbreak. In addition to the problems with the analysis techniques, handling and transport time of samples can be significant since farm locations are spread over a large geographic area. Hence, there is an urgent need for inexpensive, portable devices that can be used for rapid on-site screening of farm animals for infectious diseases.2 This device would determine disease status at the animal pen, allowing “pen-side” diagnostics.We chose to use swine influenza virus as a model veterinary pathogen for this work. We based this decision on factors including the availability of reagents, safety of researchers and the environment, and relevance to veterinary medicine. Existing swine influenza virus detection methods include PCR/RT-PCR, ELISA, and viral isolation assays. The most important parameters to compare are the sensitivity of the assay (detection limit), specificity of the assay, and the assay speed. It is difficult to compare the reported detection limits of various techniques since establishing the absolute viral concentration in the reference samples is difficult. A common measure of influenza viral concentration is the 50% tissue culture infectious dose (TCID50) or the egg infectious dose (EID50). In this paper we use estimated TCID50 ml−1 values to report our results..
Viral isolation assays are considered the reference standard assay. However, culture requires significant laboratory resources and 1–2 weeks to receive a result. ELISA, lateral flow immunoassays, and other antibody-based assays are also available for diagnosis of swine influenza.3 The assays produce a result in 20–30 minutes and while they exhibit good specificity they generally have lower sensitivity (≅104 TCID50 ml−1) than viral isolation or PCR methods.4 Reverse transcriptase-PCR (RT-PCR) and real-time RT-PCR assays have been demonstrated for the detection of swine influenza at very low concentrations (<0.1 TCID50 ml−1). While RT-PCR shows the best sensitivity of all the available assays, RT-PCR assay requires several hours to complete, expensive equipment and trained staff. In addition the high sensitivity of RT-PCR may not be necessary5 since, infected swine nasal samples have been found to have concentrations of influenza viral particles in the range 103–105 TCID50 ml−1. Therefore, by using a device incorporating the assay presented in this paper it is feasible to perform assays at a much lower cost and quicker time while meeting the detection limits required.
On-chip assays using micron-sized fluidic channels have demonstrated the ability to perform rapid and sensitive biological assays,6,7 and it has been shown that microscale electrophoretic devices can be used for the analysis of viruses.8,9 In addition, the small dimensions of microdevices allows for the creation of portable analytic devices.10,11 We have previously demonstrated the ability to use laser-based photopolymerization to create polymer structures in microfluidic chips,12,13 and chip-based immunoassays have been demonstrated that allow sensitive detection of proteins.14,15 For this work, we chose to use an assay based on recognition of viral proteins using antibodies. Immunoassays can be designed to be highly specific and can be quicker than nucleic amplification-based methods. Microfluidic electrophoretic immunoassays have been developed for the detection of proteins, for toxin detection and for the assay of disease biomarkers.14,16 These methods rely on separation of a labeled antibody from a labeled-antibody/antigen complex by virtue of differences in electrophoretic mobility, similar to capillary-based immunoassays.17–19 Separation is achieved by electrophoresis through a polyacrylamide gel in a microchannel, and antigen concentration can be determined by comparing the amount of bound antibody to the unbound antibody. The existing electrophoretic immunoassays function by separating unbound from bound antibodies. We developed an on-chip immunoassay for viral particles using open channel separation. More significantly however, we developed a more sensitive and efficient assay format using a porous polymer which can both separate and concentrate viral particles. This porous polymer structure has pores large enough (∼10 nm) for facile transport of large proteins (including antibodies), but does not allow larger (∼80 nm) viral particles to pass. Using this filtration technique, further separation or washing of the antibody complex is not needed. When the assay is used with a fluorescent antibody, the presence and concentration of virus can be determined without further separation, reducing assay complexity and time. In addition, the viral particles are reversibly concentrated and held, which opens the possibility of using secondary antibody detection schemes for signal amplification. In this work we have demonstrated that the porous polymer-based assay can concentrate swine influenza viral particles in a manner that allows rapid detection on a microchip.
Methods
Chemicals and samples
Glacial acetic acid; 3-(trimethoxysilyl)propyl acrylate; 10mM phosphate buffered saline (PBS) and 30% (37.5 : 1 acrylamide/bisacrylamide) acrylamide were purchased from Sigma-Aldrich (St. Louis, MO). AlexaFluor 488 protein labeling kit was purchased from Invitrogen (Carlsbad, CA). 2,2′-Azobis-[2-methyl-N-(2-hydroxyethyl)propionamide] (VA-086) was made by Wako Chemicals (Richmond, VA). Inactivated swine influenza (A/1972/Iowa) and inactivated bovine enterovirus were purchased from USDA NVSL (National Veterinary Services Laboratory, Ames, IA) and were dialyzed against 10 mM PBS using a Pierce (Rockford, IL) Slidalzyer dialysis membrane with 10 kDa cutoff. The viral samples were received in an inactive state, making direct measurement of the TCID50 concentration impossible. Estimated values for the TCID50 were based on the TCID50 concentration prior to inactivation as reported by USDA NVSL. Mouse anti-influenza A antibody (clone A1, product #8257) was purchased from Chemicon (Temecula, CA) and labeled using the Alexafluor 488 protein labeling kit from Invitrogen.BioRad (Hercules, CA) Tris–glycine buffer (25mM Tris, pH 8.3, 192 mM glycine), with pH modified to 8.9 using sodium hydroxide, was used to dilute all samples and as the running buffer. Viral samples were prepared by adding 5 μl of labeled antibody (∼1 mg ml−1) to the dialyzed viral stock solution and then diluting with Tris–glycine buffer to a final volume of 100 μl. Prior to introduction to the microchip, samples were incubated for 5 minutes at room temperature.
Microchip and polymer element fabrication
Both the open channel electrophoresis chip and the viral concentration and detection microchip were fabricated from fused silica substrates using standard photolithography, wet etch, and bonding techniques by Caliper Life Sciences (Mountain View, CA). A nominal microchannel depth of 25 μm and width of 100 μm was used.The chip surfaces were treated to allow adhesion of polyacrylamide to the channel walls.20 A 2 : 3 : 5 mixture of 3-(trimethoxysilyl)propyl acrylate : glacial acetic acid : water was loaded into the microchannels and incubated for 30 min at room temperature. The chip was covered to inhibit evaporation during the incubation period. The treatment solution was removed using vacuum and the channels washed sequentially with 30% acetic acid and deionized water.
The viral concentration device used previously described polymer photopatterning techniques,12 which are briefly summarized here. A polyacrylamide mixture with a final concentration of 6% polyacrylamide was made by dilution of 30% polyacrylamide with Tris–glycine buffer (pH 8.3). Following dilution, 0.2 wt% of VA86 initiator was added. The mixture was sonicated under vacuum to remove dissolved gases. The vial containing the mixture was manually agitated to dislodge gas bubbles attached to the side of the vial.
The mixture was photopolymerized by projecting the output of a 10 mW, 355 nm laser (Teem Photonics, Wellesley, MA) onto the region of the microchip where polymerization was desired. The illuminated area was shaped with a series of lenses and a mechanical slit to an approximately 150 μm × 600 μm rectangular region, and the polymerization occurred after 5–30 s of exposure. Placement of the laser beam was visualized with a CCD camera fitted with a microscope objective. A nanoporous plug is created in a small section (100 μm wide × 25 μm deep × 150 long μm) of the microchannel (Fig. 1). The dimensions of the channel determine the width and depth of the element, while the length is controlled by the size of the illuminated area. Polymerization does not result in easily discernable visual change, but formation of a polyacrylamide plug can be determined by the inability to move liquid through the channel or by close inspection with a microscope. The polymerization time was determined using a series of increasing duration exposures until complete polymerization occurred. After polymerization, the channels without a polymer element were flushed with deionized water.
 | ||
| Fig. 1 (A) Schematic of microfluidic chip. Reservoir designations: S = Sample; SW = Sample waste; B = Buffer; W = Waste. (B) A 6% polyacrylamide plug is formed in a glass microchip by projecting a shaped UV laser beam onto the acrylamide-containing channel. (C) The virus and antibody are moved to the plug region by electrophoresis. (D) Fluorescence can be measured at the membrane or as the retained species are eluted towards the waste channel. (E) Labeled antibody complexed with viral particles is detected by using epifluoresence microscopy. The virus-labeled antibody complex can be seen concentrated at the gel element. | ||
Both the open channel chip and concentration chip had internal surfaces coated with a linear polyacrylamide coating to suppress bulk electroosmotic flow. The immunoassay is performed at a pH of 8.9 and in moderate counterion concentrations (<100 mM) so that significant electroosmotic flow will occur in an open uncoated silica channel.21 A 40% linear polyacrylamide solution was used with 0.2 wt% VA-086 initiator and the chip exposed to UV light for 5 minutes using a cross-linker (CL-1000, UVP, Upland, CA). After surface treatment, the chip was flushed with deionized water and refilled with Tris–glycine buffer. Microchips containing polymer elements were stored prior to use for up to 1 month in Tris–glycine buffer at 4 °C.
Electrophoresis
The microchip was held in a custom-fabricated Delrin (DuPont) manifold with integrated ∼100 μl fluid reservoirs, as described in Hatch et al.14Platinum electrodes were placed into the reservoirs and high voltage applied using a Sandia-fabricated high voltage power supply.22 The power supply was controlled using a PC running LabVIEW (National Instruments, Austin, TX).The fluid wells are named as shown in Fig. 1, with the sample loaded in the sample (S) well. In the open channel configuration, sample is loaded for 120 s into the offset T injector towards the sample waste (SW) well using an applied potential of 400 V at the SW well while grounding the S well. A voltage of 93 V was applied at well buffer (B) to prevent sample from entering the separation channel. During the 120 s separation, the sample moves down the separation channel using 800 V at the buffer waste (BW) reservoir while grounding the B well. Voltages of 325 V at SW and 275 V at S were used to pull back sample into the side channels. The effective lengths from the wells to the intersection are B 11 mm, BW 29 mm, S 17 mm, SW 8 mm.
In the case of viral concentration using a polymer structure, a two-step procedure is used to concentrate the viruses at the element and to elute the viruses. To concentrate, a potential of 400 V is applied at SW for 120 s and well S is grounded. This causes the negatively charged sample to electrophorese to the polyacrylamide element and concentrate. To remove the virus sample, waste well (W) is put to 800 V for 4 min and SW is grounded.
Imaging and laser induced fluorescence detection
Images and movies of viral concentration were captured using an inverted fluorescence microscope (IX70, Olympus Inc.) and an Optronics Microfire CCD camera.Laser induced fluorescence (LIF) detection was used to determine the concentration of fluorescently labeled antibodies. Excitation light at 488 nm (Omnichrome argon ion laser, Melles Griot, Carlsbad, CA) was frequency modulated using a mechanical chopper (400 Hz modulation) and reflected from a dichroic mirror (XF2010, New Focus, Inc, San Jose, CA) through a 40× microscope objective (5722-AH, New Focus) that defined the detection point on the chip. Focusing and two-axis alignment of the laser beam with respect to the separation channel relied upon a custom fixture mounted on a 3-axis translation stage. The detection point was placed on the channel leading to well W at a point 1 mm from the injection cross. Fluorescence was filtered spectrally (XF3084 notch filter) and spatially before detection by a Hamamatsu H120-05 photomultiplier tube (PMT) module. The signal from the PMT was demodulated using a lock-in amplifier (SR-830DSP, Stanford Research Systems, Sunnyvale, CA) and signal was collected via a custom LabVIEW data acquisition interface (National Instruments, Austin, TX). The raw data was smoothed using an 11-point moving average using a Matlab v6.1 script (The Mathworks Inc., Natick, MA). The difference between starting fluorescence value and the maximum value was recorded as the fluorescence due to concentration of the sample.
Results
Open-channel electrophoretic immunoassay
Open channel electrophoresis was used to detect swine influenza virus by observing formation of an antibody–antigen (virus particle) complex. The open channel electrophoresis shows binding of anti-influenza antibody to swine influenza virus by the conversion of labeled antibody to a slower moving complex (Fig. 2). The area of the complex peak increases with increasing concentration of virus, as seen in Fig. 2C. The open channel results demonstrate the ability of a microfluidic immunoassay to detect the presence of swine influenza.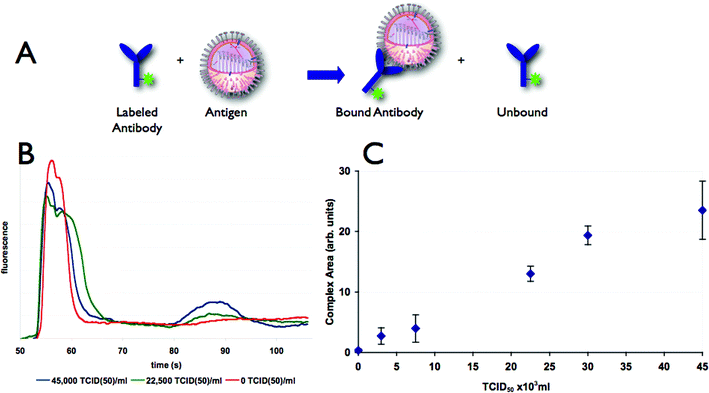 | ||
| Fig. 2 (A) Two fluorescent species are present in the channel, antibody and antibody/antigen complex. (B) Electropherograms showing separation of antibody (first peak) and antibody–virus complex (second peak) using an open channel separation. Mixtures with increasing amounts of swine influenza virus show an increase in the height of the labeled antibody–viron complex peak with a concomitant decrease in the peak height of the free labeled antibody peak. (C) The area of labeled antibody–virus complex peak increases with increasing quantity of swine influenza virus. Error bars indicate a two standard deviation range based on three replicates. | ||
However, this assay has several drawbacks. Foremost is the low sensitivity of the assay with a limit of detection (LOD) of 2750 TCID50 ml−1, where the LOD is defined as three times the standard deviation of the negative control sample. We also experienced difficulty in consistently reproducing data, with high day-to-day variations (data not shown). Possible causes include instability of the surface coating and the presence of pressure-driven flow due to reservoir fluid height differences. In our previous work with chip-based immunoassays,14,16,20 these problems were addressed by using fixed polyacrylamide gels in all of the channels. The structure of the gel suppresses hydrodynamic flow and also provides a uniform and stable surface inside the channels. However, gel-filled chips are incompatible with analysis of intact viruses, as the influenza virus has a radius of approximately 80 nm, while a 6% polyacrylamide gel has pores with an average size 8–12 nm.23,24 To improve the LOD, we implemented an integrated preconcentration-immunoassay described below.
Viral concentration enhancement and quantitation
To concentrate virus particles, we photoploymerized a porous membrane in the loading channel as depicted in Fig. 1. The porosity of gel was chosen such that the nanoporous polyacrylamide element has pores large enough to allow antibodies to pass through but small enough to exclude the influenza virus particles as they have diameters several times larger than the polymer pores. The 6% gel concentration is also a high enough concentration to give adequate mechanical rigidity for reuse of the microdevice. During the loading phase, sample is electrophoretically driven towards the porous element region, while the unbound antibodies pass through the porous element (Fig. 3A). The concentration of labeled antibody–viron complex is shown in Fig. 1, where the complex is seen as a highly fluorescent region at the edge of the gel element.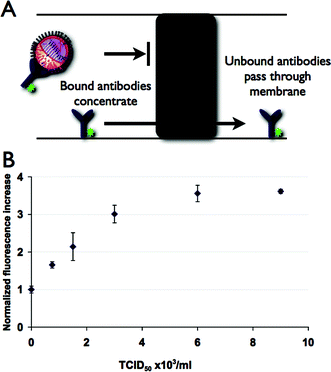 | ||
| Fig. 3 (A) Open channel or gel separations are usually required to differentiate bound and unbound antibodies, however an acrylamide membrane element allows discrimination without further separation. (B) Concentration of labeled antibody–virus complex at gel element is detected as an increase in fluorescence. For a fixed concentration time, the maximum fluorescence intensity grows as viral concentration is increased. Error bars indicate a two standard deviation range based on three replicates. | ||
After concentration, the concentrated plug of antibody–viron complex is then driven off the porous element by changing the voltage such that the sample is sent towards the waste channel. This concentrated plug is then detected at a point 1 mm down the channel using LIF. Downstream detection of the plug has the advantage of reduced background fluorescence as compared to direct measurement at the porous element due to antibody associated by non-specific adsorption to the porous element. The fluorescence intensity was found to increase with the concentration of viral particles in the sample (Fig. 3B). In addition, concentration at the porous element allowed detection of much lower concentrations of viruses as compared to the open channel electrophoretic separations. In order to verify that the presence of virus particles were not causing concentration of unbound antibody and that the antibody we used was specific for avian influenza virus we used bovine enterovirus (BEV) as a negative control. We observed no difference in fluorescence between the samples with BEV and antibody or antibody alone, demonstrating that the increase in fluorescence seen at the membrane when using the antibody and influenza virus is due to viral–antibody complexes and indicates that cross-reactivity is minimal.
Using this assay we have shown the ability to detect a range of viral concentrations 7.5 × 102 to 4.5 × 104 TCID50 ml−1, which are relevant concentrations for positive swine influenza samples,5 which have been reported to be in the range 103–105 TCID50 ml−1. The overall cycle time for the assay is less than six minutes and the assay only requires <50 μl of sample in the reservoirs. Since a very small amount of sample is consumed in each assay, repeated measurements with a small sample volume, was shown to be possible. If desired, lower concentration samples could be detected by increasing the length of the concentration step. We measured a LOD for the membrane-based assay of 610 TCID50 ml−1, more than a fourfold improvement as compared to the open channel assay.
This assay was repeated on a single chip/membrane for approximately 100 assays. The most common failure mode of the chip was accumulation of dust or other particulates at the membrane element. Field use of the chip would require filtration of samples to remove solids that would clog either the channel or membrane. The microchips described in this note were fabricated from fused silica and were reused due to cost considerations. However, the chip design is simple and could be replicated in plastic chips, which would allow the device to be used in a single-use disposable format.
Conclusions
We have demonstrated a rapid technique for the concentration and detection of influenza virus on chip. The chip architecture is simple, requiring photopolymerizing of an inexpensive polyacrylamide plug. The polyacrylamide plug serves multiple functions—(1) concentrates viral particles, (2) filters unwanted species in sample, and (3) permits separation of bound and unbound complexes, by allowing excess antibody to go through. With integrated preconcentration, a LOD of 610 TCID50 ml−1 could be obtained in less than six minutes.In this study, the virus was assayed while diluted in buffer. Practical application of this technique will have to address the effects of sample matrix on the reliability and sensitivity of the assay. Also, filtration will be required to remove large particles (> micrometer-sized) that could block a microfluidic channel, as samples collected at a farm from an animal will undoubtedly contain particles. The integration of this assay into a portable, field-ready device is possible, as it is compatible with the components developed at our laboratory by Renzi et al.22
Methods are needed to fill an urgent need for portable devices for screening cattle and other livestock and to help contain the spread of disease to other animals and people. This method shows promise for the rapid and portable detection of viruses for livestock screening applications. Future work will require testing the device against other important livestock diseases such as foot and mouth disease.
Notes and references
- N. M. Ferguson, C. A. Donnelly and R. M. Anderson, Nature, 2001, 413, 542–548 CrossRef CAS.
- Infectious Diseases of Livestock, Royal Society, London, 2002 Search PubMed.
- B. W. Lee, R. F. Bey, M. J. Baarsch and R. R. Simonson, J. Vet. Diagn. Invest., 1993, 5, 510–515 Search PubMed.
- J. A. Richt, K. M. Lager, D. F. Clouser, E. Spackman, D. L. Suarez and K. J. Yoon, J. Vet. Diagn. Invest., 2004, 16, 367–373 Search PubMed.
- P. Lekcharoensuk, K. M. Lager, R. Vemulapalli, M. Woodruff, A. L. Vincent and J. A. Richt, Emerg. Infect. Dis., 2006, 12, 787–794 Search PubMed.
- E. A. Lipman, B. Schuler, O. Bakajin and W. A. Eaton, Science, 2003, 301, 1233–1235 CrossRef CAS.
- L. Warren, D. Bryder, I. L. Weissman and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17807–17812 CrossRef CAS.
- V. Kolivoska, V. U. Weiss, L. Kremser, B. Gas, D. Blass and E. Kenndler, Electrophoresis, 2007, 28, 4734–4740 CrossRef CAS.
- L. Kremser, G. Bilek, D. Blass and E. Kenndler, J. Sep. Sci., 2007, 30, 1704–1713 CrossRef CAS.
- A. E. Herr, A. V. Hatch, D. J. Throckmorton, H. M. Tran, J. S. Brennan, W. V. Giannobile and A. K. Singh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5268–5273 CrossRef CAS.
- A. M. Skelley, J. R. Scherer, A. D. Aubrey, W. H. Grover, R. H. Ivester, P. Ehrenfreund, F. J. Grunthaner, J. L. Bada and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1041–1046 CrossRef CAS.
- D. S. Reichmuth, T. J. Shepodd and B. J. Kirby, Anal. Chem., 2004, 76, 5063–5068 CrossRef CAS.
- B. J. Kirby, D. S. Reichmuth, R. F. Renzi, T. J. Shepodd and B. W. Wiedenman, Lab Chip, 2005, 5, 184–190 RSC.
- A. V. Hatch, A. E. Herr, D. J. Throckmorton, J. S. Brennan and A. K. Singh, Anal. Chem., 2006, 78, 4976–4984 CrossRef CAS.
- A. E. Herr, A. V. Hatch, W. V. Giannobile, D. J. Throckmorton, H. M. Tran, J. S. Brennan and A. K. Singh, Ann. N. Y. Acad. Sci., 2007, 1098, 362–374 CrossRef CAS.
- A. E. Herr, D. J. Throckmorton, A. A. Davenport and A. K. Singh, Anal. Chem., 2005, 77, 585–590 CrossRef CAS.
- R. G. Nielsen, E. C. Rickard, P. F. Santa, D. A. Sharknas and G. S. Sittampalam, J. Chromatogr., 1991, 539, 177–185 CrossRef CAS.
- N. M. Schultz and R. T. Kennedy, Anal. Chem., 1993, 65, 3161–3165 CrossRef CAS.
- K. Shimura and B. L. Karger, Anal. Chem., 1994, 66, 9–15 CrossRef CAS.
- A. E. Herr and A. K. Singh, Anal. Chem., 2004, 76, 4727–4733 CrossRef CAS.
- B. J. Kirby and E. F. Hasselbrink, Jr., Electrophoresis, 2004, 25, 187–202 CrossRef CAS.
- R. F. Renzi, J. Stamps, B. A. Horn, S. Ferko, V. A. VanderNoot, J. A. A. West, R. Crocker, B. Wiedenman, D. Yee and J. A. Fruetel, Anal. Chem., 2005, 77, 435–441 CrossRef CAS.
- R. C. Lo and V. M. Ugaz, Electrophoresis, 2006, 27, 373–386 CrossRef CAS.
- V. Ugaz, Texas A & M University, Personal communication, 2007.
Footnotes |
| † The HTML version of this article has been enhanced with colour images. |
| ‡ Current address: Bioengineering Department, University of California, Berkeley, CA, 94720, USA. |
| § Current address: Biomedical Diagnostics Institute, National Centre for Sensor Research & Engineering Building, Dublin City University, Glasnevin, Dublin 9, Ireland. |
| This journal is © The Royal Society of Chemistry 2008 |